

Summary
The size of the peripheral T lymphocyte compartment is governed by complex homeostatic mechanisms that balance T cell proliferation and death. Proliferation and survival signals are mediated in part by recurrent self-peptide/MHC-TCR interactions and signaling by the common γ chain-containing cytokine receptors, including those for IL-7 and IL-15. We have previously shown that the death receptor Fas (CD95/APO-1) regulates apoptosis in response to repeated TCR stimulation whereas the BH3-only protein Bim mediates cytokine withdrawal-induced apoptosis. We therefore reasoned that these two molecules might cooperate in the regulation of homeostatic proliferation. In this study, we observe that the combined loss of Fas and Bim synergistically enhances the accumulation of T cells in lymphopenic host mice, and this is particularly pronounced for the unusual CD4−CD8−TCRαβ+ T cells that are characteristic of Fas-deficient (Faslpr/lpr) mice. Our findings demonstrate that these CD4−CD8−TCRαβ+ T cells arise from homeostatic proliferation of CD8+ T cells. These studies also underscore the profound rate of baseline T cell proliferation that likely occurs in wild-type mice even in the absence of foreign antigen, and the consequent need for its coordinated regulation by multiple death-signaling pathways.
Keywords: T lymphocyte, homeostatic proliferation, apoptosis
1. Introduction
The peripheral T lymphocyte compartment is governed by complex homeostatic mechanisms that maintain the size and diversity of the T cell repertoire while limiting the accumulation and activity of self-reactive T cells. Regulation of T cell numbers must balance the daily export of new T cells from the thymus, plus the low but significant rate of proliferation by peripheral T cells to self-antigen, with the active removal of existing T cells to prevent a continual increase in cell number. T cell homeostatic expansion requires the engagement of TCR by self-peptide/MHC complexes and γc-containing cytokine receptor-mediated signals from the IL-7 and IL-15 receptors [1-8].
By contrast, the factors that limit the homeostatic expansion of peripheral T cells are less well characterized. It has been suggested that T cell expansion following transfer to lymphopenic recipients may be restricted by competition for limiting resources, including IL-7, and access to antigen-presenting cells bearing self-peptide/MHC complexes [1, 9-11]. However, a significant portion of donor T cells continues to proliferate, as measured by BrdU incorporation, even after stable T cell numbers have been reached [12, 13]. This suggests that it is not entirely limitations on proliferation, but also active cell death that prevents further increase in T cell numbers. We have observed previously that T cells undergoing lymphopenia-induced proliferation are sensitive to Fas-mediated cell death, and Fas-deficient lpr (Faslpr/lpr) T cells accumulate to substantially greater numbers as compared with wild-type T cells following transfer into lymphopenic hosts [12]. Thus, expansion of the T cell pool in response to self-antigen/MHC>TCR signaling is counter-balanced in part by Fas-mediated cell death.
Fas-deficient lpr mice (Faslpr/lpr) manifest a profound age-dependent lymphadenopathy that includes the accumulation of both CD4+ and CD8+ T cells as well as a distinctly unusual population of polyclonal CD4−CD8−TCRαβ+ T cells that express the B cell isoform of CD45, CD45R (B220), but lack NK1.1 [14]. This phenotype is not observed in mice deficient for other death-inducing molecules, such as Bim or Bax/Bak [14-16]. Although the Faslpr/lpr genotype was originally identified 18 years ago as a retroposon-mediated disruption of the Fas gene [17], the in vivo biological process uniquely regulated by Fas has remained an enigma for many years. Little if any defect in thymic negative selection has been identified in Faslpr/lpr mice [18-23]. In addition, most studies observed little delay in the deletion of mature, peripheral Faslpr/lpr T cells in response to the administration of a single dose of antigen or following acute infection [24-26]. However, Fas does contribute to the elimination of T cells during chronic infections [25, 27]. Yet none of the studies using either acute or chronic infection models reported increased lymphadenopathy as compared with uninfected Faslpr/lpr mice or the appearance of antigen-specific CD4−CD8−TCRαβ+B220+ T cells. Thus, they do not provide an adequate explanation for the lymphadenopathy observed in Faslpr/lpr mice. This is consistent with earlier findings that Faslpr/lpr mice developed lymphadenopathy even when they were raised under germ-free or antigen-free conditions [28].
Although CD4−CD8−TCRαβ+B220+ T cells are unique to Faslpr/lpr and Fas-Ligand mutant (FasLgld/gld) mice [17, 29], understanding the origin and derivation of these T cells will likely be a valuable guide to defining the true in vivo function of Fas. Despite the progressive lymphadenopathy of Faslpr/lpr mice, the accumulation of T cells does not occur as rapidly as predicted based on the rate of proliferation determined by BrdU incorporation. We have previously observed that approximately 10-15% of CD4−CD8−TCRαβ+B220+ T cells incorporate BrdU in a 24 h period [12]. At this rate, the total number of CD4−CD8−TCRαβ+B220+ T cells would double every 7 days in the absence of any cell death. Thus, we considered that other death signals independent of FasL/Fas might also regulate the survival of T cells preserved in the absence of Fas.
In addition to cell death mediated by death receptors, initiation of apoptosis is controlled by a Bcl-2-regulated pathway that is initiated by the pro-apoptotic Bcl-2 homology domain 3 (BH3)-only proteins [30]. BH3-only proteins initiate cell death through indirect or direct activation of the pro-apoptotic Bax/Bak subgroup of the Bcl-2 family, leading to mitochondrial outer membrane permeablization, apoptosome formation, and activation of an effector caspase cascade [30]. BH3-only protein Bim (also known as Bcl2l11) is essential for T cell death during thymic negative selection and following cytokine deprivation [15, 31]. In contrast, Fas has been implicated in the deletion of T cells receiving recurrent TCR signals [32, 33]. We therefore reasoned that Fas and Bim might regulate non-overlapping death pathways to control homeostatic proliferation.
2. Results
Increased lymphadenopathy in Bim-/-Faslpr/lpr mice results in part from reduced cell death
To determine whether Fas and Bim regulate complimentary processes in homeostatic proliferation, we generated Bim-/-Faslpr/lpr mice. At 8 wk of age, as compared with wild-type mice, there was a trend toward an increase in CD4+ and CD8+ T cell numbers in Bim-/-, Faslpr/lpr, and Bim-/-Faslpr/lpr mice which achieved significance for Bim-/-Faslpr/lpr mice (Fig. 1A). In addition, Bim-/-Faslpr/lpr mice already exhibited an increase in CD4−CD8−TCRαβ+B220+ T cells as compared with Faslpr/lpr mice (Fig. 1A). At 14 weeks of age, the numbers of both CD4+ as well as CD8+ T cells were increased nearly 3-fold in Faslpr/lpr mice as compared with wild-type mice (Fig. 1B). Consistent with previous reports [15], Bim-deficient mice exhibited a modest accumulation of CD4+ (~1.5-fold) and CD8+ (~2.0-fold) T cells as compared with wild-type mice, but did not contain detectable CD4−CD8−TCRαβ+ B220+ T cells that are characteristic of Faslpr/lpr mice. Remarkably, Bim-/-Faslpr/lpr mice developed massive lymphadenopathy (Fig 1B) at a dramatically increased rate as compared with either Fas- or Bim-deficient mice. By 14 wk, Bim-/-Faslpr/lpr mice had 5-6-fold more CD4+ and CD8+ T cells than wild-type mice. This also represented approximately 2-fold more CD4+ T cells and CD8+ T cells than Faslpr/lpr mice. The number of CD4−CD8−TCRαβ+B220+ T cells was also dramatically increased (~8-fold) as compared with Faslpr/lpr mice. At 14 wk, the differences in cell number between Bim-/-Faslpr/lpr and the other three genotypes were significant for the CD4+, CD8+, and CD4−CD8−TCRαβ+B220+ T cell subsets. Thus, Fas and Bim exert non-overlapping functions in the control of basal T cell homeostasis.
Figure 1. Accelerated lymphadenopathy in Bim-/-Faslpr/lpr mice.
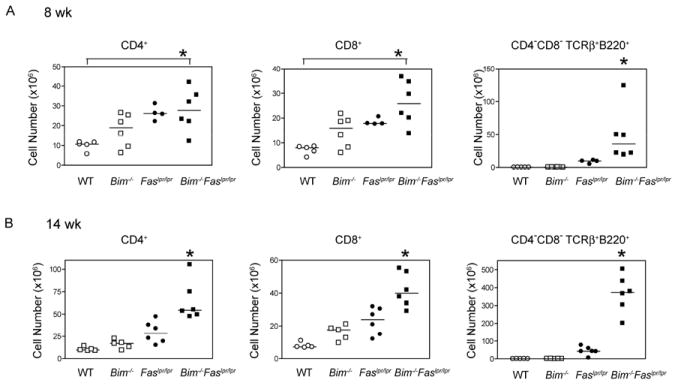
The absolute numbers of CD8+, CD4+, and CD4−CD8−TCRαβ+B220+ lymph node T cells recovered from wild-type (WT), Bim-/-, Faslpr/lpr, and Bim-/-Faslpr/lpr mice at (A) 8 wk and (B) 14 wk of age. Lymph node cells were stained for CD4, CD8, TCRβ, and CD45R (B220) and analyzed by flow cytometry. Cell numbers were calculated based on the percent positive cells. Each symbol represents an individual mouse analyzed (n=4-6 per group). Horizontal lines indicate the median number for each T cell subset. At 8 wk, differences in cell number were significant between Bim-/-Faslpr/lpr and wild-type T cells for both CD4+ and CD8+ T cells (ANOVA, Tukey post-test, *p< 0.05). At 14 wk, differences in cell number between Bim-/-Faslpr/lpr and the parental genotype T cells were significant for both CD4+ and CD8+ T cells (ANOVA, Tukey post-test, *p< 0.05). Differences in the number of CD4−CD8−TCRαβ+B220+ T cells between Bim-/-Faslpr/lpr and Faslpr/lpr were significant at both 8 and 14 wk (t test, *p<0.05).
To examine if the elevated T lymphocyte numbers in Bim-/-Faslpr/lpr mice resulted from increased proliferation, BrdU incorporation was used to determine the fraction of T cells that underwent cell cycling during a 24 h labeling period. An increased fraction of Faslpr/lpr and Bim-/-Faslpr/lpr CD4+ and CD8+ T cells incorporated BrdU as compared with wild-type and Bim-/- T cells (Fig. 2A). Consistent with our previous report [12], CD4−CD8−TCRαβ+B220+ T cells manifested a high rate of BrdU incorporation relative to CD4+ and CD8+ T cells, and this was comparable between Faslpr/lpr and Bim-/-Faslpr/lpr CD4−CD8−TCRαβ+B220+ T cells. Because the mice were not intentionally challenged with any foreign antigen, we considered that this reflected an increase in the proportion of T cells that had undergone cell cycling in response to stimulation by self-antigens.
Figure 2. Bim-/-Faslpr/lpr T cells manifest reduced apoptosis but equivalent rates of BrdU incorporation as compared with Faslpr/lpr T cells.
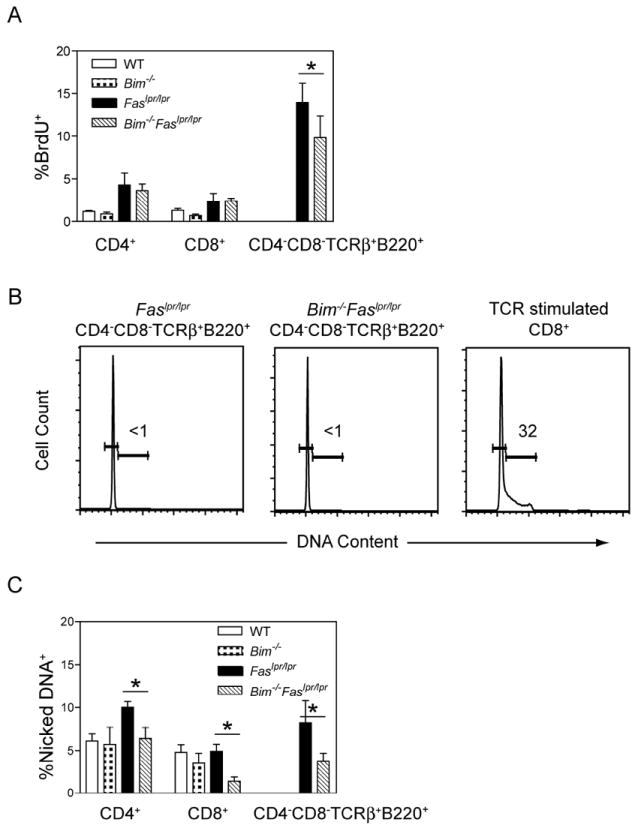
(A) Age and sex-matched wild-type (WT), Bim-/-, Faslpr/lpr, and Bim-/-Faslpr/lpr mice received four intraperitoneal injections of BrdU (1 mg each) over 24 h. Lymph node cells were stained for surface expression of CD4, CD8, CD45R (B220), and analyzed for BrdU incorporation by flow cytometry. Shown are the mean and standard deviation for the percentage of BrdU+ cells in each T cell subset from 14 wk old mice (n=4 per strain). The percentages of BrdU+ cells between Bim-/-Faslpr/lpr and Faslpr/lpr T cells were statistically different for the CD4−CD8−TCRαβ+B220+, but not for the CD4+ and CD8+ T cell subsets (ANOVA, Tukey post-test, *p<0.05). These data are representative of two experiments.
(B) Lymph node cells from age and sex-matched Faslpr/lpr and Bim-/-Faslpr/lpr mice were stained for surface expression of CD4, CD8, CD45R (B220), and TCRβ and analyzed for DNA content by Hoechst staining. Shown are representative Hoechst profiles for CD4−CD8−TCRαβ+B220+ T cells from Faslpr/lpr (n=3) and Bim-/-Faslpr/lpr mice (n=3) and for wild-type CD8+ T cells on day 3 after in vitro stimulation with plate-bound anti-CD3 and anti-CD28 antibodies. Number inserts are the percentage of cells in S/G2/M.
(C) Single cell suspensions of lymph node cells from age and sex-matched wild-type (WT), Bim-/-, Faslpr/lpr, and Bim-/-Faslpr/lpr mice were cultured at 37°C for 4 h. The cells were then stained for surface expression of CD4, CD8, CD45R (B220), and TCRβ and analyzed for the presence of nicked DNA by TUNEL assay. The fraction of cells containing nicked DNA was calculated for each T cell subset. Shown are the mean and standard deviation of the percentage of apoptotic cells in each T cell subset from 14 wk old mice (n=3-4 per strain). The percentages of apoptotic cells between Bim-/-Faslpr/lpr and Faslpr/lpr T cells were statistically different for the CD4+, CD8+, and CD4−CD8−TCRαβ+B220+ T cell subsets (ANOVA, Tukey post-test, *p<0.05). These data are representative of two experiments.
Despite exhibiting a high rate of BrdU incorporation over 24 h, when examined for DNA content at a single time point by Hoechst staining, virtually none of the CD4−CD8−TCRαβ+B220+ T cells had >2N DNA indicating that they were not in S/G2/M (Fig. 2B). We also observed that CD4−CD8−TCRαβ+B220+ T cells from lymph nodes of Faslpr/lpr mice transgenically expressing GFP under the control of the Rag promoter (Rag-GFP-transgenic Faslpr/lpr) [34] manifested very little GFP expression as compared with lymph node CD8+ T cells (Supplemental Figure 1). In these mice, transcription of GFP protein occurs in early thymocytes at the time of Rag expression and ceases when TCR rearrangement is completed. GFP protein persists in thymic emigrants but is progressively lost as they proliferate. Collectively, these observations suggested that Faslpr/lpr CD4−CD8−TCRαβ+B220+ T cells do not arise de novo in the thymus and their immediate precursors undergo several rounds of division just prior to becoming CD4−CD8−.
To investigate differences in spontaneous cell death, freshly isolated lymph node cells were analyzed by TUNEL staining. There were no differences in the percentage of apoptotic CD4+ T cells among wild-type, Faslpr/lpr, Bim-/-, and Bim-/-Faslpr/lpr mice (Fig. 2C). By contrast, Bim-/-Faslpr/lpr CD8+ T cells had a decreased proportion of apoptotic cells as compared with wild-type, Faslpr/lpr, and Bim-/- mice. Furthermore, there was a reduction in the proportion of apoptotic Bim-/-Faslpr/lpr CD4−CD8−TCRαβ+B220+ T cells as compared with the same subset from Faslpr/lpr mice. As TUNEL staining measures the proportion of apoptotic cells at a single time point, this reduction in the level of cell death summed over time would lead to a substantial increase in T cell numbers in Bim-/-Faslpr/lpr mice. Thus, decreased cell death rather than increased cell cycling likely contributed to the abnormal accumulation of T lymphocytes in Bim-/-Faslpr/lpr mice.
The synergy between Fas and Bim in T cell apoptosis might reflect the fact that they regulate different aspects of cell death during T cell homeostasis. To determine the effects of the combined mutations on T cell death, T cells were initially activated in vitro with plate-bound anti-CD3 and anti-CD28 antibodies in the presence of IL-2 for 2 days and then removed from anti-CD3 antibody stimulation and further expanded with IL-2 for an additional 3 days. On day 5, IL-2 expanded T cell blasts were restimulated anti-CD3 antibody to induce cell death. T cell blasts from both Faslpr/lpr and Bim-/-Faslpr/lpr mice, but not those from wild-type or Bim-/- mice, were resistant to TCR/CD3 restimulation-induced cell death (Fig. 3A, B). Thus, Fas, but not Bim, mediated death from TCR/CD3 restimulation.
Figure 3. Fas-deficient T cells are protected from CD3-restimulation-induced cell death whereas Bim-deficient T cells are protected from cytokine withdrawal.
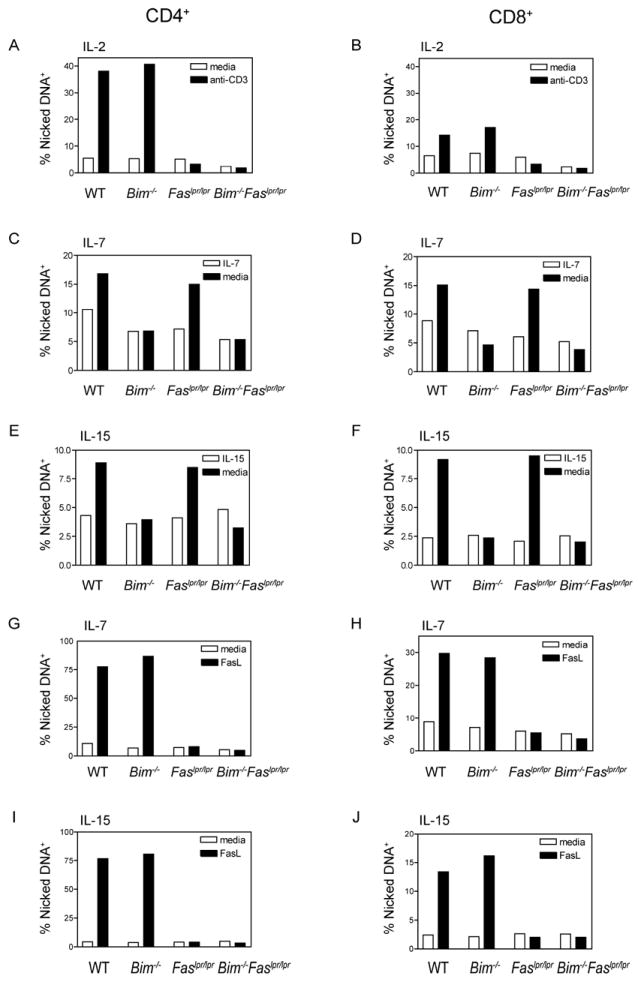
Single cell suspensions of lymph node cells from age and sex-matched wild-type (WT), Bim-/-, Faslpr/lpr, and Bim-/-Faslpr/lpr mice were stimulated with plate-bound anti-CD3 and anti-CD28 antibodies in the presence of (A, B) IL-2, (C, D, G, H) IL-7, or (E, F, I, J) IL-15. On day 2 of culture, the cells were removed from the anti-CD3/anti-CD28 antibody stimulation, washed, and cultured in the presence of IL-2, IL-7, or IL-15 consistent with the initial culture conditions. (A, B) On day 5, IL-2-cultured T cell blasts were restimulated with plate-bound anti-CD3 antibody for 6 h or left unstimulated. (C, D, E, F) T cells grown in either IL-7 or IL-15 for 5 days were washed three times to remove cytokines and then cultured in medium containing IL-7 orIL-15, respectively, or medium alone for an additional 16 h. (G, H, I, J) T cells grown in either IL-7 or IL-15 for 5 days were cultured for 2 h either with or without FLAG-tagged FasL cross-linked by anti-FLAG antibody. (A-J) The cells were then stained for surface expression of CD4, CD8, CD45R (B22O), and TCRβ and analyzed for the presence of nicked DNA by TUNEL staining. The fraction of cells containing nicked DNA was calculated for the CD4+ and CD8+ T cell subsets. These data are representative of two independent experiments (n=1 per strain).
T cells grown in the presence of IL-7 and IL-15 were tested for sensitivity to cytokine withdrawal-induced apoptosis. T cells were activated for 2 days with anti-CD3 and anti-CD28 antibodies in the presence of either IL-7 or IL-15, then removed from anti-CD3 antibody stimulation, and grown for an additional 3 days with either IL-7 or IL-15. On day 5, the T cells were washed and then returned to culture in the absence of cytokine. The absence of Bim, but not absence of Fas, protected T cells from cytokine withdrawal-induced death (Fig. 3C-F). Finally, as expected, IL-7- and IL-15-cultured wild-type and Bim-/- T cells, but not those from Faslpr/lpr or Bim-/-Faslpr/lpr mice, were sensitive to Fas-induced cell death (Fig. 3G-J). Thus, T cells from Bim-/-Faslpr/lpr mice were uniquely resistant to both TCR restimulation and cytokine withdrawal.
Fas and Bim regulate non-overlapping death pathways during lymphopenia-induced proliferation
To examine if the combined loss of Fas and Bim would act synergistically in T cell survival during homeostatic proliferation, we used the experimental model of lymphopenia-induced proliferation by adoptively transferring equal numbers of donor T cells into Rag1-deficient recipients. On day 30 post-transfer, the number of CD4+ T cells recovered from recipients that received Bim-/-Faslpr/lpr T cells was on average 7-fold higher than those found in recipients that received wild-type cells, 4.4-fold more than in mice that received Bim-/- T cells, and 3.7-fold more than in mice that received Faslpr/lpr T cells, (Fig. 4A). The increased T cell number in recipients of Bim-/-Faslpr/lpr T cells was even more striking for the CD8+ T cell subset (Fig. 4B). Recipients that received Bim-/-Faslpr/lpr T cells had on average 22-fold more CD8+ T cells than mice that received wild-type T cells, ~17-fold more than in mice that received Bim-/- T cells, and ~8-fold more than mice that received Faslpr/lpr T cells. By contrast, mice that received Bim-/- T cells had only a slight increase in CD4+ and CD8+ T cells as compared with wild-type T cells and this difference was not statistically significant.
Figure 4. Bim-/-Faslpr/lpr T cells accumulate to significantly higher levels following transfer to lymphopenic recipients.
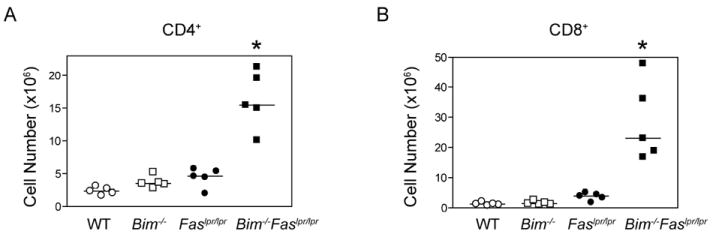
CD90.2+ Rag1-deficient mice received a single inoculum of 3.5×106 lymph node T cells from CD90.1+ wild-type, Bim-/-, Faslpr/lpr, or Bim-/-Faslpr/lpr mice. The donor mice were 5 wk of age and had equivalent proportions of CD4+ and CD8+ T cells. On day 31 post-transfer, lymph node and spleen cells from recipient mice were surfaced stained for CD4, CD8, and TCRβ and analyzed by flow cytometry. Absolute CD4+ and CD8+ T cell numbers were calculated based on the percent positive cells. Each symbol represents an individual mouse analyzed (n=5 per group). Differences in cell number between Bim-/-Faslpr/lpr and the parental genotype T cells were significant for both the CD4+ and CD8+ T cell subsets (ANOVA, Tukey post-test, *p< 0.05). These data are representative of two independent experiments.
To determine whether the greater number of Bim-/-Faslpr/lpr T cells recovered following lymphopenia-induced expansion resulted from an increased fraction of cells entering the cell cycle, we analyzed initiation of cell cycling after adoptive transfer of CFSE-labeled wild-type, Faslpr/lpr, Bim-/-, and Bim-/-Faslpr/lpr T cells into lymphopenic Rag1-deficient recipients. As shown in Figure 5, the fraction of cycling cells was not different among the four donor genotypes, although, a slight delay in the initiation of cell cycling by Bim-/-, and Bim-/-Faslpr/lpr donor T cells was occasionally observed, consistent with a previous report [35]. In vivo BrdU incorporation was used to determine whether similar cycling rates persisted at later time points after T cell transfer, beyond the number of cycles detectable by CFSE. In mice that received BrdU from day 23 to 24, there was no statistically significant difference in the fraction of cycling cells between wild-type and Faslpr/lpr donor cells or between Bim-/- and Bim-/-Faslpr/lpr donor cells (Fig. 6A), in agreement with the CFSE findings. However, Bim-/- and Bim-/-Faslpr/lpr donor cells manifested a reduction in cell cycling as compared with their counterparts with functional Bim, similar to the CFSE studies.
Figure 5. Similar cell cycle entry of wild-type, Bim-/-, Faslpr/lpr, and Bim-/-Faslpr/lpr T cells following transfer into lymphopenic recipients.
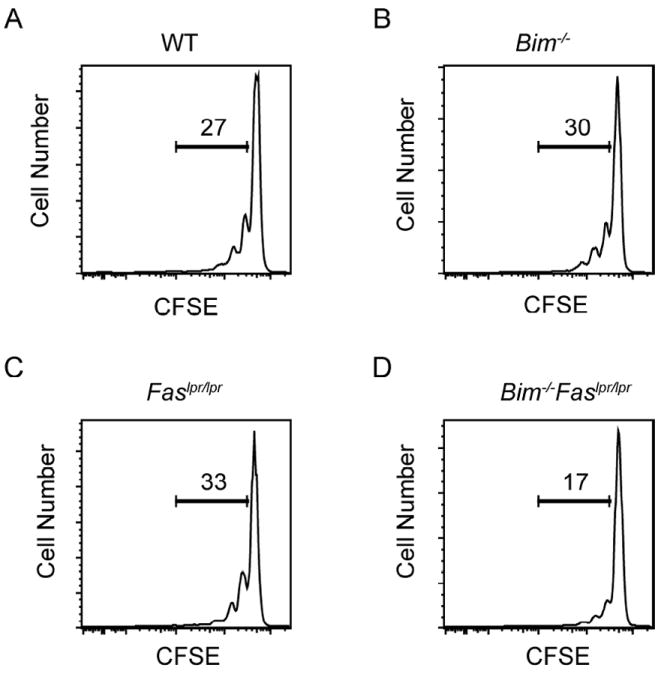
Lymph node cells from 5 wk-old wild-type (WT), Bim-/-, Faslpr/lpr, and Bim-/-Faslpr/lpr mice (n=1 per strain) were labeled with CFSE and transferred into Rag1-deficient recipients. On day 3 post-transfer, lymph node and spleen cells were stained for CD4, CD8, and TCRβ and analyzed for CFSE levels by flow cytometry. Shown are representative CFSE profiles for (A) wild-type (WT), (B) Bim-/-, (C) Faslpr/lpr, and (D) Bim-/-Faslpr/lpr T cells. The numbers in the graphs represent the percentage of cells that have undergone 1 or more cell divisions. Data are representative of two independent experiments.
Figure 6. Bim-/-Faslpr/lpr T cells undergoing lymphopenia-induced proliferation manifest decreased BrdU incorporation and a reduced proportion of apoptotic cells as compared with Faslpr/lpr T cells.
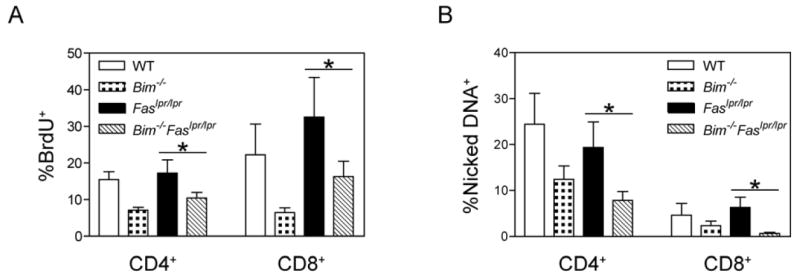
(A) Rag1-deficient recipients received four intraperitoneal injections of BrdU (1 mg each) over the 24 h period from day 23 to 24 post adoptive transfer of equivalent numbers of wild-type (WT), Bim-/-, Faslpr/lpr, or Bim-/-Faslpr/lpr lymph node T cells. On day 24, lymph node and spleen cells were isolated from the Rag1-deficient recipient mice, surfaced stained for CD4, CD8, and TCRβ, and analyzed for BrdU incorporation by flow cytometry. Shown are the means and standard deviations for the fraction of BrdU+ cells in the CD4+ and CD8+ T cell subsets (n=5 per donor strain). Differences between Bim-/-Faslpr/lpr and Faslpr/lpr donor T cells were statistically significant for both the CD4+ and CD8+ T cell subsets (ANOVA, Tukey post-test, *p< 0.05). Differences between Bim-/-Faslpr/lpr and Bim-/- donor T cells were not statistically significant. Data are representative of two independent experiments
(B) Lymph node and spleen cells were isolated from Rag1-deficient mice that received wild-type (WT), Bim-/-, Faslpr/lpr, or Bim-/-Faslpr/lpr lymph node cells 24 days previously. The cells were cultured in vitro at 37°C for 3 h, surfaced stained for CD4, CD8, and TCRβ, and analyzed for the presence of nicked DNA by TUNEL staining. The fraction of cells containing nicked DNA was calculated for each T cell subset. Shown is the mean and standard deviation (n=5 per donor strain). Differences between Bim-/-Faslpr/lpr and Faslpr/lpr donor T cells were statistically significant for both the CD4+ and CD8+ T cell subsets (ANOVA, Tukey post-test, *p< 0.05). Differences between Bim-/-Faslpr/lpr and Bim-/- donor T cells were not statistically significant. Data are representative of two independent experiments
To assess any differences in cell death of T cells undergoing lymphopenia-induced expansion, donor T cells recovered from Rag1-deficient recipients 24 days post-transfer were analyzed for apoptosis by TUNEL staining. For both CD4+ as well as CD8+ T cells, Bim-/-Faslpr/lpr T cells manifested the lowest levels of cell death of the four donor genotypes (Fig. 6B). Thus, loss of Bim further preserves Faslpr/lpr T cells that would have undergone apoptosis during lymphopenia-induced expansion. The increased expansion of Bim-/-Faslpr/lpr T cells, therefore, likely results from diminished cell death and not increased cell cycling.
CD4−CD8−TCRαβ+NK1.1− T cells arise from CD8+ T cells during lymphopenia-induced proliferation
A unique feature of the lymphadenopathy of Faslpr/lpr mice is the accumulation of polyclonal CD4−CD8−TCRαβ+B220+NK1.1− T cells [14]. Of note was that modest numbers of these CD4−CD8−TCRαβ+NK1.1− T cells were recovered from Rag1-deficient recipients that received lymph node cells from young Faslpr/lpr mice prior to the onset on lymphadenopathy (Fig. 7A). Remarkably, the number of CD4−CD8−TCRαβ+NK1.1− T cells recovered from recipients of Bim-/-Faslpr/lpr T cells was increased ~7-fold as compared with recipients of Faslpr/lpr T cells. Since the donor T cells also came from young Bim-/-Faslpr/lpr mice prior to the onset of lymphadenopathy or the accumulation of CD4−CD8−TCRαβ+B220+ T cells, the CD4−CD8−TCRαβ+ T cells likely arose from a phenotypically different precursor. Previous genetic studies have implied that Faslpr/lpr CD4−CD8−TCRαβ+B220+ T cells are derived from CD8+TCRαβ+ precursors [28, 36, 37] but the process that might induce this transition was never defined. To examine whether CD4−CD8−TCRαβ+ T cells could arise from CD8+ T cells during lymphopenia-induced proliferation, we transferred purified CD8+ T cells from lymph nodes of Bim-/-Faslpr/lpr mice into Rag1-deficient recipients (Fig. 7B). CD4−CD8−TCRαβ+NK1.1− T cells were readily detected in mice that received injections of Bim-/-Faslpr/lpr CD8+ T cells (Fig. 7C). Only a few CD4−CD8−TCRαβ+NK1.1− T cells were recovered on day 14 post-transfer, but their numbers increased with time and were 4-fold more on day 22 as compared with day 14. In contrast, when purified CD4+ T cells from lymph nodes of Faslpr/lpr or Bim-/-Faslpr/lpr mice were transferred, no increase in the numbers of CD4−CD8−TCRαβ+NK1.1− T cells was apparent up to 23 days post-transfer (Fig. 7C). A small number of CD4−CD8−TCRαβ+NK1.1− T cells were observed at day 23 post-transfer in one of the mice that received CD4+ T cells but this particular mouse also harbored a slight expansion of the CD8+ T cell contaminants. It is possible that the CD4−CD8−TCRαβ+ T cells identified following the transfer of CD8+ T cells could have arisen from the expansion of the minor contamination of CD4−CD8−TCRαβ+ T cells (3-6%) in the donor CD8+ T cell inocula. However, this appears unlikely given that the donor CD4+ T cell inocula contained similar levels of contaminating CD4−CD8−TCRαβ+ T cells as the CD8+ T cells, but no accumulation of CD4−CD8−TCRαβ+ T cells was observed in the recipients of CD4+ T cells.
Figure 7. CD4−CD8−TCRαβ+NK1.1− T cells were generated during homeostatic expansion of Faslpr/lpr CD8+, but not CD4+, T cells.
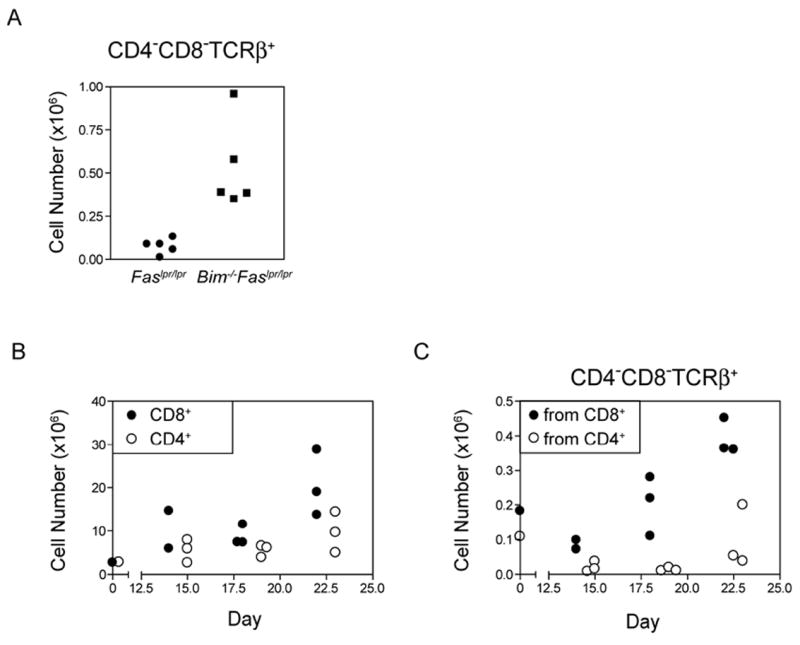
(A) Rag1-deficient mice received a single inoculum containing 3.5×106 lymph node T cells from 5 wk old Faslpr/lpr or Bim-/-Faslpr/lpr mice. On day 30 post-transfer, lymph node and spleen cells from recipient mice were surfaced stained for CD4, CD8, NK1.1, and TCRβ and analyzed by flow cytometry. Absolute cell numbers of CD4−CD8−TCRαβ+NK1.1− T cells were calculated based on the percent positive cells. (B, C) CD8+ or CD4+ T cells were purified from lymph nodes of Bim-/-Faslpr/lpr mice by negative selection and a single equal inoculum of 3×106 cells was transferred to Rag1-deficient mice (day 0). On the days indicated post-transfer, lymph node and spleen cells from recipient mice were surfaced stained for CD4, CD8, NK1.1, and TCRβ and analyzed by flow cytometry. Cell numbers were calculated based on the percent positive cells. Shown are the numbers of (B) CD8+ and CD4+ T cells and (C) CD4−CD8−TCRαβ+NK1.1− T cells recovered following the transfer of CD8+ and CD4+ T cells. These data are representative of two independent experiments for each donor population.
3. Discussion
The current observations demonstrate that the death receptor Fas and the pro-apoptotic BH3-only Bcl-2 family member, Bim, synergize in the regulation of cell survival during T cell homeostatic expansion. This was consistent in both a model system of lymphopenia-induced proliferation as well as in intact mice lacking both Fas and Bim, which had greatly enhanced lymphadenopathy and splenomegaly. In addition, lymphopenia-induced proliferation of transferred Faslpr/lpr or Bim-/-Faslpr/lpr CD8+ T cells, but not CD4+ T cells, yielded the unique CD4−CD8−TCRαβ+NK1.1− T cells that are characteristic of Fas- and Fas ligand-deficient mice. No other antigen-driven system has reported generating CD4−CD8−TCRαβ+ T cells from Faslpr/lpr CD8+ precursors. Thus, our findings support a model in which Fas and Bim cooperate to regulate non-overlapping death pathways during T cell homeostasis and also link, for the first time, homeostatic proliferation of CD8+ T cells to the generation of the unusual CD4−CD8−TCRαβ+B220+ T lymphoid cells of Faslpr/lpr mice.
The model system that defines the function of Fas in vivo should recapitulate both the increased T cell numbers as well as the appearance of CD4−CD8−TCRαβ+B220+ T cells. Previous studies on the role of Fas have examined the accumulation of T cells after various types of antigen exposure but have rarely if ever addressed the origin of the CD4−CD8−TCRαβ+ T cells in the same in vivo system. CD4−CD8−TCRαβ+B220+ T cells in Faslpr/lpr mice are polyclonal and therefore do not arise from a limited number of TCR specificities, as with CD4−CD8−NK T cells [14, 38]. Faslpr/lpr CD4−CD8−TCRαβ+B220+ T cells have been postulated to be derived from CD8+ T cell precursors based on studies showing demethylation of the CD8α gene in CD4−CD8−TCRαβ+B220+ T cells from Faslpr/lpr mice [36]. Consistent with this model, β2m-deficient Faslpr/lpr mice have a defect in positive selection of CD8+ T cells and are nearly devoid of CD4−CD8−TCRαβ+B220+ T cells despite accumulating CD4+ T cells and B cells [28, 37], whereas the loss of peripheral CD4+ T cells in MHC class II-deficient Faslpr/lpr mice does not prevent the accumulation of CD4−CD8−TCRαβ+B220+ T cells [39]. However, none of these studies demonstrated the process by which CD8+ T cells become CD4−CD8−TCRαβ+ T cells. Our findings demonstrate for the first time that CD8+ T cells give rise to CD4−CD8−TCRαβ+NK1.1− T cells in the unique environment of chronic TCR stimulation during homeostatic proliferation.
Basal homeostasis and apoptosis of T cells are dysregulated in mice lacking either Fas or Bim, yet only Fas-deficient mice develop significant lymphadenopathy [14, 15]. While loss of Bim alone results in only a ~3-fold increase in T cell numbers, it considerably augments lymphadenopathy in conjunction with loss of Fas [25, 35, 40]. Fas has been implicated in the elimination of T cells receiving recurrent TCR signals based on the prominent role of Fas in mediating the death of in vitro IL-2-cultivated T cell blasts following TCR-restimulation [32, 33]. In distinction, Bim is critical for T cell death during thymic negative selection and following cytokine deprivation as occurs when cytokine levels decline following pathogen clearance during acute infections [15, 31]. Notably, Bim is also critical for calcium flux-induced T lymphocyte apoptosis [15] and the increased expression of Bim in T cell blasts following TCR restimulation is thought to contribute to TCR restimulation-induced death, especially in CD8+ T cells [41]. As such, Bim, but not Fas, contributes to T cell elimination during the termination of acute T cell immune responses following in vitro or in vivo administration of a single dose of Staphylococcal enterotoxin B, or in vivo after acute infection with herpes simplex virus-1 [24, 42]. In contrast, Fas and Bim synergized in the contraction of the T cell response to chronic infections, such as with mouse γ-herpes virus [25]. As homeostatic proliferation is driven by both low affinity recurrent TCR stimulation in response to self-peptide/MHC complexes and certain cytokines [5], it resembles chronic antigenic stimulation but the array of self-peptides likely acts upon a much broader repertoire of T cells than occurs with a given foreign antigen.
The loss of Bim in addition to the loss of Fas greatly enhanced the appearance of CD4−CD8−TCRαβ+B220+ T cells and resulted in an increased ratio of CD4−CD8−TCRαβ+B220+ T cells to CD8+ T cells as compared with mice lacking only Fas. Since Bim-deficient mice do not accumulate CD4−CD8−TCRαβ+B220+ T cells [15], this suggests that the absence of Bim could either cause the generation of more CD4−CD8−TCRαβ+B220+ T cell precursors (naïve CD8+ T cells) and/or preferentially increase the survival of CD4−CD8−TCRαβ+B220+ T cells in Faslpr/lpr mice. Bim-deficient mice have impaired deletion of self-reactive T cells during thymic negative selection [31]. Thus, the loss of Bim may indeed result in an increased number of self-reactive CD8+ T cells in the periphery that could subsequently proliferate in response to self-peptide/MHC and become CD4−CD8−TCRαβ+B220+ T cells. Alternatively, since Bim is required for growth factor deprivation-induced apoptosis [15], loss of Bim can also enhance the persistence of CD4−CD8−TCRαβ+ B220+ T cells that arise as a consequence of the absence of Fas but are present in an environment containing insufficient cytokines. Indirect evidence suggests that CD4−CD8−TCRαβ+B220+ T cells are not long-lived in Faslpr/lpr mice. Based on their rate of BrdU incorporation over 24 h (10-15%), the number of CD4−CD8−TCRαβ+B220+ T cells in Faslpr/lpr mice would be predicted to double every 7 days, but their actual number clearly does not increase this rapidly. In addition, CD4−CD8−TCRαβ+B220+ T cells express relatively low levels of Bcl-2 as compared with CD4+ and CD8+ T cells (data not shown), and therefore may be particularly vulnerable to limiting cytokines and thus Bim-mediated cell death.
Since homeostatic proliferation is driven by self-antigens and likely occurs at a much higher rate than previously appreciated, the potential for autoimmune consequences clearly exists if the expansion of T cells is not tightly controlled. In humans, lymphopenia resulting from infection or following cancer therapy is strongly linked to autoimmunity [43-45]. Moreover, an increased basal rate of peripheral T cell proliferation has been suggested to contribute to the pathogenesis of rheumatoid arthritis [46, 47]. Similarly, nonobese diabetic (NOD) mice are lymphopenic and the compensatory lymphocyte proliferation is thought to contribute to the development of their autoimmune diabetes [48]. Even in the absence of lymphopenia, alterations in T cell homeostasis may contribute to autoimmune disease. Humans bearing Fas mutations in the autoimmune lymphoproliferative syndrome (ALPS) manifest autoimmune phenomena such as hemolytic anemia [49, 50]. Similarly, Faslpr/lpr mice on certain genetic backgrounds manifest a severe autoimmune disease characterized by autoantibody-mediated glomerulonephritis [14]. The additional loss of one or both alleles of Bim synergized with the absence of Fas to greatly enhance autoimmune pathology [25, 35, 40]. Furthermore, some patients with systemic lupus erythematosus harbor an increased proportion of CD4−CD8−TCRαβ+B220+ T cells [51] which may reflect augmented homeostatic proliferation in these patients. Thus, dysregulation of T cell homeostatic proliferation may be a prime underlying mechanism of several autoimmune syndromes.
4. Materials and Methods
Mice
Mice were bred and housed in the AAALAC-approved animal facilities of The University of Vermont College of Medicine. Original breeding pairs of C57BL/6J, B6.MRL-Faslpr/J (Faslpr/lpr), B6.129S7-Rag1tm1Mom/J (Rag1-/-), and B6.PL-Thy1a/Cy (CD90.1) mice were obtained from Jackson Laboratory (Bar Harbor, ME). B6.129S1-Bcl2l11tm1.1Ast mice (Bim-/-) were generated as previously described and backcrossed onto C57BL/6 for greater than 20 generations [15]. Bim-deficient mice homozygous for the Faslpr/lpr mutation were generated by crossing Bim-/- mice to Faslpr/lpr mice. Breeding of CD90.1 mice with Faslpr/lpr, Bim-/-, Bim-/-Faslpr/lpr mice generated mice homozygous for CD90.1. Mice transgenically expressing GFP under the control of the Rag promoter (Rag-GFP-transgenic) were the kind gift of Michel Nussenzweig (The Rockefeller University, NY, NY) [34]. Rag-GFP-transgenic mice were bred to Faslpr/lpr mice to generate Rag-GFP-transgenic Faslpr/lpr mice. All animal studies were conducted in accordance with the policies of The University of Vermont’s Animal Care and Use Committee.
Adoptive transfer of lymphocytes
5 × 106 lymph node cells from 5 wk old C57BL/6J (CD90.1) mice or the equivalent number of total T cells from age and sex-matched Faslpr/lpr, Bim-/-, Bim-/-Faslpr/lpr mice (all CD90.1) were transferred intravenously via the tail vein into Rag1-/- (CD90.2) mice. CD4+ or CD8+ T cells for adoptive transfer were purified by negative selection using Miltenyi T cell isolation kits (Auburn, CA) according to the manufacture’s protocol.
To assess entry into cell cycle following adoptive transfer, donor cells were labeled with 5(6)-carboxyfluorescein diacetate succinimidyl ester (CFSE) (Invitrogen/Molecular Probes, Carlsbad, CA) prior to transfer. Lymph node cells were washed with phosphate buffered saline containing 0.1% bovine serum albumin (PBS/0.1 % BSA), resuspended at 107 cells/ml, and incubated with 4 μM CFSE for 10 min at 37°C. Labeling was stopped by addition of ice cold PBS/0.1 % BSA. After washing, the cells were resuspended in PBS for adoptive transfer.
For the in vivo proliferation studies, mice received four intraperitoneal injections of 1 mg 5-bromo-2’-deoxyuridine (BrdU) in sterile PBS (Sigma, St. Louis, MO) during the 24 h prior to tissue harvest. Three injections were given on the day prior to tissue harvest and one injection on the day of sacrifice one hour prior to tissue harvest.
Reagents and antibodies
The following monoclonal antibodies to murine cell surface molecules were purchased from Invitrogen/Caltag Laboratories (Carlsbad, CA): FITC-conjugated CD44, PE-conjugated CD45R (B220), PE-Cy5.5-conjugated CD4, PE-Cy5.5-conjugated CD8, PE-Texas-Red-conjugated CD8α, PE-Texas-Red-conjugated CD4, Alexa 647-conjugated CD45R (B220), and Alexa 647-conjugated CD3. The following antibodies were purchased from BD Biosciences (San Jose, CA): FITC-conjugated TCRβ, PE-conjugated TCRβ, allophycocyanin-conjugated TCRβ, FITC-conjugated CD90.1, PE-conjugated CD90.1, allophycocyanin-Cy7-conjugated CD11b, FITC-conjugated NK1.1, and FITC-conjugated anti-BrdU. Lyophilized rat IgG and hamster IgG (MP Biochemicals, Solon, OH) were resuspended in PBS and stored at -80°C.
Lymphocyte preparation
Single cell suspensions of spleen and lymph nodes were prepared in RPMI 1640 (Mediatech, Inc., Herndon, VA) containing 25 mM Hepes, 5% (v/v) bovine calf serum, 5 × 10-5 M β-mercaptoethanol, 100 U/ml penicillin, and 100 U/ml streptomycin (RPMI/5% BCS). Erythrocytes in splenic suspensions were lysed with Geys solution.
Cell recovery was calculated from the absolute number of cells obtained and the percentage of CD4+, CD8+, and CD4−8−TCRβ+ T cells obtained by flow cytometry. For each Rag1-/-recipient, the spleen and eight lymph nodes (inguinal, brachial, axillary, and popliteal) were harvested. For intact mice, the spleen and all visible inguinal, brachial, axillary, cervical, and popliteal were harvested.
Cell culture
Single cell suspensions from lymph nodes were cultured in RPMI 1640 supplemented with 25 mM Hepes, 2500 μg/ml glucose, 10 μg/ml folate, 110.04 μg/ml pyruvate, 5 × 10-5 M 2-ME, 292.3 μg/ml glutamine, 100 U/ml penicillin, 100 U/ml streptomycin, and 5% (v/v) FBS. T cells were stimulated with 10 μg/ml plastic-bound anti-CD3 antibody (145-2C11) plus soluble anti-CD28 antibody (1:1000 dilution of ascites) and either 50 U/ml human rIL-2 (Cetus), 10 ng/ml murine rIL-7 (Peprotech, Rocky Hill, NJ), or 20 ng/ml human rIL-15 (Peprotech). On day 2, T cells were removed from anti-CD3 antibody stimulation and expanded for an additional 3 days in the presence of rIL-2, rIL-7, or rIL-15. To test for TCR/CD3 restimulation-induced apoptosis, IL-2 expanded T cell blasts were cultured either with or without plate-bound anti-CD3 antibody for 6 h. To test for cytokine withdrawal induced apoptosis, IL-7 and IL-15 grown T cells were washed and returned to culture in the absence of cytokine. To test for sensitivity to Fas-mediated death, day 5 T cell blasts were cultured with 250 ng/ml FLAG tagged recombinant FasL (Alexis Biochemicals, Farmingdale, NY) plus anti-FLAG antibody (Sigma, St Louis, MO) for 2 h.
Flow cytometry
For direct staining, single cell suspensions (1 × 106) were washed with cold (4°C) PBS and then incubated with Live Dead Fixable Blue Stain (Invitrogen, Carlsbad, CA) in PBS for 30 min at 4°C. The cells were washed with cold PBS containing 1% (w/v) BSA fraction V (Sigma, St. Louis, MO) (PBS/1% BSA) and then incubated with a mixture of unconjugated rat IgG and hamster IgG for 30 min at 4°C. After washing with PBS/1% BSA, the cells were incubated with the appropriate antibodies in PBS/1% BSA, washed, and fixed with freshly made 1% (v/v) methanol-free formaldehyde (Ted Pella Inc., Redding, CA) in PBS/1% BSA. Flow cytometry was performed on an LSRII (BD Biosciences, San Jose, CA) calibrated with DNA check beads. Analysis was performed using FlowJo software (Tree Star, Inc, Ashland, OR).
Staining for DNA-incorporated BrdU was performed as previously described [12]. In brief, cells from BrdU-pulsed mice were incubated with Live Dead stain (Invitrogen) followed by unconjugated rat and hamster IgG as described above. The cells were stained for TCRβ, CD4, and CD8 expression, and then fixed with 70% ethanol (-20°C) followed by 1% methanol-free formaldehyde. The cells were permeablized in PBS containing 1% methanol-free formaldehyde and 0.01% Tween20 overnight at 4°C. After washing, the cells were incubated with 50 Kunitz units of DNase I (Sigma, St. Louis, MO) in 0.15 M NaCl (pH=5) containing 4.2 mM MgCl2 for 15 min at 37°C. After washing, the cells were incubated with anti-BrdU FITC (BD Biosciences, San Diego, CA), washed, and fixed in 1% methanol-free formaldehyde in PBS/1% BSA.
Apoptotic cell death was examined by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) [12]. Briefly, cells were incubated with unconjugated rat and hamster IgG, stained for expression of cell surface molecules, and then fixed with 1% methanol-free formaldehyde followed by 70% ethanol. After washing, the TUNEL reaction was performed by incubating cells in 50 μl of reaction mix containing 10 U of terminal deoxribosyl transferase (TdT), 2.5 mM cobalt chloride in 1x TdT buffer (Roche, Indianapolis, IN), and 0.2 pmol/μl of FITC-dUTP (Roche, Indianapolis, IN,) in sterile distilled water for 1 hour at 37°C. The cells were washed twice and fixed in 1% methanol free formaldehyde. Murine thymocytes were included as a positive control for apoptotic cells.
DNA content was examined by Hoechst staining. Cells were incubated with Live Dead stain (Invitrogen) followed by unconjugated rat and hamster IgG as described above. The cells were then stained for TCRβ, CD45R, NK1.1, CD4, and CD8 expression using antibodies in PBS/1% BSA, fixed with 70% ethanol (-20°C) followed by 1% methanol-free formaldehyde. The cells were then incubated in 5 μg/ml Hoechst 33342 (Invitrogen/Molecular Probes, Eugene, OR) for 20 min. After washing, the cells were resuspended in PBS/1% BSA and stored at 4°C until analysis.
Acknowledgments
The authors would like to thank Jennifer Russell for technical assistance and the members of the Vermont Center for Immunology and Infectious Disease for helpful discussions.
This work was supported by NIH/NIAID (AI36333 and AI0797112 to R.C.B.), the Australian NHMRC (461221 to A.S. and P.B., IRIISS grant 361646 to A.S., and CDA to P.B.), the Victorian State Government (OIS grant to A.S. and P.B.), Cancer Cancel Victoria (to P.B.), and the Leukemia and Lymphoma Society (SCOR grant 7015 to A.S. and P.B).
Footnotes
The authors have no conflicting financial or commercial interests.
References
- 1.Ernst B, Lee DS, Chang JM, Sprent J, Surh CD. The peptide ligands mediating positive selection in the thymus control T cell survival and homeostatic proliferation in the periphery. Immunity. 1999;11:173–181. doi: 10.1016/s1074-7613(00)80092-8. [DOI] [PubMed] [Google Scholar]
- 2.Goldrath AW, Bevan MJ. Low-affinity ligands for the TCR drive proliferation of mature CD8+ T cells in lymphopenic hosts. Immunity. 1999;11:183–190. doi: 10.1016/s1074-7613(00)80093-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ku CC, Murakami M, Sakamoto A, Kappler J, Marrack P. Control of homeostasis of CD8+ memory T cells by opposing cytokines. Science. 2000;288:675–678. doi: 10.1126/science.288.5466.675. [DOI] [PubMed] [Google Scholar]
- 4.Schluns KS, Kieper WC, Jameson SC, Lefrancois L. Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nature Immunol. 2000;1:426–432. doi: 10.1038/80868. [DOI] [PubMed] [Google Scholar]
- 5.Surh CD, Sprent J. Homeostasis of naive and memory T cells. Immunity. 2008;29:848–862. doi: 10.1016/j.immuni.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 6.Takeda S, Rodewald HR, Arakawa H, Bluethmann H, Shimizu T. MHC class II molecules are not required for survival of newly generated CD4+ T cells, but affect their long-term life span. Immunity. 1996;5:217–228. doi: 10.1016/s1074-7613(00)80317-9. [DOI] [PubMed] [Google Scholar]
- 7.Tanchot C, Lemonnier FA, Perarnau B, Freitas AA, Rocha B. Differential requirements for survival and proliferation of CD8 naive or memory T cells. Science. 1997;276:2057–2062. doi: 10.1126/science.276.5321.2057. [DOI] [PubMed] [Google Scholar]
- 8.Viret C, Wong FS, Janeway CA., Jr Designing and maintaining the mature TCR repertoire: the continuum of self-peptide:self-MHC complex recognition. Immunity. 1999;10:559–568. doi: 10.1016/s1074-7613(00)80055-2. [DOI] [PubMed] [Google Scholar]
- 9.Barthlott T, Kassiotis G, Stockinger B. T cell regulation as a side effect of homeostasis and competition. J Exp Med. 2003;197:451–460. doi: 10.1084/jem.20021387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dummer W, Ernst B, LeRoy E, Lee D, Surh C. Autologous regulation of naive T cell homeostasis within the T cell compartment. J Immunol. 2001;166:2460–2468. doi: 10.4049/jimmunol.166.4.2460. [DOI] [PubMed] [Google Scholar]
- 11.Troy AE, Shen H. Cutting edge: homeostatic proliferation of peripheral T lymphocytes is regulated by clonal competition. J Immunol. 2003;170:672–676. doi: 10.4049/jimmunol.170.2.672. [DOI] [PubMed] [Google Scholar]
- 12.Fortner KA, Budd RC. The death receptor Fas (CD95/APO-1) mediates the deletion of T lymphocytes undergoing homeostatic proliferation. J Immunol. 2005;175:4374–4382. doi: 10.4049/jimmunol.175.7.4374. [DOI] [PubMed] [Google Scholar]
- 13.Goldrath AW, Bogatzki LY, Bevan MJ. Naive T cells transiently acquire a memory-like phenotype during homeostasis-driven proliferation. J Exp Med. 2000;192:557–564. doi: 10.1084/jem.192.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cohen PL, Eisenberg RA. Lpr and gld: single gene models of systemic autoimmunity and lymphoproliferative disease. Annu Rev Immunol. 1991;9:243–269. doi: 10.1146/annurev.iy.09.040191.001331. [DOI] [PubMed] [Google Scholar]
- 15.Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Kontgen F, Adams JM, Strasser A. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286:1735–1738. doi: 10.1126/science.286.5445.1735. [DOI] [PubMed] [Google Scholar]
- 16.Rathmell JC, Lindsten T, Zong WX, Cinalli RM, Thompson CB. Deficiency in Bak and Bax perturbs thymic selection and lymphoid homeostasis. Nature Immunol. 2002;3:932–939. doi: 10.1038/ni834. [DOI] [PubMed] [Google Scholar]
- 17.Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356:314–317. doi: 10.1038/356314a0. [DOI] [PubMed] [Google Scholar]
- 18.Kotzin BL, Babcock SK, Herron LR. Deletion of potentially self-reactive T cell receptor specificities in L3T4-, Lyt-2- T cells of lpr mice. J Exp Med. 1988;168:2221–2229. doi: 10.1084/jem.168.6.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mountz JD, Smith TM, Toth KS. Altered expression of self-reactive TCR Vb regions in autoimmune mice. J Immunol. 1990;144:2158–2159. [PubMed] [Google Scholar]
- 20.Singer PA, Balderas RS, McEvilly RJ, Bobardt M, Theofilopoulos AN. Tolerance-related V b clonal deletions in normal CD4-8-, TCR-ab+ and abnormal lpr and gld cell populations. J Exp Med. 1989;170:1869–1877. doi: 10.1084/jem.170.6.1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou T, Bluethmann H, Eldridge J, Brockhaus M, Berry K, Mountz JD. Abnormal thymocyte development and production of autoreactive T cells in T cell receptor transgenic autoimmune mice. J Immunol. 1991;147:466–474. [PubMed] [Google Scholar]
- 22.Kishimoto H, Surh CD, Sprent J. A role for Fas in negative selection of thymocytes in vivo. J Exp Med. 1998;187:1427–1438. doi: 10.1084/jem.187.9.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsumoto K, Yoshikai Y, Asano T, Himeno K, Iwasaki A, Nomoto K. Defect in negative selection in lpr donor-derived T cells differentiating in non-lpr host thymus. J Exp Med. 1994;173:127–136. doi: 10.1084/jem.173.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hildeman DA, Zhu Y, Mitchell TC, Bouillet P, Strasser A, Kappler J, Marrack P. Activated T cell death in vivo mediated by proapoptotic Bcl-2 family member Bim. Immunity. 2002;16:759–767. doi: 10.1016/s1074-7613(02)00322-9. [DOI] [PubMed] [Google Scholar]
- 25.Hughes PD, Belz GT, Fortner KA, Budd RC, Strasser A, Bouillet P. Apoptosis regulators Fas and Bim cooperate in the shutdown of chronic immune responses and prevention of autoimmunity. Immunity. 2008;28:1–9. doi: 10.1016/j.immuni.2007.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mixter PF, Russell JQ, Budd RC. Delayed kinetics of T lymphocyte anergy and deletion in lpr mice. J Autoimmunity. 1994;7:697–710. doi: 10.1006/jaut.1994.1055. [DOI] [PubMed] [Google Scholar]
- 27.Strasser A, Harris AW, Huang DC, Krammer PH, Cory S. Bcl-2 and Fas/APO-1 regulate distinct pathways to lymphocyte apoptosis. EMBO J. 1995;14:6136–6147. doi: 10.1002/j.1460-2075.1995.tb00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maldonado MA, Eisenberg RA, Roper E, Cohen PL, Kotzin BL. Greatly reduced lymphoproliferation in lpr mice lacking major histocompatibility complex class I. J Exp Med. 1995;181:641–648. doi: 10.1084/jem.181.2.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takahashi T, Tanaka M, Brannan CI, Jenkins NA, Copeland NG, Suda T, Nagata S. Generalized lymphoproliferative disease in mice, caused by a point mutation in the Fas ligand. Cell. 1994;76:969–976. doi: 10.1016/0092-8674(94)90375-1. [DOI] [PubMed] [Google Scholar]
- 30.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 31.Bouillet P, Purton JF, Godfrey DI, Zhang L-C, Coultas L, Puthalakath H, Pellegrini M, Cory S, Adams JM, Strasser A. BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes. Nature. 2002;415:922–926. doi: 10.1038/415922a. [DOI] [PubMed] [Google Scholar]
- 32.Alderson MR, Tough TW, Davis-Smith T, Braddy S, Falk B, Schooley KA, Goodwin RG, Smith CA, Ramsdell F, Lynch DH. Fas ligand mediates activation-induced cell death in human T lymphocytes. J Exp Med. 1995;181:71–77. doi: 10.1084/jem.181.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brunner T, Mogil RJ, LaFace D, Yoo NJ, Mahboubi A, Echeverri F, Martin SJ, Force WR, Lynch DH, Ware CF, et al. Cell-autonomous Fas (CD95)/Fas-ligand interaction mediates activation-induced apoptosis in T-cell hybridomas. Nature. 1995;373:441–444. doi: 10.1038/373441a0. [DOI] [PubMed] [Google Scholar]
- 34.Yu W, Nagaoka H, Jankovic M, Misulovin Z, Suh H, Rolink A, Melchers F, Meffre E, Nussenzweig M. Continued RAG expression in late stages of B cell development and no apparent re-induction after immunization. Nature. 1999;400:682–687. doi: 10.1038/23287. [DOI] [PubMed] [Google Scholar]
- 35.Weant AE, Michalek RD, Khan IU, Holbrook BC, Willingham MC, Grayson JM. Apoptosis regulators Bim and Fas function concurrently to control autoimmunity and CD8+ T cell contraction. Immunity. 2008;28:218–230. doi: 10.1016/j.immuni.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 36.Landolfi MM, Van Houten N, Russell JQ, Scollay R, Parnes JR, Budd RC. CD2-CD4-CD8- lymph node T lymphocytes in MRL lpr/lpr mice are derived from a CD2+CD4+CD8+ thymic precursor. J Immunol. 1993;151:1086–1096. [PubMed] [Google Scholar]
- 37.Mixter PF, Russell JQ, Durie FH, Budd RC. Decreased CD4-CD8- TCR-ab+ cells in lpr/lpr mice lacking beta 2-microglobulin. J Immunol. 1995;154:2063–2074. [PubMed] [Google Scholar]
- 38.Herron LR, Eisenberg RA, Roper E, Kakkanaiah VN, Cohen PL, Kotzin BL. Selection of the T cell receptor repertoire in Lpr mice. J Immunol. 1993;151:3450–3459. [PubMed] [Google Scholar]
- 39.Jevnikar AM, Grusby MJ, Glimcher LH. Prevention of nephritis in major histocompatiblity complex class II-deficient MRL-lpr mice. J Exp Med. 1994;179:1137–1143. doi: 10.1084/jem.179.4.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hutcheson J, Scatizzi JC, Siddiqui AM, Haines GK, III, Wu T, Li Q-Z, Davis LS, Mohan C, Perlman H. Combined deficiency of proapoptotic regulators Bim and Fas results in the early onset of systemic autoimmunity. Immunity. 2008;28:208–217. doi: 10.1016/j.immuni.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 41.Snow AL, Oliveira JB, Zheng L, Dale JK, Fleischer TA, Lenardo MJ. Critical role for BIM in T cell receptor restimulation-induced death. Biology Direct. 2008;3:34–51. doi: 10.1186/1745-6150-3-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pellegrini M, Belz G, Bouillet P, Strasser A. Shutdown of an acute T cell immune response to viral infection is mediated by the proapoptotic Bcl-2 homology 3-only protein Bim. Proc Natl Acad Sci USA. 2003;100:14175–14180. doi: 10.1073/pnas.2336198100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abu-Shakra M, Buskila D, Ehrenfeld M, Conrad K, Shoenfeld Y. Cancer and autoimmunity: autoimmune and rheumatic features in patients with malignancies. Ann Rheum Dis. 2001;60:433–441. doi: 10.1136/ard.60.5.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arkwright PD, Abinun M, Cant AJ. Autoimmunity in human primary immunodeficiency diseases. Blood. 2001;99:2694–2702. doi: 10.1182/blood.v99.8.2694. [DOI] [PubMed] [Google Scholar]
- 45.Reveille JD. The changing spectrum of rheumatic disease in human immunodeficiency virus infection. Semin Arth Rheum. 2000;30:147–166. doi: 10.1053/sarh.2000.16527. [DOI] [PubMed] [Google Scholar]
- 46.Goronzy JJ, Weyand CM. Thymic function and peripheral T-cell homeostasis in rheumatoid arthritis. Trends Immunol. 2001;22:251–255. doi: 10.1016/s1471-4906(00)01841-x. [DOI] [PubMed] [Google Scholar]
- 47.Koetz K, Bryl E, Spickschen K, O’Fallon WM, Goronzy JJ, Weyand CM. T cell homeostasis in patients with rheumatoid arthritis. Proc Natl Acad Sci USA. 2000;97:9203–9208. doi: 10.1073/pnas.97.16.9203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.King C, Ilic A, Koelsch K, Sarvetnick N. Homeostatic expansion of T cells during immune insufficiency generates autoimmunity. Cell. 2004;117:265–277. doi: 10.1016/s0092-8674(04)00335-6. [DOI] [PubMed] [Google Scholar]
- 49.Sneller MC, Wang J, Dale JK, Strober W, Middelton LA, Choi Y, Fleisher TA, Lim MS, Jaffe ES, Puck JM, Lenardo MJ, Straus SE. Clinical, immunologic, and genetic features of an autoimmune lymphoproliferative syndrome associated with abnormal lymphocyte apoptosis. Blood. 1997;89:1341–1348. [PubMed] [Google Scholar]
- 50.Straus SE, Sneller M, Lenardo MJ, Puck JM, Strober W. An inherited disorder of lymphocyte apoptosis:the autoimmune lymphoproliferative syndrome. Ann Int Med. 1999;130:591–601. doi: 10.7326/0003-4819-130-7-199904060-00020. [DOI] [PubMed] [Google Scholar]
- 51.Shivakumar S, Tsokos GC, Datta SK. T cell receptor alpha/beta expressing double-negative (CD4-/CD8-) and CD4+ T helper cells in humans augment the production of pathogenic anti-DNA autoantibodies associated with lupus nephritis. J Immunol. 1989;143:103–112. [PubMed] [Google Scholar]