

Abstract
CCAAT enhancer binding protein-delta (C/EBPδ) is a transcription factor that regulates inflammatory processes mediating bystander neuronal injury and CNS autoimmune inflammatory disease. The mechanism of the involvement of C/EBPδ in these processes remains to be determined. Here, we examined the cellular source(s) and mechanisms by which C/EBPδ may be involved in an animal model of multiple sclerosis. Mice deficient in C/EBPδ expression exhibited less severe clinical disease than wild-type littermates in response to induction of experimental autoimmune encephalomyelitis (EAE) by vaccination with a myelin oligodendrocyte glycoprotein (MOG) fragment. This reduction in EAE severity was associated with a significant alteration in the complement of major CNS T-helper (Th) cell subtypes throughout disease, manifest as reduced ratios of Th17 cells to regulatory T-cells (Tregs). Studies in bone marrow chimeric mice indicated that C/EBPδ expression by peripherally derived immune cells mediates C/EBPδ involvement in EAE. Follow up in vitro and in vivo examination of dendritic cell (DC) mediated Th-cell development suggests that C/EBPδ suppresses DC expression of interleukin-10 (IL-10), favoring Th17 over Treg development. In vitro and in vivo blockade of IL-10 signaling attenuated the effect of reduced C/EBPδ expression by DCs on Th17:Treg ratios. These findings identify C/EBPδ as an important DC transcription factor in CNS autoimmune inflammatory disease by virtue of its capacity to alter the Th17:Treg balance in an IL-10 dependent fashion.
Introduction
Multiple sclerosis (MS) is a CNS inflammatory disease, probably involving a myelin-specific autoimmune attack. The dominant animal model of MS, experimental autoimmune encephalomyelitis (EAE), is induced by vaccination with myelin antigens. Our recent mapping of inflammatory foci throughout EAE suggested initiation of CNS pathology at meningeal surfaces of the CNS followed by recruitment of circulating T-helper (Th) cells and antigen-presenting cells (APCs) (Brown and Sawchenko, 2007). While various T-cell subtypes can mediate EAE (Huseby et al., 2001), functional APCs are indispensable for disease induction (Greter et al., 2005). The dendritic cell (DC) is an APC that facilitates CNS T-cell entry, reactivation, and differentiation in EAE (Bailey et al., 2007).
Examination of T-cell development in EAE has defined Th-cell subsets. The characterization of interleukin-17 (IL-17) expressing T-cells, so-called Th17 cells, indicated they were the prime mediators of EAE (Langrish et al., 2005). However, subsequent work suggests that interferon-γ producing Th1 cells are also involved (Domingues et al., 2010). Th-cell differentiation is highly regulated; for example, Th1 cells retard the development of Th17 cells, which share developmental cues with T-regulatory cells (Tregs) that suppress destructive autoinflammation. In the presence of TGFβ, differentiation of Tregs proceeds at the expense of Th17 cells, and Tregs can suppress both Th1 and Th17 cell activity. However, if TGFβ is present with IL-6, Th17 development ensues (Bettelli et al., 2006). Therefore, IL-6 production would be expected to regulate both Treg and Th17 cell differentiation. CNS Tregs suppress immune responses by secreting IL-10 (Lavasani et al., 2010), and DC secretion of IL-10 may also promote Treg development (Rutella et al., 2006) and directly suppress inflammatory Th-cells (Kao et al., 2010). Consequently, regulation of IL-6 and/or IL-10 could directly impact Th-cell function and development.
Multiple DC factors upregulate IL-6 (Lu et al., 2009), including toll-like receptor (TLR) activation (Waldner et al., 2004) and exposure to proinflammatory cytokines such as IL-1, TNFα (Matzinger, 2002), and IL-17 (Steiner et al., 2003). Proinflammatory cytokines are upregulated by transcription factors that play pivotal roles in inflammatory responses. Many cytokine promoter regions have binding sites for the basic-leucine zipper (bZIP) C/EBP family of transcriptional regulators (Wedel and Ziegler-Heitbrock, 1995). In the CNS, C/EBPα, β, δ, and C/EBP homologous protein are the most abundantly expressed isoforms of this family (Sandhir and Berman, 2010). These transcription factors have been implicated in inflammatory processes accompanying neurodegenerative diseases (Cardinaux et al., 2000; Ejarque-Ortiz et al., 2007) and the sequelae of brain injury (Cortés-Canteli et al., 2004). One family member, C/EBPδ, is upregulated in mouse models of brain injury (Sandhir and Berman, 2010) and in Alzheimer's disease (Li et al., 2004).
Recently, C/EBPδ was defined as a major regulator of IL-6 production (Litvak et al., 2009). This bZIP transcription factor mediates IL-17 receptor signaling (Shen et al., 2006) and upregulates IL-6 in the context of APC exposure to TNFα and IL-1 (Juan et al., 1993; Ruddy et al., 2004). However, C/EBPδ can also suppress gene transcription (Lacorte et al., 1997a). Therefore, C/EBPδ may upregulate inflammatory and/or suppress anti-inflammatory cytokine production in response to inflammatory insults. C/EBPδ mRNA is overexpressed in the MS-afflicted CNS (Lock et al., 2002; Tzartos et al., 2008), and mononuclear cells isolated from CSF of MS patients have increased C/EBP binding sites (Brynedal et al., 2010). Additionally, microarray analysis of peripheral blood mononuclear cells from MS patients showed increased levels of C/EBPδ associated with increasing disease activity and progression (Riveros et al., 2010). As the evidence summarized above suggested that C/EBPδ expression by DCs may regulate CNS Th-cell differentiation, we examined the role of C/EBPδ in EAE, where the balance between Th17 and Treg cells is an important determinant of disease severity (Korn et al., 2007).
Materials and Methods
Mice.
Female C57BL/6 mice, 6–8 weeks of age, were purchased from the Animal Resource Centre. C/EBPδ−/− (C57BL/6 background) mice were a kind gift from Dr. Esta Sterneck (NCI, Fredrick, MD) (Sterneck et al., 1998). 2D2 transgenic mice (C57BL/6 background) were a kind gift from Dr. Vijay Kuchroo (Harvard Medical School, Boston, MA) (Korn et al., 2007). Hemizygous transgenic mice expressing GFP under control of the chicken β-actin promoter and cytomegalovirus enhancer were initially purchased from Jackson Laboratories. A colony was then established and maintained on a C57B/6 background (Vallières and Sawchenko, 2003). All mice were maintained on a 12/12 h light/dark cycle with standard chow and water ad libitum available. All procedures were approved by the animal welfare committee at the Salk Institute for Biological Studies or the Garvan Institute–St Vincent's Hospital Animal Ethics Committee.
Induction of EAE.
EAE was induced and clinically scored as previously described (Brown and Sawchenko, 2007) with minor variation. Briefly, mice were immunized with 100 μg of MOG35–55 peptide in complete Freund's adjuvant supplemented with 1 mg M. tuberculosis (H37Ra; Difco) in a 100 μl total volume, over two sites, one in each flank, subcutaneously. All animals received 200 ng (i.p.) of pertussis toxin (List Biologicals) on the day of immunization and 2 d later.
LPS treatment of wild-type and C/EBPδ−/− mice.
Wild-type (wt) and C/EBPδ−/− animals were treated with LPS (50 μg/kg; Escherichia coli 055:B5, Sigma, in 100 μl, i.p.) and killed 4 h later. Mononuclear cells from spleens were harvested and isolated as described below.
Generation of polyclonal anti-C/EBPδ antibody.
Immunohistochemistry performed using commercially available anti-C/EBPδ antisera revealed significant nonspecific staining in C/EBPδ−/− animals, indicating that these sera were unsuitable for further use. Therefore, we generated a rabbit polyclonal anti-C/EBPδ peptide antiserum. A peptide fragment corresponding to amino acids 7–32 of C/EBPδ (SDLSPVRGTPWPTEPAAFYEPGRVDK-NH2 of C/EBPδ) was generously synthesized by Dr. Jean Rivier (The Salk Institute) using solid phase methodology. It was purified by HPLC and characterized using mass spectrometry. The antiserum against C/EBPδ was raised in rabbits immunized against the synthetic peptide coupled to keyhole limpet hemocyanin using a protocol detailed previously (Vaughan et al., 1989). Antiserum validation studies demonstrated a single, ∼28 KDa band on Western blots of nuclear extracts from LPS-stimulated astrocytes and no immunolabeling in sections of multiple tissues from C/EBPδ−/− animals treated with LPS, or in the EAE-afflicted CNS (data not shown).
Immunohistochemistry.
Dual immunoperoxidase labeling was performed as previously described (Brown and Sawchenko, 2007), with minor variations. Briefly, tissue sections were incubated in anti-C/EBPδ (1:50,000) in 2% donkey serum and 0.3% Triton X-100, followed by biotinylated secondary antiserum (raised in donkey; 1:200; Jackson ImmunoResearch) and avidin-biotin complex and then developed using a nickel-enhanced diaminobenzidine chromogen, yielding a black precipitate. Sections were then incubated with mouse-derived anti-CD11c (1:2000; clone N418; eBioscience), anti-MHC class II (1:2000; clone IBL-5/22; Millipore Bioscience Research Reagents), or anti-GFAP (1:2000, clone 2.2B10; Zymed) overnight under the same conditions as for the initial primary antibody and processed and developed in DAB without nickel-enhancement to yield a brown reaction product.
In situ hybridization.
The C/EBPδ riboprobe was generated as described by Reyes et al. (2003). In situ hybridization was performed using 35S-labeled sense (control) and antisense cRNA probes. Slides were digested with 0.1–10 μg/ml proteinase K for 30 min at 37°C. Probes were labeled to specific activities of 1–3 × 109 dpm/μg and applied to the slide at concentrations of 107 cpm/ml, overnight at 56°C in a solution containing 50% formamide, 0.3 m NaCl, 10 mm Tris, 1 mm EDTA, 0.05% tRNA, 10 mm dithiothreitol, 1 Denhardt's solution, and 10% dextran sulfate, after which they were treated with 20 μg/ml ribonuclease A for 30 min at 37°C and washed in 15 mm NaCl/1.5 mm sodium citrate at 65–68°C. Slides were then dehydrated and exposed to x-ray films (β-Max; Eastman Kodak) for 24 h. They were coated with Eastman Kodak NTB-2 liquid emulsion and exposed at 4°C for 15–30 d, as determined by the strength of signal on film. Slides were developed with Eastman Kodak D-19 and fixed with Eastman Kodak rapid fixer.
Isolation of spleen and CNS mononuclear cells.
CNS and spleen were removed from mice and cut into 2 mm pieces before digestion with collagenase D (2.5 mg/ml; Roche Applied Science) and DNase I (1 mg/ml; Roche Applied Science) in R10 media (RPMI 1640 medium; Life Technologies), supplemented with 10% (v/v) FCS (GeneTex), 2 mm l-glutamine (Life Technologies), 100 U/ml penicillin (Life Technologies), 100 μg/ml streptomycin (Life Technologies), and 50 μm 2-mercaptoethanol (Sigma) for 45 min at 37°C. Tissues were then passed through a cell strainer (100 μm; BD Biosciences), followed by a Percoll gradient (70%/37%; Pharmacia) and centrifugation at 350 × g.
Generation of bone marrow-derived DCs.
Bone marrow from wt and C/EBPδ−/− mice was harvested by flushing femurs, and cells were diluted to a concentration of 3 × 106/ml and cultured in presence of 300 ng/ml recombinant murine fms-like tyrosine kinase 3 ligand (FLT3; ProTech) as described previously (Naik et al., 2010). After 9 d in culture with FLT3, non-adherent and loosely adherent cells were removed from the flask with PBS at room temperature, replated without FLT3 and cultured for 4 h in the presence or absence of MOG35–55 peptide (10 μg/ml; ProTech). To generate matured DCs, cells were treated for 4 h with LPS (10 μg/ml; Sigma). Cells were and washed twice with cold PBS-5%FCS buffer before use as described previously (Zozulya et al., 2009).
In vitro T-cell assays.
Bone marrow-derived DCs (BMDCs) were used as DCs in an in vitro MOG antigen-specific T-cell activation and differentiation assay as described by Bailey et al. (2007) with minor modifications. Briefly, 1 × 107 CD4+ cells from the lymph nodes of 2D2 TCR mice were isolated with negative and positive magnetic selection according to the manufacturer's instructions (CD4+ T cell isolation kit; Milteny Biotec). BMDCs were cocultured with 2D2 CD4+ cells at a DC:T-cell ratio of 1:5 in presence of TGFβ alone (2 ng/ml; PeroTech) or TGFβ and IL-6 (20 ng/ml; PeroTech) or vehicle. For IL-10R blockade, anti-IL-10R (20 ng/ml; NA/LE rat anti-mouse CD210; 1B1.3a, BD Pharminogen) or isotype antibody (20 ng/ml; NA/LE rat IgG1, k isotype control; R3–34, BD Pharminogen) or vehicle was present throughout culture. Each condition was performed in triplicate in 96-U-well plates (Costar Corning) for 5 d at 37°C in 5% CO2 humidified atmosphere. Cells were then processed as described for flow cytometry analysis.
Intracerebral injection of BMDCs.
2D2 mice were subjected to injection of wt or C/EBPδ−/− MOG-presenting BMDCs (2.5 × 105) into the frontal cortex at position 1.70 mm rostral to bregma, 1.5 mm lateral to the midline, and 1.60 mm beneath the dura, as described previously (Zozulya et al., 2009). These injections were compared with ones involving matured, nonantigen-pulsed BMDCs and acellular vehicle control injections. To block IL-10 signaling, recipient mice received injections of anti-IL-10R (250 μg/mouse, i.p., every other day) commencing the day before DC injection. At day 5, brain mononuclear cells were harvested as above for examination.
Cell labeling for FACS analysis.
Cells from in vitro coculture experiments or isolated from tissues were prepared for FACS analysis as described previously (Korn et al., 2007). Briefly, for T-cell phenotype determination, cells were stimulated with phorbol 12-myristate 13-acetate (PMA; 50 ng/ml, Sigma), ionomycin (1 μg/ml, Sigma), and monensin (GolgiStop 1 μl/ml; BD Biosciences) at 37°C, in a humidified 5% CO2 atmosphere for 4 h before surface antibody labeling. For examination of DCs, cells were labeled with anti-CD45, anti-CD3, anti-CD11c, anti-CD11b, and anti-B220, and cell types were identified as per Bailey et al. (2007). For antigen-specific T-cell phenotype analysis, cells were labeled for anti-CD45, anti-CD3, anti-CD4, and anti-Vβ11, followed by fixation and permeabilization using Cytofix/Cytoperm and Perm/Wash buffer from BD Biosciences according to the manufacturer's instructions. Cells were incubated with antibodies for IL-17, IFNγ, and FOXP3 (1:100) for 20 min for intracellular labeling before flow cytometric analysis.
For C/EBPδ intracellular labeling, cells were incubated with rabbit-derived C/EBPδ antiserum or normal rabbit serum as an isotype control (both at 1:50,000) for 30 min. This was followed by incubation with FITC-conjugated-anti-rabbit IgG (1:200; Jackson ImmunoResearch Laboratories) at 4°C for 20 min. Cells were then washed twice. All flow cytometric analyses were performed on a LSRII cytometer (BD Biosciences) and were analyzed with FACSDiva software (BD Biosciences) or FlowJo software (TreeStar).
Generation of bone marrow chimerae.
Bone marrow transplantation was performed as previously described (Vallières and Sawchenko, 2003). Bone marrow was harvested as above. Recipient mice were exposed to 10 gray total body irradiation using a cobalt-60 source, administered in two equal fractions 3 h apart to minimize gastrointestinal toxicity. Approximately 24 h later, the animals were injected intravenously with 1 × 107 bone marrow cells freshly collected from donor mice. Irradiated mice transplanted with this suspension were housed in autoclaved cages and treated with antibiotics (Sulfatrim; 175 mg/250 ml of drinking water given for 3 d before and 4 weeks after irradiation).
Multiplex cytokine immunoassay.
A cytometric bead array immunoassay (BD Bioscience) was used in accordance with the manufacturer's protocol to measure the levels of inflammatory cytokines (IL-1β, IL-2, IL-4, IL-6, TGFβ, IL-10, IL-12, IL-23) in culture supernatants from T cell in vitro assays. Data were analyzed with the FCAP array software (BD Bioscience).
IL-10 and C/EBPδ qPCR.
Total RNA was isolated from splenocytes or BMDCs using TRIZOL and Pure-link RNA mini Kit (Invitrogen) and from cell cultures using RNeasy extraction kit (Qiagen). The synthesis of cDNA was performed using reverse transcriptase (RT), avian myeloblastis virus (Roche). RT-PCR was performed using the SensiMix Sybr Kit (Bioline). The RNA expression of C/EBPδ [forward (F): 5′-GCTCAAGCGCGAACCCGACT-3′, reverse (R): 5′-GCCGCCAAGCTCACCACTGT-3′] and IL-10 (F: 5′-TGAATTCCCTGGGTGAGAAG-3′, R: 5′-GCCTTGCTCTTATTTTCACAGG-3′) with product sizes of 99 and 88 bp respectively was normalized to β-actin (F: 5′-TGACAGGATGCAGAAGGAGATTACTG-3′, R: 5′-CCACCGATCCACACAGAGTACTTG-3′) with a product size of 101 bp. RT-negative controls were used to control for the amplification of DNA. Amplification and data analysis were performed on a LightCycler 480 II and associated software (Roche).
Statistical analysis.
To determine whether differences between groups were significant, Mann–Whitney U test was performed. Where repeated measures were made over time, as occurred in the assessment of EAE disease, repeated-measures ANOVA was performed. p < 0.05 was considered significant. All analysis was performed using STATA version 11.0 (StataCorp).
Results
C/EBPδ is upregulated in DCs and astrocytes in EAE
The disposition of central C/EBPδ expression in EAE was characterized histochemically in cohorts of mice (n = 6) killed at 10 d (preclinical) or 21 d (peak disease) after vaccination; tissue from untreated controls was processed similarly for comparison. Mice in the preclinical phase of EAE development (10 d) displayed marked and consistent upregulation of C/EBPδ mRNA expression in the meninges and perivascular regions, areas in which DCs have been reported to reside (Bulloch et al., 2008) and which showed no evidence of positive labeling in nonvaccinated controls (Fig. 1A). At the peak of disease (21 d), C/EBPδ mRNA expression in these same areas was increased, and was also seen in isolated cells in the CNS parenchyma (Fig. 1A). To identify the cell type(s) expressing C/EBPδ protein, dual immunolabeling with cell-specific markers was used.
Figure 1.
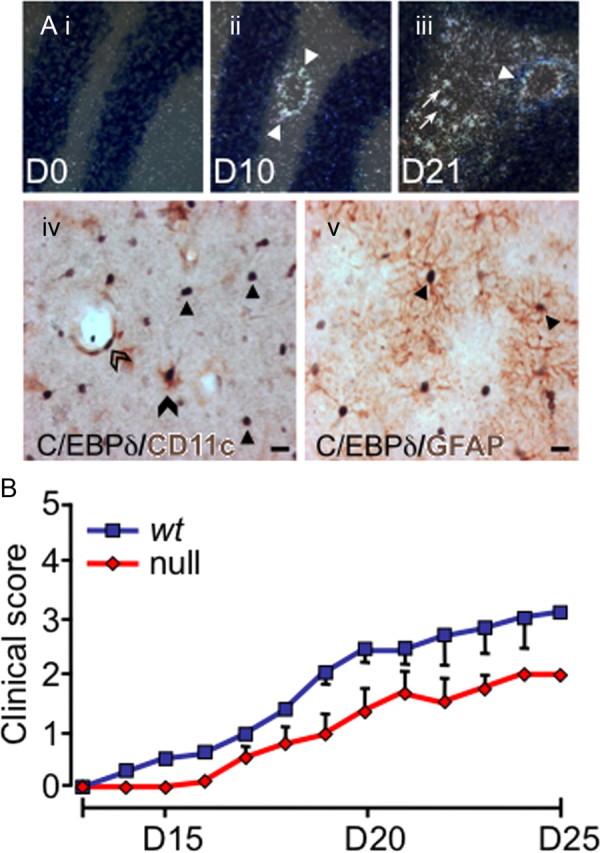
C/EBPδ is expressed centrally in EAE, and EAE severity is reduced in C/EBPδ-null mutants. Ai–iii, C/EBPδ is upregulated throughout EAE. Wild-type animals were killed at day 10 (D10; preclinical EAE), and day 21 (D21; peak disease), and compared with unvaccinated (day 0) animals (n = 3/group). Representative sections of the cerebellum are shown. Before vaccination (D0), C/EBPδ mRNA is not detectable. At D10, C/EBPδ is expressed in cells associated with the cerebral vasculature (arrowheads). C/EBPδ is further upregulated in the perivascular region (arrowhead) and is present in parenchymal cells (arrows) at day 21 (D21). iv, Dual immunohistochemical labeling demonstrates that C/EBPδ protein is expressed in DCs and astrocytes in EAE. Dual staining for CD11c (brown) and C/EBPδ (black) was performed on spinal cord sections from D21 EAE mice. DCs expressing nuclear C/EBPδ (black) and cytoplasmic/surface CD11c (brown) are seen in the CNS parenchyma (filled arrowhead) and perivascular (open arrowhead) regions. Many C/EBPδ-positive nuclei are not associated with CD11c coexpression (arrows). Substituting anti-CD11c for anti-MHC class II gave a similar pattern of single and dual staining (data not shown). v, Dual labeling for the astrocyte-specific marker GFAP (brown) and C/EBPδ (black) identify remaining C/EBPδ+CD11c− cells as astrocytes (arrows). Scale bars, 10 μm. B, EAE severity is reduced in C/EBPδ-deficient mice. Animals were vaccinated with MOG35–55 and clinical scores monitored daily. The clinical severity of disease was reduced by one full grade in C/EBPδ−/− (n = 6 per group; p = 0.019; repeated-measures ANOVA). Error bars represent SEM.
Using a validated, high-titer C/EBPδ antiserum (see Materials and Methods), we performed dual immunohistochemical analysis of wt animals (n = 3 per time point). In both healthy and EAE-affected animals, C/EBPδ labeling of hippocampal neurons was observed, as previously described (Sterneck et al., 1998) (data not shown). In early EAE, but not in healthy mice, there was specific C/EBPδ staining of CD11c+ cell nuclei associated with the cerebral vasculature. Later in disease, labeled C/EBPδ+CD11c+ vascular cells persisted and was accompanied by C/EBPδ+ labeling of astrocytes, identified on the basis of costaining for glial fibrillary acidic protein (GFAP; Fig. 1A). Substituting anti-MHC class II for anti-CD11c yielded a similar pattern and degree of colabeling, indicating that CD11c+ cells were capable of presenting antigen (Fig. 1A). These data are consistent with a potential role for C/EBPδ in the progression of EAE.
EAE disease severity is dependent on C/EBPδ expression
To determine whether C/EBPδ is indeed active in the development of EAE, clinical disease progression was monitored in syngeneic wt and C/EBPδ−/− mice. In replicate experiments, C/EBPδ−/− animals displayed a significant reduction in EAE disease severity (n = 6/group; Fig. 1B), of >1 full clinical grade at the peak of disease (day 21). As EAE-induced changes in central C/EBPδ expression were confined to resident astrocytes and bone marrow (BM)-derived DCs, we sought to determine whether C/EBPδ derived from BM, the CNS, or both compartments contributed to EAE disease progression.
Reduction of C/EBPδ in BM-derived cells is responsible for reduced EAE disease
In EAE, the initial influx of Th-cells to the CNS is mediated by circulation-derived DCs (Greter et al., 2005). As two CNS cell types were found to produce C/EBPδ (Fig. 1A), we generated BM chimerae to parse the contribution of C/EBPδ expression by resident astrocytes and peripherally derived hemopoietic cells. Four groups of chimeric animals were produced, representing the parametric combinations of BM transfer within and between wt and C/EBPδ-null mutants (i.e., wt marrow→wt recipient, wt→null, null→null, and null→wt). In peripheral blood collected 6 weeks after transplantation, the profile of peripheral blood mononuclear cells expressing GFP was virtually indistinguishable from that of donor animals (Fig. 2A). At this time point, chimeric animals and controls were vaccinated with MOG. EAE induction was associated with clear evidence of transplanted cells migrating to the CNS (Fig. 2B). In these animals, dual label immunohistochemistry revealed no C/EBPδ expression by CD11c+ cells in the CNS of mice receiving C/EBPδ−/− BM, while C/EBPδ was expressed in astrocytes of all wt bone marrow recipient mice (data not shown). When mice were grouped according to the recipient mouse genotype, there was no significant difference in disease (Fig. 2C). However, animals transplanted with C/EBPδ−/− BM, like mice with global C/EBPδ−/− deficiency, displayed a full clinical grade reduction in EAE disease severity, regardless of the genotype of the recipients (Fig. 2D). These data suggested that C/EBPδ expressed in circulation-derived CD11c+ mononuclear cells mediate the actions of C/EBPδ in EAE. However, it was important to exclude significant production of C/EBPδ protein by other CNS APCs in EAE.
Figure 2.
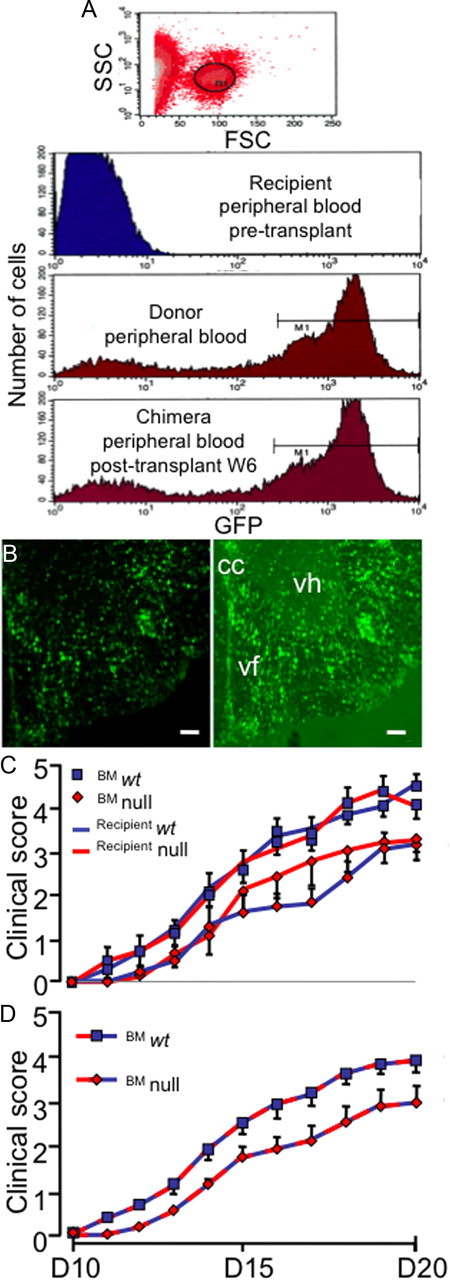
Absent C/EBPδ expression in peripheral immune cells is responsible for reduced EAE in null mice. A, Six weeks after irradiation and BMT, most circulating cells are of donor origin. BM cells from ubiquitous GFP-expressing animals were transferred to irradiated wt mice. High forward scatter cells were gated and assessed for GFP expression (R1). This was undetectable in peripheral blood of recipients before transplantation, while most peripheral blood mononuclear cells of donor animals expressed GFP. Six weeks (W6) after BMT the profile of mononuclear cell GFP expression in recipient mice was similar to that of donors (n = 6). B, Fluorescence image (right) of ventral spinal cord from a mouse at day 21 (D21) after EAE induction (clinical score = 3) showing infiltration of transplanted GFP cells into the cord. Infiltrating cells displayed morphological features of both lymphocytes and APCs. Left, Infiltrating, bone-marrow derived, GFP-fluorescent cells. Right, Fluorescence overlaid with dark field to provide anatomical detail (cc, central canal; vf, ventral funiculus; vh, ventral horn). Scale bars, 100 μm. C, Chimeric animals were generated and EAE induced 6 weeks later and animals clinically scored as a measure of disease. There was no significant difference in disease between wt and C/EBPδ−/− (null) animals when circulating mononuclear cells were C/EBPδ+/+ (n = 9 per group; p = 0.54; pooled from 2 independent experiments). Similarly, there was no difference in EAE severity when wt and C/EBPδ−/− hosts had circulating mononuclear cells that were C/EBPδ−/− (n = 9 per group; p = 0.25; pooled from 2 independent experiments). D, Reduced C/EBPδ expression in circulating mononuclear cells is responsible for reduced EAE in C/EBPδ−/− animals. As the differences in EAE severity appeared dependent on the genotype of circulating mononuclear cells, data were pooled based on the genotype of donor bone marrow. Animals whose circulating monocytes were C/EBPδ−/− displayed a significant reduction in EAE disease (n = 18 per group; p = 0.0003; repeated-measures ANOVA). All error bars represent SEM.
CD11c+ DCs, alone, express C/EBPδ early in the EAE-afflicted CNS
For Th-cells to gain entry to the CNS and initiate EAE, fully functional DCs are required (Greter et al., 2005). As DC-mediated T-cell entry into the CNS occurs early in the course of EAE, we examined the C/EBPδ production of subsets of CNS inflammatory cells just before disease onset, using a FACS-intracellular staining protocol using our anti-C/EBPδ antiserum. Using this methodology we could clearly distinguish C/EBPδ labeling in wt LPS-stimulated splenocytes (Fig. 3). Additionally, we confirmed these cells increased C/EBPδ mRNA expression in response to LPS treatment (Fig. 3B) (Alam et al., 1992). Similar specific staining was absent in identically treated C/EBPδ−/− and vehicle-treated splenocytes, as well as in LPS-treated splenocytes labeled with isotype control antibody (Fig. 3A). Using this assay, we next examined mononuclear cells isolated from the spinal cords of MOG-vaccinated animals early in EAE disease, when DCs are thought to facilitate T-cell entry into the CNS (Fig. 4) (Greter et al., 2005). We identified CNS CD45+ cell subsets, including microglia, T-cells, B-cells, DCs, and macrophages, by gating on surface markers CD45, CD3, B220, CD11c, and CD11b (Bailey et al., 2007). There was clearly identifiable C/EBPδ labeling of CD11c+ DCs (Fig. 4Biii) with no identifiable expression in other potential APCs, including B-cells, microglia, macrophages, and T-cells (Fig. 4B). As DCs were the dominant BM-derived cell expressing C/EBPδ early in EAE, we next wished to determine whether altered DC expression of C/EBPδ is associated with altered Th-cell development as we had postulated.
Figure 3.

Validation of an intracellular staining method for detecting C/EBPδ in bone marrow-derived mononuclear cells. A, C/EBPδ was detected by intracellular protein labeling. To validate an intracellular FACS staining method, we demonstrated that intracellular C/EBPδ labeling was not present in C/EBPδ−/− splenocytes treated with LPS (diagonal black lines), which could not be discriminated from similarly treated wt splenocytes labeled with control antibody (solid gray). However, wt splenocytes treated with LPS clearly demonstrated C/EBPδ labeling (black outline) compared with these controls. B, To confirm that LPS treatment was likely to lead to increased C/EBPδ protein, mRNA expression was assayed 4 h after activation of splenocytes with LPS. Quantitative rtPCR demonstrated significant upregulation of C/EBPδ mRNA in wt splenocytes compared with vehicle-treated cells (black). C/EBPδ mRNA could not be amplified (nd, not detected) from C/EBPδ−/− (null) splenocytes (white). *p < 0.05.
Figure 4.

Dendritic cells are the only CNS APCs expressing C/EBPδ protein in EAE. CNS mononuclear cells were isolated from EAE animals on day 10, before disease onset. C/EBPδ-specific labeling was demonstrated in CD3−CD11c+ cells indicating a DC phenotype. A, CD45+ cell subsets of the CNS, including microglia, T-cells, B-cells, DCs, and macrophages, were identified by gating on surface markers CD45, CD3, B220, CD11c, and CD11b (Bailey et al., 2007). B, C/EBPδ protein expression was not detected in microglia that were identified on the basis of their intermediate CD45 expression (i; CD45int). Of the cells expressing higher levels of CD45 (CD45hi), only DCs (iii; CD45hiCD3−CD11c+) were found to express C/EBPδ protein, while T-cells (ii; CD45hiCD3+), B-cells (iv; CD45hiCD3−CD11c−B220+CD11b−), and macrophages (v; CD45hiCD3−CD11c−B220−CD11b+) did not.
C/EBPδ −/− mice have reduced Th17, and enhanced Treg, development throughout EAE
While DCs are necessary for CNS Th-cell recruitment in EAE (Greter et al., 2005), EAE also requires T-cells (Brown and Sawchenko, 2007). In addition to facilitating T-cell entry into the CNS, DCs directly activate and participate in T-cell differentiation (Bailey et al., 2007). Changes in the proportions of Th-cell phenotypes in the CNS over the course of EAE have been described (Korn et al., 2007), and involve a transition from Th17 cell predominance at preclinical and peak disease to increased Treg proportions at later stages. As the differentiation of these two Th-cell phenotypes are closely related (Bettelli et al., 2006) and mediated by DCs (Bailey et al., 2007), we asked whether C/EBPδ is active in this branch of Th-cell development. We therefore induced EAE in wt and C/EBPδ−/− animals and determined the proportion of Th17 and Treg cells at and after the peak of clinical disease, when their relative numbers differ substantially (Korn et al., 2007). In replicate experiments, C/EBPδ−/− mice displayed significantly reduced Th17 cell proportions at peak disease and significantly enhanced Treg cell proportions in later disease, with a significantly reduced Th17:Treg ratio at both stages examined (n = 24, 12 wt and 12 C/EBPδ−/−; Fig. 5). Combined with the previous findings, this result suggested that C/EBPδ production by DCs modulates Th17/Treg development. However, creation of BM chimerae necessarily alters the C/EBPδ expression capacity of all BM derivatives, including Th-cells. We therefore wished to confirm that isolated modulation of C/EBPδ expression in DCs similarly altered Th17/Treg development, in vivo.
Figure 5.

C/EBPδ−/− animals have fewer Th17 and more Treg cells in the CNS over the course of EAE. wt and C/EBPδ−/− (null) mice were vaccinated with MOG, and CNS mononuclear cells were isolated near the peak of disease (day 18; n = 8) and at partial recovery (day 25; n = 16). CNS mononuclear cells were surface stained with anti-CD45, anti-CD3, and anti-CD4 and then labeled for intracellular FOXP3, IL-17, and INFγ. Data represent the pooled results of two independent experiments. The proportions of Th17 to Treg cells in wt mice were comparable to those reported in murine EAE (Korn et al., 2007). C/EBPδ−/− animals had fewer Th17 cells at peak disease and more Tregs during the recovery phase of EAE (Rec), with the resultant Th17:Treg ratios being significantly reduced at both time points (p = 0.02, p = 0.03). Error bars represent SEM. *p < 0.05.
C/EBPδ expression in DCs mediates Th17/Treg development
To examine the effect of isolated modulation of C/EBPδ expression in CNS DCs, we generated both plasmacytoid and myeloid DCs (Brasel et al., 2000) from the BM of wt and C/EBPδ−/− mice by culturing in the presence of fms-like tyrosine kinase 3 ligand (FLT3) (Naik et al., 2010). After 9 d in culture, >95% of CD45+ cells were CD11c+ and there were no significant differences in the proportion of plasmacytoid or myeloid DCs derived from wt and C/EBPδ−/− BM. Following in vitro differentiation, CD11c+ BMDCs were antigen-pulsed with MOG and then matured with LPS. LPS maturation was not associated with differential expression of markers of maturation in wt versus C/EBPδ −/− DCs (data not shown). These antigen-pulsed, matured DCs were then injected, with appropriate controls, into the right frontal cortex (coordinates given in Materials and Methods) of congenic C/EBPδ+/+ Vβ11 TCR transgenic 2D2 mice. In these mice, between 50 and 90% of circulating T-cells have T-cell receptors that respond to MOG presented by DCs (Bettelli et al., 2006). Therefore, they will be activated when DCs present MOG antigen to them. Injection of MOG antigen-pulsed, matured DCs into the 2D2 mouse brain lead to the recruitment, activation, and differentiation of antigen-specific Th-cells, as others have shown (Zozulya et al., 2009). While there was no significant difference in the number of antigen-specific Th-cells recruited by injected wt and C/EBPδ−/− BMDCs (p = 0.58), C/EBPδ−/− BMDCs biased against Th17 differentiation, favoring Treg development. This resulted in a significant decrease in the Th17:Treg ratio (n = 9; Fig. 6A), with no significant differences in Th1 differentiation (Fig. 6B). Nonantigen-pulsed, matured DCs, and vehicle injection controls showed significantly less recruitment and activation of antigen specific T-cells (data not shown). Having demonstrated that isolated variation of C/EBPδ expression in DCs significantly modulates Th17:Treg development in the CNS, we wished to examine this process in vitro to elucidate potential mechanisms by which DC expression of C/EBPδ might influence Th-cell development.
Figure 6.

Expression of C/EBPδ by CD11c+ DCs directs Th17/Treg cell differentiation in vitro and in vivo. A, 2D2 transgenic mice that received intracerebral injections of BMDCs from wt and C/EBPδ−/− (null) BM had similar numbers of CD3+, CD4+, and Vβ11+ Th-cells recruited to the CNS (p = 0.58). Matured, MOG antigen-pulsed BMDCs were cocultured with MOG-reactive wt 2D2 Th-cells (CD3+CD4+) under Treg and Th17 polarizing conditions. Under vehicle control conditions there was no significant difference in the development of Th17, Treg, or Th1 T-cells directed by wt versus C/EBPδ−/− BMDCs (p = 0.46). With Treg- and Th17-polarizing conditions there was a significant reduction in the Th17:Treg ratio directed by C/EBPδ−/− BMDCs (p = 0.04; p = 0.005 n = 9 per group). In EAE (Fig. 5) there was also a significantly decreased Th17:Treg ratio in vivo between wt and C/EBPδ−/− animals (p = 0.02). When BMDCs were injected into the CNS of 2D2 animals, differentiation of MOG antigen-specific Th-cells was again skewed by C/EBPδ−/− BMDCs, leading to significantly a reduced Th17:Treg ratio (p = 0.02; n = 6). B, For both in vivo and in vitro groups there were no significant differences in Th1 development when cells were activated in vitro (control, p = 0.29; TGFβ, p = 0.67, or TGFβ and IL-6, p = 0.60) or in vivo (p = 0.2). *p < 0.05; all error bars represent SEM.
DC expression of C/EBPδ regulates Th17:Treg development via IL-10
To examine mechanisms by which C/EBPδ might regulate Th-cell development, we established an in vitro system of DC-mediated Th-cell activation. MOG antigen-pulsed matured wt and C/EBPδ−/− BMDCs were incubated for 5 d with CD4+ T-cells isolated from congenic C/EBPδ+/+ 2D2 animals under Th17- (IL-6 and TGFβ) and Treg (TGFβ)-polarizing conditions. Cell-conditioned media produced over the culture period was stored at −80°C for later analysis, and cultured mononuclear cells were surface- and intracellularly labeled for FACS analysis as above. Similar to the in vivo model, three separate experiments in vitro demonstrated a significant decrease in the Th17:Treg ratio mediated by C/EBPδ−/− BMDCs (n = 9; Fig. 6A). In the stored supernatants, we determined the concentration of multiple cytokines using multiplex bead-based immunoassays. After 5 d in culture, there were no significant differences between wt and C/EBPδ−/− supernatants in the levels of several key cytokines known to mediate Th development including IL-6, TGFβ, IL-12, IL-23, and IL-1β (data not shown). However, IL-10 concentrations tended to be elevated in C/EBPδ−/− BMDC cultures under Th17-polarizing conditions, although this failed to reach significance (n = 9; p = 0.15). As C/EBPδ is known to interact with the IL-10 promoter (Liu et al., 2006), we examined IL-10 mRNA expression in BMDCs exposed to LPS (Fig. 7A). This confirmed that IL-10 mRNA expression is significantly upregulated in C/EBPδ−/− BMDCs compared with wt, and that the relatively reduced IL-10 expression in C/EBPδ+/+ BMDCs coincided with upregulation of C/EBPδ mRNA (Fig. 7A). To determine whether IL-10 mediated the effects of reduced DC expression of C/EBPδ on Th-cell development, we inhibited IL-10 signaling by the addition of a well characterized IL-10 receptor-blocking antibody (O'Farrell et al., 1998). Under the same in vitro conditions, IL-10 receptor blockade reversed the effect of reduced DC expression of C/EBPδ on Th17:Treg development (n = 9; Fig. 7B). This indicated that increased IL-10 production by DCs is at least partially responsible for the effect of C/EBPδ deficiency, and that C/EBPδ might repress IL-10 production.
Figure 7.

Expression of C/EBPδ by DCs modulates Th17:Treg ratios via IL-10. A, Total mRNA was isolated 2 h after LPS or vehicle stimulation of MOG antigen-pulsed wt and C/EBPδ−/− (null) BMDCs, and qPCR assessment of IL-10 and C/EBPδ mRNA was performed. LPS treatment led to a significant increase in C/EBPδ mRNA, which was not detectible (nd) in C/EBPδ−/− BMDCs. Upon treatment with LPS, both C/EBPδ−/− and wt BDMCs significantly upregulated IL-10 mRNA (p = 0.046). However, C/EBPδ−/− BMDCs produced significantly more IL-10 mRNA than wt under the same stimulation conditions (p = 0.046). B, In vitro and in vivo experiments in Figure 6A were repeated using Th17-polarizing conditions, with, and without, IL-10 receptor blockade (IL-10R ab). In vitro incubation with isotype control antibody showed again that C/EBPδ−/− BMDCs mediated a significantly reduced Th17:Treg ratio (p = 0.005). Blockade of IL-10 signaling significantly increased the Th17:Treg ratio mediated by C/EBPδ−/− DCs (p = 0.01; n = 9), but did not completely negate the effect of C/EBPδ deficiency in DCs. wt or C/EBPδ−/− MOG antigen-pulsed, matured DCs were injected into the CNS of 2D2 mice undergoing isotype or specific IL-10R antibody treatment every other day. Blocking IL-10 signaling again lead to a significant increase in the Th17:Treg ratio generated by C/EBPδ−/− BMDCs (p = 0.012), which did not differ significantly from that induced by isotype antibody-treated wt BMDCs in vivo (p = 0.78). Data represent n = 9; NS, p > 0.2; *p < 0.05; all error bars represent SEM.
IL-10 mediates DC-C/EBPδ modulation of Th17:Treg development in vivo
To confirm that IL-10 mediates the effects of C/EBPδ expression by DCs in vivo, we used the same IL-10 receptor blocking antibody in 2D2 animals that received CNS injections of wt or C/EBPδ−/− MOG antigen-pulsed, matured, BMDCs, as above. Here again, IL-10 receptor blockade by intraperitoneal injection of antibody significantly attenuated the effects of reduced C/EBPδ expression by DCs (n = 9; Fig. 7B), leading to return of the Th17:Treg ratio close to that supported by wt BMDCs. These in vitro and in vivo results strongly suggest that C/EBPδ modulates DC expression of IL-10, which in turn regulates Th17:Treg phenotype development.
Discussion
Here, we demonstrate in EAE that C/EBPδ expression by DCs influences Th17 versus Treg differentiation, with little or no impact upon Th1 development, via an IL-10-dependent mechanism. With reduced expression of C/EBPδ, DC IL-10 mRNA is upregulated and presumably IL-10 protein is secreted locally. This leads to relatively decreased Th17, and enhanced Treg, differentiation, associated with reduced EAE disease severity. These findings may have implications for DC function in a number of CNS inflammatory disorders, as well as other neurological disorders in which the immune system is engaged.
Our initial assessment of the known literature suggested that altered IL-6 production would mediate altered Th-cell differentiation due to reduced C/EBPδ expression. However, the link between IL-6 and regulation of Th17 and Treg development has become less clear (Fujimoto et al., 2011). Indeed, excessive IL-6 production associated transgenic overexpression leads to increased Treg numbers (Fujimoto et al., 2011), and there are IL-6-independent pathways for Th17 development (Kimura et al., 2007). However, overexpression of IL-10 by antigen-presenting cells is known to completely inhibit EAE induction (Cua et al., 1999). Furthermore, DCs with upregulated IL-10 expression promote Treg development (Monti et al., 2004; Cai et al., 2009; Gregori et al., 2010; Poncini et al., 2010; Yoshiki et al., 2010). Such mechanisms have been described for immunosuppressive tumor-associated DCs (Monti et al., 2004; Fujii et al., 2005; McCarter et al., 2007), skin DCs (Yoshiki et al., 2010), and IL-10 producing DCs that mediate immune tolerance (Gregori et al., 2010). Our data now suggest that the regulatory nature of IL-10 production by DCs is mediated, at least in part, by C/EBPδ. Supporting this notion is the finding that DC regulatory function mediated by IL-10 requires MyD88 and TLR4 (Arora et al., 2010), both of which are regulated by C/EBPδ (Lu et al., 2009).
Our data suggest that upregulation of C/EBPδ leads to suppression of IL-10 secretion. Consistent with this finding, the C/EBP family and C/EBPδ, specifically, repress gene transcription associated with inflammation (Okazaki et al., 2002). For example, C/EBPδ directly mediates TNFα-induced suppression of gene expression (Lacorte et al., 1997a) and is upregulated by IL-1β (Lacorte et al., 1997b). Both TNFα and IL-1β are potent proinflammatory cytokines, with the later involved in Th17 phenotype development (Ben-Sasson et al., 2009). Therefore, it seems likely that increased IL-10 mRNA in C/EBPδ-deficient DCs is due to lack of transcriptional repression by C/EBPδ, although this remains to be demonstrated directly. Conversely, upregulation of C/EBPδ in DCs is likely to lead to decreased IL-10 secretion, resulting in the skewing of Th-responses away from the Treg and toward the Th17 phenotype, thus facilitating EAE progression.
In addition to EAE, there are a number of other CNS pathologies that necessarily engage the immune system, which in turn regulates their outcome. These include traumatic spinal cord injury and Alzheimer's disease, in both of which increased C/EBPδ expression has been documented (Li et al., 2004; De Biase et al., 2005). Additionally, the immune complement of cells that regulate pathology in both Alzheimer's disease and spinal cord injury have been defined as circulating CD45hi CD11b+ CD11c+ monocyte-derived cells, a similar phenotype to that which we find expresses C/EBPδ. In both these diseases, blood-derived DCs mediate beneficial outcomes via upregulation of IL-10 (Schwartz and Shechter, 2010). The regulation of IL-10 by C/EBPδ in DCs raises the possibility that therapeutic modulation of peripheral DC expression of C/EBPδ could favorably influence clinical outcomes in traumatic spinal cord injury, Alzheimer's disease, and MS.
While C/EBPδ deficiency in DCs was associated with a relatively modest reduction in the clinical severity of EAE, this effect was significant and comparable in magnitude to the degree of reduction of peak EAE disease seen in IL-17 knock-out mice (Komiyama et al., 2006). Therapeutic neutralization of IL-17 in animals (Hofstetter et al., 2005) in which EAE was induced using the same method used in the present experiments similarly reduced disease. A technical concern with our study derives from the possibility that transplanted BM-derived cells, present in the CNS before EAE induction, might themselves significantly affect disease progression. However, the number of donor-derived cells that populate the CNS within 6 weeks of transplantation is small (Vallières and Sawchenko, 2003). Moreover, direct CNS injection of C/EBPδ−/− DCs into wt animals produced similar Th17:Treg developmental changes as EAE induction did in C/EBPδ−/− mice (Figs. 5–7). These data suggest a minimal influence of CNS chimeric cells before disease induction. A second potential concern is that the very low antiserum concentration used to detect C/EBPδ may have allowed some lower-expressing cell types to escape detection. Indeed, increased C/EBPδ mRNA expression has been described in microglia and astrocytes upon stimulation with LPS (Ejarque-Ortiz et al., 2010). We were able to demonstrate C/EBPδ expression in astrocytes (Fig. 1), but saw no clear evidence of such in microglia. It may be noted that Ejarque-Ortiz et al. used a microglial cell line, leaving open to question the capacity of primary microglia to express C/EBPδ. Additionally, present experiments in BM chimerae indicated that maintained microglial and/or astrocyte capacity to express C/EBPδ did not promote disease when peripheral DCs lacked C/EBPδ (Fig. 2). Also, when T-cells were capable of producing C/EBPδ in vitro and in vivo, it was DC expression of C/EBPδ that skewed Th17:Treg ratios (Fig. 6). Therefore, any capacity of cell types other than DCs to produce C/EBPδ protein does not appear to significantly impact Th17:Treg differentiation in the situations we examined.
Overall, the present findings suggest that DC production of C/EBPδ mediates Th-cell differentiation and impacts upon autoimmune disease severity. The proinflammatory effects of C/EBPδ expression by DCs raise the possibility that numerous DC subtypes may regulate immunity by this mechanism, perhaps explaining the conflicting data regarding plasmacytoid DCs inducing both Treg (Bailey-Bucktrout et al., 2008) and opposing Th17 cell phenotypes (Isaksson et al., 2009). It is therefore possible that production of C/EBPδ by DCs, rather than DC phenotype, directs Th-immune responses. If this holds true, it raises the possibility of modulating C/EBPδ expression in DCs to target specific CNS and systemic immune responses in an antigen-specific manner.
Footnotes
SpinalCure/NRMA, Multiple Sclerosis Research Australia, the National Multiple Sclerosis Society USA, and a New South Wales Health Research and Development Infrastructure Grant supported this work. D.A.B holds a National Health and Medical Research Council Career Development Fellowship and was awarded the inaugural SpinalCure Australia Senior Research Fellowship. P.E.S. is supported by NIH Grant NS-21182 and the Clayton Medical Research Foundation. We would like to thank Dr. Cecile King and Professor Jonathon Sprent for their insightful review and suggestions in preparing this manuscript.
The authors declare no competing financial interests.
References
- Alam T, An MR, Papaconstantinou J. Differential expression of three C/EBP isoforms in multiple tissues during the acute phase response. J Biol Chem. 1992;267:5021–5024. [PubMed] [Google Scholar]
- Arora M, Poe SL, Oriss TB, Krishnamoorthy N, Yarlagadda M, Wenzel SE, Billiar TR, Ray A, Ray P. TLR4/MyD88-induced CD11b(+)Gr-1(int)F4/80(+) non-migratory myeloid cells suppress Th2 effector function in the lung. Mucosal Immunol. 2010;3:578–593. doi: 10.1038/mi.2010.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey SL, Schreiner B, McMahon EJ, Miller SD. CNS myeloid DCs presenting endogenous myelin peptides ‘preferentially’ polarize CD4(+) T(H)-17 cells in relapsing EAE. Nat Immunol. 2007;8:172–180. doi: 10.1038/ni1430. [DOI] [PubMed] [Google Scholar]
- Bailey-Bucktrout SL, Caulkins SC, Goings G, Fischer JA, Dzionek A, Miller SD. Cutting edge: central nervous system plasmacytoid dendritic cells regulate the severity of relapsing experimental autoimmune encephalomyelitis. J Immunol. 2008;180:6457–6461. doi: 10.4049/jimmunol.180.10.6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Sasson SZ, Hu-Li J, Quiel J, Cauchetaux S, Ratner M, Shapira I, Dinarello CA, Paul WE. IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc Natl Acad Sci U S A. 2009;106:7119–7124. doi: 10.1073/pnas.0902745106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Brasel K, De Smedt T, Smith JL, Maliszewski CR. Generation of murine dendritic cells from flt3-ligand-supplemented bone marrow cultures. Blood. 2000;96:3029–3039. [PubMed] [Google Scholar]
- Brown DA, Sawchenko PE. Time course and distribution of inflammatory and neurodegenerative events suggest structural bases for the pathogenesis of experimental autoimmune encephalomyelitis. J Comp Neurol. 2007;502:236–260. doi: 10.1002/cne.21307. [DOI] [PubMed] [Google Scholar]
- Brynedal B, Khademi M, Wallström E, Hillert J, Olsson T, Duvefelt K. Gene expression profiling in multiple sclerosis: a disease of the central nervous system, but with relapses triggered in the periphery? Neurobiol Dis. 2010;37:613–621. doi: 10.1016/j.nbd.2009.11.014. [DOI] [PubMed] [Google Scholar]
- Bulloch K, Miller MM, Gal-Toth J, Milner TA, Gottfried-Blackmore A, Waters EM, Kaunzner UW, Liu K, Lindquist R, Nussenzweig MC, Steinman RM, McEwen BS. CD11c/EYFP transgene illuminates a discrete network of dendritic cells within the embryonic, neonatal, adult, and injured mouse brain. J Comp Neurol. 2008;508:687–710. doi: 10.1002/cne.21668. [DOI] [PubMed] [Google Scholar]
- Cai G, Wang H, Qin Q, Zhang J, Zhu Z, Liu M, Shen Q. Amelioration of myocarditis by HVEM-overexpressing dendritic cells through induction of IL-10-producing cells. Cardiovasc Res. 2009;84:425–433. doi: 10.1093/cvr/cvp219. [DOI] [PubMed] [Google Scholar]
- Cardinaux JR, Allaman I, Magistretti PJ. Pro-inflammatory cytokines induce the transcription factors C/EBPbeta and C/EBPdelta in astrocytes. Glia. 2000;29:91–97. [PubMed] [Google Scholar]
- Cortés-Canteli M, Wagner M, Ansorge W, Pérez-Castillo A. Microarray analysis supports a role for ccaat/enhancer-binding protein-beta in brain injury. J Biol Chem. 2004;279:14409–14417. doi: 10.1074/jbc.M313253200. [DOI] [PubMed] [Google Scholar]
- Cua DJ, Groux H, Hinton DR, Stohlman SA, Coffman RL. Transgenic interleukin 10 prevents induction of experimental autoimmune encephalomyelitis. J Exp Med. 1999;189:1005–1010. doi: 10.1084/jem.189.6.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Biase A, Knoblach SM, Di Giovanni S, Fan C, Molon A, Hoffman EP, Faden AI. Gene expression profiling of experimental traumatic spinal cord injury as a function of distance from impact site and injury severity. Physiol Genomics. 2005;22:368–381. doi: 10.1152/physiolgenomics.00081.2005. [DOI] [PubMed] [Google Scholar]
- Domingues HS, Mues M, Lassmann H, Wekerle H, Krishnamoorthy G. Functional and pathogenic differences of Th1 and Th17 cells in experimental autoimmune encephalomyelitis. PLoS One. 2010;5:e15531. doi: 10.1371/journal.pone.0015531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejarque-Ortiz A, Medina MG, Tusell JM, Pérez-González AP, Serratosa J, Saura J. Upregulation of CCAAT/enhancer binding protein beta in activated astrocytes and microglia. Glia. 2007;55:178–188. doi: 10.1002/glia.20446. [DOI] [PubMed] [Google Scholar]
- Ejarque-Ortiz A, Gresa-Arribas N, Straccia M, Mancera P, Solà C, Tusell JM, Serratosa J, Saura J. CCAAT/enhancer binding protein delta in microglial activation. J Neurosci Res. 2010;88:1113–1123. doi: 10.1002/jnr.22272. [DOI] [PubMed] [Google Scholar]
- Fujii S, Nishimura MI, Lotze MT. Regulatory balance between the immune response of tumor antigen-specific T-cell receptor gene-transduced CD8 T cells and the suppressive effects of tolerogenic dendritic cells. Cancer Sci. 2005;96:897–902. doi: 10.1111/j.1349-7006.2005.00124.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto M, Nakano M, Terabe F, Kawahata H, Ohkawara T, Han Y, Ripley B, Serada S, Nishikawa T, Kimura A, Nomura S, Kishimoto T, Naka T. The influence of excessive IL-6 production in vivo on the development and function of Foxp3+ regulatory T cells. J Immunol. 2011;186:32–40. doi: 10.4049/jimmunol.0903314. [DOI] [PubMed] [Google Scholar]
- Gregori S, Tomasoni D, Pacciani V, Scirpoli M, Battaglia M, Magnani CF, Hauben E, Roncarolo MG. Differentiation of type 1 T regulatory cells (Tr1) by tolerogenic DC-10 requires the IL-10-dependent ILT4/HLA-G pathway. Blood. 2010;116:935–944. doi: 10.1182/blood-2009-07-234872. [DOI] [PubMed] [Google Scholar]
- Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, Laufer T, Noelle RJ, Becher B. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med. 2005;11:328–334. doi: 10.1038/nm1197. [DOI] [PubMed] [Google Scholar]
- Hofstetter HH, Ibrahim SM, Koczan D, Kruse N, Weishaupt A, Toyka KV, Gold R. Therapeutic efficacy of IL-17 neutralization in murine experimental autoimmune encephalomyelitis. Cell Immunol. 2005;237:123–130. doi: 10.1016/j.cellimm.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Huseby ES, Liggitt D, Brabb T, Schnabel B, Ohlén C, Goverman J. A pathogenic role for myelin-specific CD8(+) T cells in a model for multiple sclerosis. J Exp Med. 2001;194:669–676. doi: 10.1084/jem.194.5.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaksson M, Ardesjö B, Rönnblom L, Kämpe O, Lassmann H, Eloranta ML, Lobell A. Plasmacytoid DC promote priming of autoimmune Th17 cells and EAE. Eur J Immunol. 2009;39:2925–2935. doi: 10.1002/eji.200839179. [DOI] [PubMed] [Google Scholar]
- Juan TS, Wilson DR, Wilde MD, Darlington GJ. Participation of the transcription factor C/EBP delta in the acute-phase regulation of the human gene for complement component C3. Proc Natl Acad Sci U S A. 1993;90:2584–2588. doi: 10.1073/pnas.90.7.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao JY, Zhang M, Miller MJ, Mills JC, Wang B, Liu M, Eaton KA, Zou W, Berndt BE, Cole TS, Takeuchi T, Owyang SY, Luther J. Helicobacter pylori immune escape is mediated by dendritic cell-induced Treg skewing and Th17 suppression in mice. Gastroenterology. 2010;138:1046–1054. doi: 10.1053/j.gastro.2009.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura A, Naka T, Kishimoto T. IL-6-dependent and -independent pathways in the development of interleukin 17-producing T helper cells. Proc Natl Acad Sci U S A. 2007;104:12099–12104. doi: 10.1073/pnas.0705268104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, Bäckström BT, Sobel RA, Wucherpfennig KW, Strom TB, Oukka M, Kuchroo VK. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med. 2007;13:423–431. doi: 10.1038/nm1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacorte JM, Beigneux A, Parant M, Chambaz J. Repression of apoC-III gene expression by TNFalpha involves C/EBPdelta/NF-IL6beta via an IL-1 independent pathway. FEBS Lett. 1997a;415:217–220. doi: 10.1016/s0014-5793(97)01127-7. [DOI] [PubMed] [Google Scholar]
- Lacorte JM, Ktistaki E, Beigneux A, Zannis VI, Chambaz J, Talianidis I. Activation of CAAT enhancer-binding protein delta (C/EBPdelta) by interleukin-1 negatively influences apolipoprotein C-III expression. J Biol Chem. 1997b;272:23578–23584. doi: 10.1074/jbc.272.38.23578. [DOI] [PubMed] [Google Scholar]
- Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavasani S, Dzhambazov B, Nouri M, Fåk F, Buske S, Molin G, Thorlacius H, Alenfall J, Jeppsson B, Weström B. A novel probiotic mixture exerts a therapeutic effect on experimental autoimmune encephalomyelitis mediated by IL-10 producing regulatory T cells. PLoS One. 2010;5:e9009. doi: 10.1371/journal.pone.0009009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Strohmeyer R, Liang Z, Lue LF, Rogers J. CCAAT/enhancer binding protein delta (C/EBPdelta) expression and elevation in Alzheimer's disease. Neurobiol Aging. 2004;25:991–999. doi: 10.1016/j.neurobiolaging.2003.10.016. [DOI] [PubMed] [Google Scholar]
- Litvak V, Ramsey SA, Rust AG, Zak DE, Kennedy KA, Lampano AE, Nykter M, Shmulevich I, Aderem A. Function of C/EBPdelta in a regulatory circuit that discriminates between transient and persistent TLR4-induced signals. Nat Immunol. 2009;10:437–443. doi: 10.1038/ni.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YW, Chen CC, Tseng HP, Chang WC. Lipopolysaccharide-induced transcriptional activation of interleukin-10 is mediated by MAPK- and NF-kappaB-induced CCAAT/enhancer-binding protein delta in mouse macrophages. Cell Signal. 2006;18:1492–1500. doi: 10.1016/j.cellsig.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, Langer-Gould A, Strober S, Cannella B, Allard J, Klonowski P, Austin A, Lad N, Kaminski N, Galli SJ, Oksenberg JR, Raine CS, Heller R, Steinman L. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002;8:500–508. doi: 10.1038/nm0502-500. [DOI] [PubMed] [Google Scholar]
- Lu YC, Kim I, Lye E, Shen F, Suzuki N, Suzuki S, Gerondakis S, Akira S, Gaffen SL, Yeh WC, Ohashi PS. Differential role for c-Rel and C/EBPbeta/delta in TLR-mediated induction of proinflammatory cytokines. J Immunol. 2009;182:7212–7221. doi: 10.4049/jimmunol.0802971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- McCarter MD, Baumgartner J, Escobar GA, Richter D, Lewis K, Robinson W, Wilson C, Palmer BE, Gonzalez R. Immunosuppressive dendritic and regulatory T cells are upregulated in melanoma patients. Ann Surg Oncol. 2007;14:2854–2860. doi: 10.1245/s10434-007-9488-3. [DOI] [PubMed] [Google Scholar]
- Monti P, Leone BE, Zerbi A, Balzano G, Cainarca S, Sordi V, Pontillo M, Mercalli A, Di Carlo V, Allavena P, Piemonti L. Tumor-derived MUC1 mucins interact with differentiating monocytes and induce IL-10highIL-12low regulatory dendritic cell. J Immunol. 2004;172:7341–7349. doi: 10.4049/jimmunol.172.12.7341. [DOI] [PubMed] [Google Scholar]
- Naik SH, O'Keeffe M, Proietto A, Shortman HH, Wu L. CD8+, CD8-, and plasmacytoid dendritic cell generation in vitro using flt3 ligand. Methods Mol Biol. 2010;595:167–176. doi: 10.1007/978-1-60761-421-0_10. [DOI] [PubMed] [Google Scholar]
- O'Farrell AM, Liu Y, Moore KW, Mui AL. IL-10 inhibits macrophage activation and proliferation by distinct signaling mechanisms: evidence for Stat3-dependent and -independent pathways. EMBO J. 1998;17:1006–1018. doi: 10.1093/emboj/17.4.1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki K, Li J, Yu H, Fukui N, Sandell LJ. CCAAT/enhancer-binding proteins beta and delta mediate the repression of gene transcription of cartilage-derived retinoic acid-sensitive protein induced by interleukin-1 beta. J Biol Chem. 2002;277:31526–31533. doi: 10.1074/jbc.M202815200. [DOI] [PubMed] [Google Scholar]
- Poncini CV, Giménez G, Pontillo CA, Alba-Soto CD, de Isola EL, Piazzón I, Cappa SM. Central role of extracellular signal-regulated kinase and Toll-like receptor 4 in IL-10 production in regulatory dendritic cells induced by Trypanosoma cruzi. Mol Immunol. 2010;47:1981–1988. doi: 10.1016/j.molimm.2010.04.016. [DOI] [PubMed] [Google Scholar]
- Reyes TM, Walker JR, DeCino C, Hogenesch JB, Sawchenko PE. Categorically distinct acute stressors elicit dissimilar transcriptional profiles in the paraventricular nucleus of the hypothalamus. J Neurosci. 2003;23:5607–5616. doi: 10.1523/JNEUROSCI.23-13-05607.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riveros C, Mellor D, Gandhi KS, McKay FC, Cox MB, Berretta R, Vaezpour SY, Inostroza-Ponta M, Broadley SA, Heard RN, Vucic S, Stewart GJ, Williams DW, Scott RJ, Lechner-Scott J, Booth DR, Moscato P. A transcription factor map as revealed by a genome-wide gene expression analysis of whole-blood mRNA transcriptome in multiple sclerosis. PLoS One. 2010;5:e14176. doi: 10.1371/journal.pone.0014176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruddy MJ, Wong GC, Liu XK, Yamamoto H, Kasayama S, Kirkwood KL, Gaffen SL. Functional cooperation between interleukin-17 and tumor necrosis factor-alpha is mediated by CCAAT/enhancer-binding protein family members. J Biol Chem. 2004;279:2559–2567. doi: 10.1074/jbc.M308809200. [DOI] [PubMed] [Google Scholar]
- Rutella S, Danese S, Leone G. Tolerogenic dendritic cells: cytokine modulation comes of age. Blood. 2006;108:1435–1440. doi: 10.1182/blood-2006-03-006403. [DOI] [PubMed] [Google Scholar]
- Sandhir R, Berman NE. Age-dependent Response of CCAAT/Enhancer Binding Proteins following Traumatic Brain Injury in Mice. Neurochem Int. 2010;56:188–193. doi: 10.1016/j.neuint.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz M, Shechter R. Systemic inflammatory cells fight off neurodegenerative disease. Nat Rev Neurol. 2010;6:405–410. doi: 10.1038/nrneurol.2010.71. [DOI] [PubMed] [Google Scholar]
- Shen F, Hu Z, Goswami J, Gaffen SL. Identification of common transcriptional regulatory elements in interleukin-17 target genes. J Biol Chem. 2006;281:24138–24148. doi: 10.1074/jbc.M604597200. [DOI] [PubMed] [Google Scholar]
- Steiner GE, Newman ME, Paikl D, Stix U, Memaran-Dagda N, Lee C, Marberger MJ. Expression and function of pro-inflammatory interleukin IL-17 and IL-17 receptor in normal, benign hyperplastic, and malignant prostate. Prostate. 2003;56:171–182. doi: 10.1002/pros.10238. [DOI] [PubMed] [Google Scholar]
- Sterneck E, Paylor R, Jackson-Lewis V, Libbey M, Przedborski S, Tessarollo L, Crawley JN, Johnson PF. Selectively enhanced contextual fear conditioning in mice lacking the transcriptional regulator CCAAT/enhancer binding protein delta. Proc Natl Acad Sci U S A. 1998;95:10908–10913. doi: 10.1073/pnas.95.18.10908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, Fugger L. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. 2008;172:146–155. doi: 10.2353/ajpath.2008.070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallières L, Sawchenko PE. Bone marrow-derived cells that populate the adult mouse brain preserve their hematopoietic identity. J Neurosci. 2003;23:5197–5207. doi: 10.1523/JNEUROSCI.23-12-05197.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan JM, Rivier J, Corrigan AZ, McClintock R, Campen CA, Jolley D, Voglmayr JK, Bardin CW, Rivier C, Vale W. Detection and purification of inhibin using antisera generated against synthetic peptide fragments. Methods Enzymol. 1989;168:588–617. doi: 10.1016/0076-6879(89)68044-5. [DOI] [PubMed] [Google Scholar]
- Waldner H, Collins M, Kuchroo VK. Activation of antigen-presenting cells by microbial products breaks self tolerance and induces autoimmune disease. J Clin Invest. 2004;113:990–997. doi: 10.1172/JCI19388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedel A, Ziegler-Heitbrock HW. The C/EBP family of transcription factors. Immunobiology. 1995;193:171–185. doi: 10.1016/s0171-2985(11)80541-3. [DOI] [PubMed] [Google Scholar]
- Yoshiki R, Kabashima K, Sakabe J, Sugita K, Bito T, Nakamura M, Malissen B, Tokura Y. The mandatory role of IL-10-producing and OX40 ligand-expressing mature Langerhans cells in local UVB-induced immunosuppression. J Immunol. 2010;184:5670–5677. doi: 10.4049/jimmunol.0903254. [DOI] [PubMed] [Google Scholar]
- Zozulya AL, Ortler S, Lee J, Weidenfeller C, Sandor M, Wiendl H, Fabry Z. Intracerebral dendritic cells critically modulate encephalitogenic versus regulatory immune responses in the CNS. J Neurosci. 2009;29:140–152. doi: 10.1523/JNEUROSCI.2199-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]