

Conspectus
Reactions that convert carbon–hydrogen (C–H) bonds into carbon–carbon (C–C) or carbon–heteroatom (C–Y) bonds are attractive tools for organic chemists, potentially expediting the synthesis of target molecules through new disconnections in retrosynthetic analysis. Despite extensive inorganic and organometallic study of the insertion of homogeneous metal species into unactivated C–H bonds, practical applications of this technology in organic chemistry are still rare. Only in the past decade have metal-catalyzed C–H functionalization reactions become more widely utilized in organic synthesis.
Research in the area of homogeneous transition metal–catalyzed C–H functionalization can be broadly grouped into two subfields. They reflect different approaches and goals and thus have different challenges and opportunities. One approach involves reactions of completely unfunctionalized aromatic and aliphatic hydrocarbons, which we refer to as “first functionalization.” Here the substrates are nonpolar and hydrophobic and thus interact very weakly with polar metal species. To overcome this weak affinity and drive metal-mediated C–H cleavage, chemists often use hydrocarbon substrates in large excess (for example, as solvent). Because highly reactive metal species are needed in first functionalization, controlling the chemoselectivity to avoid over-functionalization is often difficult. Additionally, because both substrates and products are comparatively low-value chemicals, developing cost-effective catalysts with exceptionally high turnover numbers that are competitive with alternatives (including heterogeneous catalysts) is challenging. Although an exciting field, first functionalization is beyond the scope of this Account.
The second subfield of C–H functionalization involves substrates containing one or more pre-existing functional groups, termed “further functionalization.” One advantage of this approach is that the existing functional group (or groups) can be used to chelate the metal catalyst and position it for selective C–H cleavage. Precoordination can overcome the paraffin nature of C–H bonds by increasing the effective concentration of the substrate so that it needn't be used as solvent. From a synthetic perspective, it is desirable to use a functional group that is an intrinsic part of the substrate so that extra steps for installation and removal of an external directing group can be avoided. In this way, dramatic increases in molecular complexity can be accomplished in a single stroke through stereo- and site-selective introduction of a new functional group. Although reactivity is a major challenge (as with first functionalization), the philosophy in further functionalization differs—the major challenge is developing reactions that work with predictable selectivity in intricately functionalized contexts on commonly occurring structural motifs.
In this Account, we focus on an emergent theme within the further functionalization literature: the use of commonly occurring functional groups to direct C–H cleavage through weak coordinations. We discuss our motivation for studying Pd-catalyzed C–H functionalization assisted by weakly coordinating functional groups and chronicle our endeavors to bring reactions of this type to fruition. Through this approach, we have developed reactions with a diverse range of substrates and coupling partners, with the broad scope likely stemming from higher reactivity of the less stable cyclopalladated intermediates held in place by weak coordinations.

1. Introduction
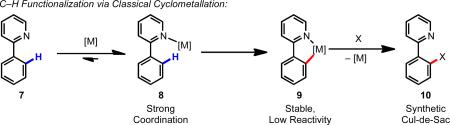
Transition metal–catalyzed C–H functionalization reactions can be grouped into two distinct subfields depending on whether the substrate is a completely unfunctionalized hydrocarbon (“first functionalization”) or a small molecule bearing at least one pre-existing functional group (“further functionalization”) (Scheme 1).1 Within the realm of further functionalization, one strategy for controlling site-selectivity is using the pre-existing functional group(s) to coordinate the metal and position it for selective C–H cleavage (Scheme 2). Traditionally this has been accomplished by using nitrogen-, sulfur-, or phosphorous-containing directing groups (e.g., pyridine, oxazoline, sulfide, and phosphine), which are strong σ-donors and/or π-acceptors. We refer to the formation of thermodynamically stable five- or six-membered metallacycle with this type of substrate as “classical “cyclometallation” (Eqs. 1 and 2), and the majority of directed C–H activation reactions since the 1950s fall into this category. One disadvantage of this approach is that the strong directing groups mentioned above are synthetically restrictive, either because they must be installed then removed, or because they are a permanent part of the substrate. Another problem is that directed C–H functionalization typically proceeds via five-membered metallacycle intermediates; when alternative carbon skeletons are sought, the inability to utilize remotely located directing groups can be synthetically restrictive. Finally, strongly chelating substrates give cyclometallated intermediates that are thermodynamically stable, and thus are less reactive in the subsequent functionalization step, which limits the range of nucleophiles and electrophiles with which they can react.
 |
(1) |
 |
(2) |
Scheme 1.

Comparison of “first functionalization” and “further functionalization”.1
Scheme 2.

C–H cleavage promoted by a proximal directing group (DG).
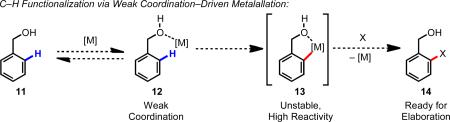
Cyclometallation using weakly coordinating directing groups (e.g., ketones, carboxylic acids, and ethers), on the other hand, has been less actively studied. With weakly coordinating directing groups, the resulting metallacycles are less thermodynamically stable and are not necessarily isolable. Pioneering work in the 1990s showed that Ru(0) 2, 3 and Rh(I)4 could coordinate with ketones and esters to trigger oxidative addition of the C–H bond to the metal center, as the first step in novel catalytic C–C bond-forming reactions (Scheme 3), setting the stage for continued studies to improve the scope and practicality of these reactions with Ru(0) catalysts.
Scheme 3.

Ru(0)-catalyzed C–H functionalization of arylketones.2
Pd(II) is arguably a more desirable metal to use in developing a diverse collection of C–H functionalization reactions through a completely different redox catalysis compared to the aforementioned Ru(0) and Rh(I) catalyzed reactions. In particular, highly versatile [Pd(II)–R1] species is generated following the C–H cleavage (which is redox-neutral with concomitant loss of HX). In aryl halide chemistry with Pd(0) catalysts, it has been shown that this type of intermediate can participate in the different functionalization reactions depicted in Scheme 4. Thus, one would imagine that analogous Pd(II)-catalyzed C–H functionalization reactions could be carried out by employing nucleophilic coupling partners via Pd(II)/Pd(0) catalysis or electrophilic coupling partners via Pd(II)/Pd(IV) or Pd(II)2/Pd(III)2 catalysis. With Pd(II), however, the C–H cleavage step has traditionally required strongly coordinating directing groups, which are disadvantageous for the reasons outlined above.
Scheme 4.

Versatile reactivity of [Pd(II)–aryl] and [Pd(II)–alkyl] intermediates.5
This Account describes tactics that our laboratory has implemented to overcome the limitations posed by classical cyclometallation in order to promote reactivity with new classes of weakly coordinating directing groups. To execute this chemistry, our group has pursued two complementary strategies: (1) utilizing common, synthetically versatile functional groups directly and (2) developing practical auxiliaries for C–H functionalization that can readily be installed or removed. By centering our research in Pd catalysis on this concept, not only have we been able to achieve unprecedented substrate scope, but we have also taken advantage of the high reactivity of the cyclopalladated intermediates to discover a diverse collection of new C–C and C–Y bond–forming reactions. For our laboratory, this idea has served as a crucial platform for reaction discovery and for studying ligand-controlled catalytic reactions.
2. Historical Perspective on Cyclometallation
The term “cyclometallation” was first introduced by Trofimenko,6 and reactions of this type involving cleavage of C(sp2)–H and C(sp3)–H bonds by late transition metals to form defined [M–R] species have been known for several decades, with examples of well characterized cyclometallated complexes appearing in the literature as early as the 1960s (Figure 1). These inner-sphere processes are known to proceed by a variety of different mechanisms depending on the metal species, the substrate, and the reaction conditions, including oxidative addition, electrophilic activation, concerted metallation/deprotonation, and σ-bond metathesis.1, 7 Of the many examples, those involving aromatic rings (so-called “ortho–metallation”) have been the most widely studied. In 1963, Klieman and Dubeck reported that NiCp2 (Cp = cyclopentadienyl) reacted with azobenzene to form nickelacycle 19.8 Subsequently Cope and Siekman observed similar reactivity between azobenzene and Pd(II) and Pt(II) salts, giving complexes 20 and 21, respectively.9 Bennet and Milner observed that ortho-C–H bonds of triphenylphospine ligands underwent oxidative addition to give [Ir(III)–H] species 22,10 and other metals were found to exhibit analogous reactivity with both phosphines and phosphonites (23 and 24).11, 12
Figure 1.

Early examples of well defined metallacycles using strong directing groups.8-10
With Pd(II), a large number of directing groups have been reported, the vast majority of which contain strongly coordinating nitrogen, phosphorous, or sulfur atoms (Figure 2).13 One exception is complex 29, in which Pd(II) is coordinated to oxygen; however, it is important to clarify that this type of substrate is unusually reactive because of the electron-donating properties of the nitrogen atom lone pair. The stability of these intermediates allows for detailed characterization. These palladacycles were found to react with alkenes, alkynes, CO, halogens, and organometallic reagents,13 a far broader range of reactivity than was observed with other metals.
Figure 2.

Palladacycles using representative traditional directing groups.13
For substrates bearing alkyl groups in close proximity to strong directing groups, cyclopalladation via C(sp3)–H cleavage was also established with a several different metals including Pd(II) (Figure 3). Activated benzylic C(sp3)–H bonds could be cleaved in the presence of strong directing groups (31 and 32). In a pioneering report in 1978, Shaw and coworkers also found that the oxime derived from phenyl tert-butyl ketone could also undergo cyclopalladation to give complex 33 through cleavage of an unactivated β-methyl C(sp3)–H bond.14
Figure 3.

Taken together, this body of work provided important early precedent for the development of directed C–H functionalization reactions based on Pd(II)/Pd(0) and Pd(II)/Pd(IV) catalysis.
3. The Renaissance of Pd(II)-Catalyzed C–H Functionalization
3.1 Early Work and Perspectives on Catalysis
Pd(II)-catalyzed C–H functionalization reactions have been studied for the past several decades, since the pioneering work on Pd(II)-catalyzed C(sp2)–H olefination of simple arenes by Fujiwara and Moritani in the late 1960s (Scheme 6).15 This early work demonstrated the impressive reactivity of Pd(II) in activating C–H bonds of simple aromatic hydrocarbons, but the practical utility of this chemistry in organic synthesis has been limited due to the need for excess arene substrate (often as solvent), the lack of positional selectivity with mono-substituted arenes, and the poor reactivity with electron-deficient arenes. Formation of the [Ar–Pd(II)] intermediate in this reaction is thought to proceed by a Friedel–Crafts-type electrophilic palladation mechanism.
Scheme 6.

C–H olefination of benzene along a Pd(II)/Pd(0) catalytic cycle.15
Seminal work using cyclometallation to control regioselectivity and enhance the rate of C–H cleavage was reported by Fahey in 1971 in the Pd(II)-catalyzed ortho-C(sp2)–H chlorination of azobenzene (37) (Scheme 7a).16 Reaction of the putative palladacycle with Cl2 leads to a high–oxidation state Pd(III) or Pd(IV) intermediate, which then undergoes C–Cl reductive elimination to release the product. Another important finding came in 1984 when Tremont and Rahman found that acetanilide substrate 40 could undergo ortho-methylation with MeI in good yields, and they demonstrated that TONs up to 10 could be achieved by using AgOAc as an oxidant (Scheme 7b).17
Scheme 7.

Pd(II)-catalyzed ortho-C–H functionalization of substrates bearing directing groups.16, 17
3.2 Resurgence of Interest
During the past decade, a number of groups have revisited Pd-catalyzed C–H functionalization and have developed a collection of promising new reactions (Scheme 8). Sanford reported a C–H acetoxylation protocol using a series of different nitrogen-containing directing groups and PhI(OAc)2 as the oxidant.18 Using a removable chiral oxazoline auxiliary, our group disclosed the first example of diastereoselective C(sp3)–H iodination (47→49),19 a method that represents a potentially powerful approach for synthesizing all-carbon quaternary stereocenters. Later the same year, Daugulis reported Pd(II)-catalyzed C(sp2)–H arylation using acetanilide-type substrates (50) and aryl iodides.20 All of these reactions are proposed to proceed through Pd(II)/Pd(IV) or Pd(II)2/Pd(III)2 catalysis.
Scheme 8.

ortho-C–H functionalization via Pd(II)/Pd(IV) or Pd(II)2/Pd(III)2 catalysis.18-20
Our research laboratory went on to develop the first example of C–H/R–M cross-coupling via Pd(II)/Pd(0) catalysis (Scheme 9).21 The presence of 1,4-benzoquinone (BQ) was found to be crucial for promoting reductive elimination from putative intermediate 56. Batch-wise addition of the organotin reagent lowered the rate of homocoupling such that Pd(II)-mediated C–H cleavage could take place.
Scheme 9.

Pd(II)-catalyzed C–H/R–BX2 cross-coupling with oxazoline substrates.21
4. Early Inspiration and Motivation for Studying Weak Coordination–Driven C–H Functionalization
The classical cyclometallation/functionalization approach has proven to be indispensable for designing and developing new Pd(II)-catalyzed C–H activation reactions. In the preceding section, examples of Pd(II)-catalyzed C–H functionalization reactions were described in which the substrate contained a strong directing group, which served both to promote C–H cleavage and to control regioselectivity. However, this approach can be disadvantageous for the reasons outlined in Section 1.
In reflecting upon these limitations, we looked to classical substrate-directable reactions in organic synthesis for inspiration (Scheme 10).22-26 In addition to their reliability and efficiency, a common feature that these reactions share is that they employ simple, commonly encountered functional groups (e.g., carboxylic acids, amines, and alcohols) to control the stereo- or regiochemical course of the reaction through weak coordinative interactions. Collectively, these classical substrate-directable reactions also cover a range of carbon skeletons in which the directing atom lies in different spatial relationships to the other reactive sites allowing for intricate functional group patterns to be constructed. Hence, we reasoned that by developing C–H functionalization chemistry that similarly utilizes ubiquitous functional groups, we could overcome many of the problems discussed above. One key to this approach would be devising tactics to functionalize remotely located C–H bonds (those positioned 5–6 bonds away from the directing atom).
Scheme 10.

Classical substrate-directable reaction in organic synthesis.23-25
At the same time, we recognized that removable auxiliaries that mask more common functional groups, including the Evans oxazolidinone (69),27 Myers pseudoephedrin amide (70),28 and Ellman N-tert-butanesulfinyl imine (71)29 auxiliaries (Figure 4), often imbue organic substrates with unique reactivity, and that developing widely versatile auxiliaries of this type may enable Pd(II)-catalyzed C–H functionalization to reach new heights.
Figure 4.

Versatile auxiliaries for organic synthesis.
In both of the scenarios, we were keen to target weakly coordinating functional groups (Figure 5). We hypothesized that if we could rely on geometry to trigger a kinetically favorable C–H cleavage event, we could then harness the enhanced reactivity of the metallated intermediate to improve the range of coupling partners that could be used, and hence increase the number of new reactions that could be developed. We were also inspired in these endeavors by other areas outside of the realm of transition metal catalysis in which weak coordination has been successfully exploited to control the reactivity and selectivity of chemical transformations, including directed lithiation, Lewis acid catalysis, and organocatalysis. With respect to lithiation chemistry, Beak and Sniekus described the complex-induced proximity effect (CIPE),30 the essence of which is controlling the selectivity of a reaction by positioning the reactive species in specified spatial orientations and driving the reaction through high effective molarity. This example reinforced our understanding that the key in our endeavors would not necessarily be the strength of coordination to the directing group but the distance and geometry between the metal and the target C–H bond.
Figure 5.

Representative weakly coordinating substrates.
At the outset, we reasoned that through pursuing reactions of this type, we would enable new synthetic disconnections in retrosynthetic analysis. For instance, commonly occurring heterocyclic ring systems could rapidly be constructed using either a C–H activation/C–Y cyclization reaction, or alternatively intervening electrophiles or nuceophiles could be incorporated (Scheme 11). In designing a synthetic route to aliphatic carboxylic acids bearing a chiral center at the β-position (91), rather than resorting to traditional asymmetric conjugate addition to an α,β-unsaturated ester (92), following C–H functionalization logic, one could start with a simple aliphatic acid bearing no other functionality (76) (Scheme 12). Applying this logic in a sequential sense, molecules like pivalic acid (93) could be used for the construction of all-carbon quaternary stereocenters (Scheme 13). This approach is not only step-economical, but it also allows pools of feedstock chemicals, like aliphatic acids, to be drastically advanced in molecular complexity in a modicum of chemical transformations, which stands in contrast to the gradual or incremental manner that traditional synthetic methods build complexity.
Scheme 11.

New retrosynthetic disconnections for heterocyclic ring formation.
Scheme 12.

A new retrosynthetic disconnection for β-functionalization.
Scheme 13.

Hypothetical synthetic route to chiral building blocks containing all-carbon quaternary stereocenters from pivalic acid using sequential C(sp3)–H functionalization.
Following this thinking, we also set our sights on other synthetic transformations that have proven to be broadly useful and decided to develop C–H activation reactions that would mirror these reactions. For instance, Pd(II)-catalyzed cross-coupling of C–H bonds with organometallic reagents is especially appealing considering the broad synthetic applications of traditional Pd(0)-catalyzed cross-coupling reactions.
The fundamental challenge in using the functional groups in Figure 5 (or auxiliaries thereof) to direct C–H cleavage with Pd(II) is that in many cases, the functional groups form comparatively low-energy interactions with Pd(II). In these cases, traditional cyclometalation to form a robust isolable intermediate is less likely to occur, so new chemical methods need to be developed to take advantage of the coordination chemistry of these substrates.
5. Implementation
5.1 Approach
Many of the ideal functional groups to direct Pd(II)-mediated C–H functionalization form weaker interactions than do the traditional directing groups used to promote cylclopalladation, and they are less rigid, possessing more conformational degrees of freedom. Though the reactivity is far less established, there are significant advantages to using these types of substrates from a synthetic perspective. However, for developing metal-catalyzed C–H activation reactions, having an appreciably high rate of C–H cleavage is crucial for choreographing a catalytic cycle. For instance, as discussed in Section 3, in Pd(II)-catalyzed C–H/R–M cross-coupling reactions, one of the major problems is the undesired reaction between Pd(II) and the organometallic reagents, which leads to homocoupling and ultimately to catalyst death. This side reaction becomes predominant if C–H cleavage is not rapid. In considering how to extend Pd(II)-mediated C–H activation reactions to new substrate classes, including those in Figure 5, we needed new methodology in which C–H palladation would be promoted by weak coordination analogous to CIPE chemistry (Figure 6). However, when we initiated our work in this area, examples of catalytic reactions of this type were rare.31, 32
Figure 6.

(a) Lithiation through the CIPE and (b) a hypothetical example of Pd(II)-mediated C–H cleavage through weak coordination.
From our oxazoline chemistry19 we learned several key lessons that were pertinent to our endeavor to develop C–H functionalization reactions using weakly coordinating directing groups. We had originally targeted oxazoline as a directing group because of its capacity for effecting stereoinduction, and because it could be easily installed and removed, and thus could be viewed as a carboxylic acid surrogate. During our work, oxazoline showed particularly high reactivity as a directing group, with C(sp3)–H cleavage taking place at room temperature. We thus sought to understand the root of this reactivity through conformational/geometrical analysis. In the square planar pre-transition state coordination structure 99, the target C–H bond is lined up directly adjacent to acetate anion (in other words, the dihedral angle between the C–H bond and the Pd(II)–OAc bond is at its minimum), which is thought to be critical during the presumptive concerted metallation/deprotonation transition state, which can involve could potentially involve internal (100) or external (101) base (Scheme 14).
Scheme 14.

Precise geometrically positioning of the target methyl group triggers a room temperature C–H cleavage event from square-planar pretransition state coordination structure 99.19
5.2 Boc-directed C(sp3)–H functionalization
Given the dominant role that distance and geometry effects played in the Pd(II)-mediated C–H functionalization using oxazoline as the directing group, we wondered whether weaker coordinating oxy functional groups could be used provided that the geometry was correct. Our efforts began using N-tert-butoxycarbonyl (Boc) directing groups to activate and functionalize C(sp3)–H bonds (Scheme 15).33 We were attracted to the possibility of using Boc directing groups because of their synthetic utility as protecting groups, and also because the work would serve as a stepping stone to access other weakly coordinating directing groups. To our delight, after an extensive survey of reaction conditions, we found that Boc-protected methylamines could be acetoxylated at the α-position in good yield under mild conditions.
Scheme 15.

Boc-directed C(sp3)–H acetoxylation of α-methyl groups.33
5.3 Carboxylate-directed C–H functionalization
Our research group has long been interested in Pd(II)-catalyzed C–H functionalization with carboxylic acid substrates, which we viewed as a simplified version of our oxazoline chemistry, without the added operational burden of installation and removal of the directing group. Early work by Miura and coworkers established a Pd(II)-catalyzed ortho-C(sp2)–H olefination protocol for benzoic acids.31 Sen,34 Sames,35 and Chang36 achieved moderate efficiency in carboxylate-directed C(sp3)–H hydroxylation using Pt(II) catalysts. We initiated our research efforts in this area with the aim of developing generally applicable methods to enhance reactivity in order to expand the cope of both substrate and coupling partners. Our early efforts, however, were largely unsuccessful, and we began to wonder whether the limited reactivity could be attributed to an undesired coordination geometry between the metal and the substrate (Figure 7). We hypothesized that Pd(II) was binding the carboxylate group in a κ2 fashion, which was effectively sequestering Pd(II) away from the target β-C–H bond. Pd(II) is known to adopt a variety of coordination modes with carboxylates, and in this case, if the equilibrium between κ1 and κ2 coordination strongly favored κ2 coordination, one would expect C–H cleavage to be prohibitively sluggish (Scheme 16). Using other metals, such as Rh(I) and Ir(I), ortho-C–H cleavage with benzoic acids has been reported.37 In these cases, a κ2 to a κ1 transition takes place forming C–H metallated complexes (112), in which the carboxylate is coordinated to the metal as an X-type ligand. Following the logic of our hypothesis, it would seem that in the case of Pd(II), there exists an energetic preference for the complex to remain in a κ2 configuration.
Figure 7.

κ2 coordination structures.33
Scheme 16.

Possible carboxylate binding modes.
If this coordination geometry situation were indeed the problem, one solution would be to devise a means of shifting the equilibrium between κ1 and κ2 coordination. We wondered whether a hard Lewis acidic metal might preferentially bind to the carboxylate and reorient Pd(II) into the desired spatial relationship with the C–H bond. Initial efforts in our laboratory focused on ortho-C–H iodination of benzoic acids.38 After surveying a variety of conditions, we found to our delight, that countercations, such as Na+, K+, NR4+, or even protonated DMF, were capable of promoting Pd(II)-mediated ortho-C–H cleavage in carboxylic acid substrates (Scheme 17). Interestingly, even simple table salt (NaCl) could be used as the cation source.
Scheme 17.

Dramatic countercation effect in the ortho-C–H iodination of benzoic acids.
On the basis of 1H NMR spectroscopy and X-ray crystallography,39 we have formulated a working model to explain the observed reactivity patterns. In this model, the countercation binds to the carboxylate group in a κ2 coordination mode, which induces Pd(II) to coordinate with the unhindered oxygen lone pair in a κ1 fashion (Scheme 18). In this case, the oxygen atom serves as an L-type ligand. When this coordination structure is assembled in the pre-transition state, it triggers C–H cleavage through a process closely resembling the CIPE in lithiation chemistry. Importantly the Lewis basicity of the sodium–carboxylate lone pairs is still enhanced relative to an unactivated carbonyl (e.g., of an ester of ketone) due to both resonance effects and electrostatic effects. Later we found that the dramatic influence of countercations also held in other reactions of carboxylic acid substrates (as discussed in the following section).
Scheme 18.

Evidence for proposed coordination structures in counteraction-promoted C–H cleavage by Pd(II).39
Based on this enhanced reactivity, we were able to achieve Pd(II)-catalyzed C–H/R–M cross-coupling (Scheme 19).40 While yields in our initial report were generally low and the substrate scope was limited, we nevertheless established an important precedent that C–H/R–M cross-coupling could be carried out using substrates lacking strong directing groups. Our C–H functionalization protocol was found to be compatible with CH3–B(OH)2 (128). Other alkyl boron reagents probed to be problematic, likely because of competitive β-hydride elimination. Importantly, aliphatic acids (93) could also be used as substrates in this C–H/R–M cross-coupling protocol, giving the corresponding functionalized products (130) in low yields. Contemporaneous to our work, Daugulis reported an important study on ortho-C–H arylation of benzoic acids using aryl halides and Pd(II) or Pd(0) catalysts.41
Scheme 19.

C–H/R–BX2 cross-coupling of carboxylic acid substrates.40
5.4 Versatile auxiliaries for C–H functionalization
Compared to C(sp2)–H cleavage, C(sp3)–H cleavage with weak directing groups poses a unique challenge because the system is conformationally less rigid and because the metal cannot engage with target C–H bond via an initial π-orbital interaction. The low reactivity of carboxylate directing groups in C(sp3)–H functionalization motivated us to engineer a versatile auxiliary that would mimic the weak coordinative affinity of a carboxylate while offering more facile C(sp3)–H cleavage. In the long term, we hoped that our endeavors would one day lead to the identification of a directing group that exhibited superb reactivity, selectivity, and scope (both in terms of substrates and reaction partners) and that could be readily installed and removed—in essence, an “Evans auxiliary for C–H functionalization”.
We first sought to develop an amide directing group with an acidic N–H bond (analogous to the acidic carboxylate O–H bond), where the Pd–substrate interaction would be strengthened because of the presence of a nucleophilic nitrogen atom.42 We ultimately found that N-methoxy amides (131) gave drastically improved efficiency in C(sp3)–H/R–M cross-coupling, even when alkyl boronic acid were used as coupling partners (Scheme 20).
Scheme 20.

C–H/R–BX2 cross-coupling of carboxylic acid substrates.40
In this reaction, several possible pre-transition state coordination structures exist (137–142, Figure 8). In the context of C(sp2)–H activation, Wang and coworkers obtained a crystal structure of a cyclopalladated intermediate where the amide was coordinated through the an X-type nitrogen (corresponding to structure 139).43 However, care must be taken in interpreting this result, since it is possible that any thermodynamically stable intermediates are the result of reorganization following C–H cleavage. Ongoing efforts in our laboratory are focused on teasing out the mechanistic nuances of this and related directing groups.
Figure 8.

Possible pre-transition state coordination structures with electron-deficient N-substituted amide directing groups.
Later, in our efforts to develop the first example of intermolecular Pd(0)-catalyzed C(sp3)–H arylation with aryliodides, we designed a class of N-aryl amide directing groups, wherein the increased reactivity stems from the presence of electron-withdrawing substituents on the aromatic ring which increase the acidity of the N–H bond (Scheme 21).44 We hypothesize that these amides can be deprotonated under weakly basic conditions, binding to Pd(II) in one of the possible κ1 coodination modes in Figure 8 to facilitate C–H cleavage. Notably, this type of directing group has proven to be effective in a remarkably broad range of reactions, and can readily be installed and removed. This work thus represents an important step forward in the search for truly enabling C–H functionalization auxiliaries.
Scheme 21.

Weakly coordinating acidic N-aryl amide auxiliaries.
5.5 Other versatile functional groups
Based on the lessons learned from carboxylic acids, we sought to use the weak coordination concept to employ amines and alcohols and directing groups. After extensive reaction optimization, we were ultimately able to achieve reactivity with phenethyltriflamides (147)45, 46 and phenethyl alcohols (150) (Scheme 22). 47, 48 The reactions shown in Scheme 22 showcase how these versatile substrates can be used employed for expedient heterocycle synthesis. In the case of the phenethylamine substrates, attenuating the binding affinity of the substrates through attachment of the trifluoromethanesulfonyl group is critical for reactivity.
Scheme 22.

Applications of remote, weakly coordinating directing groups.
5.6 Diversity of reaction partners
As discussed above, a major advantage of using weakly coordinating substrates for Pd(II)-catalyzed C–H activation is the relatively high reactivity of the palladated intermediate. Because of the higher reactivity, a range of different nucleophiles and electrophiles can be successfully used as reaction partners. This has led to the discovery and development of several new Pd(II)-catalyzed C–H functionalization reactions. For instance with benzoic acid substrates, C–H iodination,38 hydroxylation,49 carboxylation,39 alkylation,50 and arylation40 reactions have been reported by our group, using several different redox manifolds (Scheme 23a). For particularly challenging transformations such as ortho-C–H amination51 and fluorination,52 where a fast rate of C–H cleavage is crucial because of interference from the reaction partners (i.e., amines and fluorinating reagents), use of the versatile electron-deficient N-aryl amide auxiliary described above, has been found to facilitate the reaction (Scheme 23b).
Scheme 23.

Diverse reactivity in C–H functionalization using weakly coordinating substrates.
By drawing on this approach, we have designed a weakly coordinating auxiliary for sulfonamides to enable rapid lead diversification in medicinal chemistry. For example, Celebrex analog 167, can undergo six categorically distinct Pd(II)-catalyzed C–H functionalization reactions (Scheme 24).
Scheme 24.

Divergent C–H functionalization of drug candidates.53
6. Future Outlook
As the field moves forward, weakly coordinating directing groups will play an important role in the development of ligand-controlled metal-catalyzed C–H functionalization, including enantio- and position-selective reactions, as well as ligand-accelerated reactions. Directing groups that coordinate strongly to the metal can facilitate rapid C–H cleavage, but by the same token, can cause the substrate to outcompete potential ligands for empty coordination sites on the metal. Our group has found success using substrate/ligand combinations with matched coordinative affinity. In particular, we have had success in developing enantioselective C–H functionalization reactions using [Pd(II)–amino acid] catalysts, first using diphenyl pyridyl substrates (which are weakly coordinative due to steric bulk) and later diphenylacetic acids (Scheme 25).54, 55 We found that this same class of ligands was successful in promoting position-selective C–H olefination,56 and in accelerating C–H functionalization reactions (Scheme 26).5
Scheme 25.

Scheme 26.

Ligand-accelerated Pd(II)-catalyzed C–H olefination.5
Weak coordinating directing groups have also recently emerged as a promising approach for achieving high reactivity in C–H functionalization with other metals, including Rh(III) and Ru(II). Though we cannot highlight examples here due to space constraints, we believe that the use of weakly coordinating functional groups will be generally applicable in enabling the discovery of many new metal-catalyzed C–H functionalization reactions.
6. Conclusions
Utilizing substrates with weakly coordinating directing groups for Pd(II)-catalyzed C–H functionalization is a powerful approach for developing synthetically versatile reactions. In our experience this approach enables unprecedented breadth in the functionalization step, owing to the higher reactivity of the putative cyclopalladated intermediates. We have pursued two primary strategies with weak coordination: (1) utilizing commonly occurring functional groups and (2) developing practical auxiliaries that can readily be installed and removed. In the future, we expect this approach to play an instrumental role in the study of ligand-controlled catalytic C–H functionalization reactions with Pd(II) and other metals.
Scheme 5.

Different catalytic manifolds in Pd-catalyzed C–H functionalization.
Acknowledgements
We are indebted to all present and former group members for their invaluable contributions to the work described herein. We acknowledge TSRI, the NSF (CHE-1011898), the NIH (NIGMS, 1 R01 GM084019-02), and Pfizer for financial assistance. Additional support was provided through the NSF Center for Stereoselective C–H Functionalization (CHE-0943980). Individual awards and fellowships were granted by TSRI, the NSF GRFP, the NDSEG Fellowship program, and the Skaggs-Oxford Scholarship program (K.M.E.); the Chinese Government Scholarship Council (T.-S.M.); and Bristol-Myers-Squibb (M.W.). TSRI Manuscript no. 21344.
Biography
Keary Engle received his B.Sc. from the University of Michigan in 2007, then spent the following as a Fulbright Scholar with Prof. Manfred Reetz at the at the MPI für Kolenforschung. He subsequently moved to the laboratory of Prof. Jin-Quan Yu at TSRI, where is currently and NSF and NDSEG predoctoral fellow.
Tiang-Sheng Mei earned his B.Sc. in Chemistry from Lanzhou University in 2001. While there, he carried out research under the supervision of Prof. Yu-Lin Li. In 2005, he began graduate studies under Prof. Jin-Quan Yu at Brandeis University, where he earned his M.Sc. He subsequently relocated to TSRI, where he is presently pursuing his Ph.D.
Masayuki Wasa earned his B.Sc. degree with Highest Honors from Brandeis University in 2006. He is currently pursuing his Ph.D. at TSRI with Prof. Jin-Quan Yu, where he is a Bristol-Myers Squibb Fellow.
Jin-Quan Yu received his B.Sc. in Chemistry from East China Normal University and his M.Sc. from the Guangzhou Institute of Chemistry. In 2000, he obtained his Ph.D. at the University of Cambridge with Prof. J. B. Spencer. Following time as a junior research fellow at Cambridge, he joined the laboratory of Prof. E. J. Corey at Harvard University as a postdoctoral fellow. He then began his independent career at Cambridge (2003–2004), before moving to Brandeis University (2004–2007), and finally to TSRI, where he is currently Professor of Chemistry.
References
- 1.Engle KM, Yu J-Q. Transition Metal–Catalyzed C–H Functionalization: Synthetically Enabling Reactions for Building Molecular Complexity. Weinheim, Germany; Wiley: 2011. [Google Scholar]
- 2.Murai S, Kakiuchi F, Sekine S, Tanaka Y, Kamatani A, Sonoda M, Chatani N. Efficient catalytic addition of aromatic carbon-hydrogen bonds to olefins. Nature. 1993;366:529–531. [Google Scholar]
- 3.Trost BM, Imi K, Davies IW. Elaboration of Conjugated Alkenes Initiated by Insertion into a Vinylic C-H Bond. J. Am. Chem. Soc. 1995;117:5371–5372. [Google Scholar]
- 4.Lenges CP, Brookhart M. Addition of Olefins to Aromatic Ketones Catalyzed by Rh(I) Olefin Complexes. J. Am. Chem. Soc. 1999;121:6616–6623. doi: 10.1021/ja066509x. [DOI] [PubMed] [Google Scholar]
- 5.Engle KM, Wang D-H, Yu J-Q. Ligand-Accelerated C–H Activation Reactions: Evidence for a Switch of Mechanism. J. Am. Chem. Soc. 2010;132:14137–14151. doi: 10.1021/ja105044s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trofimenko S. Cyclopalladation reaction. Inorg. Chem. 1973;12:1215–1221. [Google Scholar]
- 7.Labinger JA, Bercaw JE. Understanding and exploiting C–H bond activation. Nature. 2002;417:507–514. doi: 10.1038/417507a. [DOI] [PubMed] [Google Scholar]
- 8.Kleiman JP, Dubeck M. The Preparation of Cyclopentadienyl [o-(Phenylazo)Phenyl]Nickel. J. Am. Chem. Soc. 1963;85:1544–1545. [Google Scholar]
- 9.Cope AC, Siekman RW. Formation of Covalent Bonds from Platinum or Palladium to Carbon by Direct Substitution. J. Am. Chem. Soc. 1965;87:3272–3273. [Google Scholar]
- 10.Bennett MA, Milner DL. Chlorotris(triphenylphosphine)iridium(I): an example of hydrogen transfer to a metal from a co-ordinated ligand. Chem. Commun. (London) 1967:581–582. [Google Scholar]
- 11.Keim W. New [sigma]-bonded rhodium(I) complexes containing a metal--carbon bond. I. J. Organomet. Chem. 1968;14:179–184. [Google Scholar]
- 12.Knoth WH, Schunn RA. Ligand-metal hydrogen transfer in phosphite complexes. J. Am. Chem. Soc. 1969;91:2400. [Google Scholar]
- 13.Ryabov AD. Cyclopalladated Complexes in Organic Synthesis. Synthesis. 1985:233–252. [Google Scholar]
- 14.Constable AG, McDonald WS, Sawkins LC, Shaw BL. Palladation of dimethylhydrazones, oximes, and oxime O-allyl ethers: crystal structure of [Pd3(ON = CPriPh)6]. J. Chem. Soc., Chem. Commun. 1978:1061–1062. [Google Scholar]
- 15.Fujiwara Y, Moritani I, Matsuda M, Teranishi S. Aromatic substitution of olefin. IV Reaction with palladium metal and silver acetate. Tetrahedron Lett. 1968;9:3863–3865. [Google Scholar]
- 16.Fahey DR. The coordination-catalyzed ortho-halogenation of azobenzene. J. Organomet. Chem. 1971;27:283–292. [Google Scholar]
- 17.Tremont SJ, Rahman H. u. Ortho-alkylation of acetanilides using alkyl halides and palladium acetate. J. Am. Chem. Soc. 1984;106:5759–5760. [Google Scholar]
- 18.Dick AR, Hull KL, Sanford MS. A Highly Selective Catalytic Method for the Oxidative Functionalization of C–H Bonds. J. Am. Chem. Soc. 2004;126:2300–2301. doi: 10.1021/ja031543m. [DOI] [PubMed] [Google Scholar]
- 19.Giri R, Chen X, Yu J-Q. Palladium-Catalyzed Asymmetric Iodination of Unactivated C–H Bonds under Mild Conditions. Angew. Chem. Int. Ed. 2005;44:2112–2115. doi: 10.1002/anie.200462884. [DOI] [PubMed] [Google Scholar]
- 20.Daugulis O, Zaitsev VG. Anilide ortho-Arylation by Using C–H Activation Methodology. Angew. Chem. Int. Ed. 2005;44:4046–4048. doi: 10.1002/anie.200500589. [DOI] [PubMed] [Google Scholar]
- 21.Chen X, Li J-J, Hao X-S, Goodhue CE, Yu J-Q. Palladium-Catalyzed Alkylation of Aryl C-H Bonds with sp3 Organotin Reagents Using Benzoquinone as a Crucial Promoter. J. Am. Chem. Soc. 2006;128:78–79. doi: 10.1021/ja0570943. [DOI] [PubMed] [Google Scholar]
- 22.Hoveyda AH, Evans DA, Fu GC. Substrate-directable chemical reactions. Chem. Rev. 1993;93:1307–1370. [Google Scholar]
- 23.Katsuki T, Sharpless KB. The first practical method for asymmetric epoxidation. J. Am. Chem. Soc. 1980;102:5974–5976. [Google Scholar]
- 24.Noyori R. Asymmetric Catalysis: Science and Opportunities (Nobel Lecture). Angew. Chem. Int. Ed. 2002;41:2008–2022. [PubMed] [Google Scholar]
- 25.Zhang W, Yamamoto H. Vanadium-Catalyzed Asymmetric Epoxidation of Homoallylic Alcohols. J. Am. Chem. Soc. 2006;129:286–287. doi: 10.1021/ja067495y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown JM. Tilden Lecture. Selectivity and mechanism in catalytic asymmetric synthesis. Chem. Soc. Rev. 1993;22:25–41. [Google Scholar]
- 27.Evans DA, Bartroli J, Shih TL. Enantioselective aldol condensations. 2. Erythro-selective chiral aldol condensations via boron enolates. J. Am. Chem. Soc. 1981;103:2127–2129. [Google Scholar]
- 28.Myers AG, Yang BH, Chen H, Gleason JL. Use of Pseudoephedrine as a Practical Chiral Auxiliary for Asymmetric Synthesis. J. Am. Chem. Soc. 1994;116:9361–9362. [Google Scholar]
- 29.Ellman JA, Owens TD, Tang TP. N-tert-Butanesulfinyl Imines: Versatile Intermediates for the Asymmetric Synthesis of Amines. Acc. Chem. Res. 2002;35:984–995. doi: 10.1021/ar020066u. [DOI] [PubMed] [Google Scholar]
- 30.Whisler MC, MacNeil S, Snieckus V, Beak P. Beyond Thermodynamic Acidity: A Perspective on the Complex-Induced Proximity Effect (CIPE) in Deprotonation Reactions. Angew. Chem. Int. Ed. 2004;43:2206–2225. doi: 10.1002/anie.200300590. [DOI] [PubMed] [Google Scholar]
- 31.Miura M, Tsuda T, Satoh T, Pivsa-Art S, Nomura M. Oxidative Cross-Coupling of N-(2'-Phenylphenyl)benzene- sulfonamides or Benzoic and Naphthoic Acids with Alkenes Using a Palladium-Copper Catalyst System under Air. J. Org. Chem. 1998;63:5211–5215. [Google Scholar]
- 32.Hennings DD, Iwasa S, Rawal VH. Anion-Accelerated Palladium-Catalyzed Intramolecular Coupling of Phenols with Aryl Halides. J. Org. Chem. 1997;62:2–3. doi: 10.1021/jo961876k. [DOI] [PubMed] [Google Scholar]
- 33.Wang D-H, Hao X-S, Wu D-F, Yu J-Q. Palladium-Catalyzed Oxidation of Boc-Protected N-Methylamines with IOAc as the Oxidant: A Boc-Directed sp3 C–H Bond Activation. Org. Lett. 2006;8:3387–3390. doi: 10.1021/ol061384m. [DOI] [PubMed] [Google Scholar]
- 34.Kao L-C, Sen A. Platinum(II) catalysed selective remote oxidation of unactivated C-H bonds in aliphatic carboxylic acids. J. Chem. Soc., Chem. Commun. 1991:1242–1243. [Google Scholar]
- 35.Dangel BD, Johnson JA, Sames D. Selective Functionalization of Amino Acids in Water: A Synthetic Method via Catalytic C–H Bond Activation. J. Am. Chem. Soc. 2001;123:8149–8150. doi: 10.1021/ja016280f. [DOI] [PubMed] [Google Scholar]
- 36.Lee JM, Chang S. Pt-Catalyzed sp3 C–H bond activation of o-alkyl substituted aromatic carboxylic acid derivatives for the formation of aryl lactones. Tetrahedron Lett. 2006;47:1375–1379. [Google Scholar]
- 37.Kisenyi JM, Sunley GJ, Cabeza JA, Smith AJ, Adams H, Salt NJ, Maitlis PM. The cyclometallation of benzoic acid to give rhodium, iridium, and osmium C,O-benzoates. X-Ray structure determination of the dibenzoate [(C5Me5)Rh(OOCPh)2(H2O)]. J. Chem. Soc., Dalton Trans. 1987:2459–2466. [Google Scholar]
- 38.Mei T-S, Giri R, Maugel N, Yu J-Q. Pd(II)-Catalyzed Monoselective ortho-Halogenation of C-H Bonds Assisted by Counter Cations: A Complementary Method to Directed ortho-Lithiation. Angew. Chem. Int. Ed. 2008;47:5215–5219. doi: 10.1002/anie.200705613. [DOI] [PubMed] [Google Scholar]
- 39.Giri R, Yu J-Q. Synthesis of 1,2- and 1,3-Dicarboxylic Acids via Pd(II)-Catalyzed Carboxylation of Aryl and Vinyl C–H Bonds. J. Am. Chem. Soc. 2008;130:14082–14083. doi: 10.1021/ja8063827. [DOI] [PubMed] [Google Scholar]
- 40.Giri R, Maugel N, Li J-J, Wang D-H, Breazzano SP, Saunders LB, Yu J-Q. Palladium-Catalyzed Methylation and Arylation of sp2 and sp3 C–H Bonds in Simple Carboxylic Acids. J. Am. Chem. Soc. 2007;129:3510–3511. doi: 10.1021/ja0701614. [DOI] [PubMed] [Google Scholar]
- 41.Chiong HA, Pham Q-N, Daugulis O. Two Methods for Direct ortho-Arylation of Benzoic Acids. J. Am. Chem. Soc. 2007;129:9879–9884. doi: 10.1021/ja071845e. [DOI] [PubMed] [Google Scholar]
- 42.Wang D-H, Wasa M, Giri R, Yu J-Q. Pd(II)-Catalyzed Cross-Coupling of sp3 C–H Bonds with sp2 and sp3 Boronic Acids Using Air as the Oxidant. J. Am. Chem. Soc. 2008;130:7190–7191. doi: 10.1021/ja801355s. [DOI] [PubMed] [Google Scholar]
- 43.Wang G-W, Yuan T-T, Li D-D. One-Pot Formation of C–C and C–N Bonds through Palladium-Catalyzed Dual C–H Activation: Synthesis of Phenanthridinones. Angew. Chem. Int. Ed. 2011;50:1380–1383. doi: 10.1002/anie.201005874. [DOI] [PubMed] [Google Scholar]
- 44.Wasa M, Engle KM, Yu J-Q. Pd(0)/PR3-Catalyzed Intermolecular Arylation of sp3 C–H Bonds. J. Am. Chem. Soc. 2009;131:9886–9887. doi: 10.1021/ja903573p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li J-J, Mei T-S, Yu J-Q. Synthesis of Indolines and Tetrahydroisoquinolines from Arylethylamines by Pd(II)-Catalyzed C–H Activation Reactions. Angew. Chem. Int. Ed. 2008;47:6452–6455. doi: 10.1002/anie.200802187. [DOI] [PubMed] [Google Scholar]
- 46.Mei T-S, Wang X, Yu J-Q. Pd(II)-Catalyzed Amination of C–H Bonds Using Single-Electron or Two-electron Oxidants. J. Am. Chem. Soc. 2009;131:10806–10807. doi: 10.1021/ja904709b. [DOI] [PubMed] [Google Scholar]
- 47.Lu Y, Wang D-H, Engle KM, Yu J-Q. Pd(II)-Catalyzed Hydroxyl-Directed C–H Olefination Enabled by Monoprotected Amino Acid Ligands. J. Am. Chem. Soc. 2010;132:5916–5921. doi: 10.1021/ja101909t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang X, Lu Y, Dai H-X, Yu J-Q. Pd(II)-Catalyzed Hydroxyl-Directed C–H Activation/C–O Cyclization: Expedient Construction of Dihydrobenzofurans. J. Am. Chem. Soc. 2010;132:12203–12205. doi: 10.1021/ja105366u. [DOI] [PubMed] [Google Scholar]
- 49.Zhang Y-H, Yu J-Q. Pd(II)-Catalyzed Hydroxylation of Arenes with 1 atm of O2 or Air. J. Am. Chem. Soc. 2009;131:14654–14655. doi: 10.1021/ja907198n. [DOI] [PubMed] [Google Scholar]
- 50.Zhang Y-H, Shi B-F, Yu J-Q. Palladium(II)-Catalyzed ortho Alkylation of Benzoic Acids with Alkyl Halides. Angew. Chem. Int. Ed. 2009;48:6097–6100. doi: 10.1002/anie.200902262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yoo EJ, Ma S, Mei T-S, Chan KSL, Yu J-Q. Pd-Catalyzed Intermolecular C–H Amination with Alkylamines. J. Am. Chem. Soc. 2011;133:7652–7655. doi: 10.1021/ja202563w. [DOI] [PubMed] [Google Scholar]
- 52.Chan KSL, Wasa M, Wang X, Yu J-Q. Palladium(II)-Catalyzed Selective Monofluorination of Benzoic Acids Using a Practical Auxiliary: A Weak-Coordination Approach. Angew. Chem. Int. Ed. 2011 doi: 10.1002/anie.201102985. DOI: 10.1002/anie.201102985. [DOI] [PubMed] [Google Scholar]
- 53.Dai H-X, Stepan AF, Plummer MS, Zhang Y-H, Yu J-Q. Divergent C–H Functionalizations Directed by Sulfonamide Pharmacophores: Late-Stage Diversification as a Tool for Drug Discovery. J. Am. Chem. Soc. 2011;133:7222–7228. doi: 10.1021/ja201708f. [DOI] [PubMed] [Google Scholar]
- 54.Shi B-F, Maugel N, Zhang Y-H, Yu J-Q. Pd-Catalyzed Enantioselective Activation of C(sp2)–H and C(sp3)–H Bonds Using Monoprotected Amino Acids as Chiral Ligands. Angew. Chem. Int. Ed. 2008;47:4882–4886. doi: 10.1002/anie.200801030. [DOI] [PubMed] [Google Scholar]
- 55.Shi B-F, Zhang Y-H, Lam JK, Wang D-H, Yu J-Q. Pd(II)-Catalyzed Enantioselective C–H Olefination of Diphenylacetic Acids. J. Am. Chem. Soc. 2010;132:460–461. doi: 10.1021/ja909571z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang D-H, Engle KM, Shi B-F, Yu J-Q. Ligand-Enabled Reactivity and Selectivity in a Synthetically Versatile Aryl C-H Olefination. Science. 2010;327:315–319. doi: 10.1126/science.1182512. [DOI] [PMC free article] [PubMed] [Google Scholar]