

Abstract
AMP-activated protein kinase (AMPK) is a critical monitor of cellular energy status and also controls processes related to tumor development, including cell cycle progression, protein synthesis, cell growth and survival. Therefore AMPK as an anti-cancer target has received intensive attention recently. It has been reported that the anti-diabetic drug metformin and some natural compounds, such as quercetin, genistein, capsaicin and green tea polyphenol epigallocatechin gallate (EGCG), can activate AMPK and inhibit cancer cell growth. Indeed, natural products have been the most productive source of leads for the development of anti-cancer drugs but perceived disadvantages, such as low bioavailability and week potency, have limited their development and use in the clinic. In this study we demonstrated that synthetic EGCG analogs 4 and 6 were more potent AMPK activators than metformin and EGCG. Activation of AMPK by these EGCG analogs resulted in inhibition of cell proliferation, up-regulation of the cyclin-dependent kinase inhibitor p21, down-regulation of mTOR pathway, and suppression of stem cell population in human breast cancer cells. Our findings suggest that novel potent and specific AMPK activators can be discovered from natural and synthetic sources that have potential to be used for anti-cancer therapy in the clinic.
Keywords: EGCG analogs, AMP-activated protein kinase, cancer stem cells, breast cancer, cell proliferation
1. Introduction
AMP-activated protein kinase (AMPK) is a serine/threonine protein kinase, which serves as an metabolic sensor and plays a key role in cellular energy homeostasis in all eukaryotic cells1. AMPK is a heterotrimeric protein complex composed of three subunits: a catalytic (α) subunit and two regulatory (β and γ) subunits. AMPK is activated through phosphorylation of Thr-172 on the α subunit under multiple conditions including: (i) an energy-depletion stress, such as ATP level declines and AMP level increases 2-3; (ii) hypoxia 4; (iii) hormone and cytokines such as adiponectin and leptin 5-6; (iv) cellular kinases including liver kinase B1 (LKB1, a primary upstream kinase of AMPK) 7-8, Calmodulin-dependent protein kinase kinase (CaMKK) 9 and proto-oncogene tyrosine-protein kinase Src (c-Src) 10; (v) the anti-diabetic medicine metformin (see below). Once activated, AMPK switches on catabolic pathway and activates ATP-generating processes including the uptake and oxidation of glucose and fatty acids. Activated AMPK also switches off anabolic pathways, which consume ATP, including protein fatty acid and cholesterol syntheses. Activation of AMPK also induces cell cycle arrest and inhibits cell proliferation in malignant cells through multiple mechanisms including up-regulation of the p53-p21 axis 11 and down-regulation of the mammalian target of rapamycin (mTOR) pathway 12-13. Investigation into the regulation and bio-functions of AMPK in cancer field has been intensively conducted. Accumulating evidence suggests that targeting AMPK may be a promising therapeutic option for cancer therapy.
Metformin, an anti-diabetic drug, could also activate AMPK, raising a hypothesis that metformin may reduce the risk of cancer in patients with type 2 diabetes through activation of AMPK pathway 14. It has been reported that metformin not only inhibits the tumor cell growth in both cell lines 15-16 and mouse models 17, but also selectively kills cancer stem cells 18. Besides metformin, green tea polyphenol EGCG (Figure 1A) demonstrated ability to induce apoptosis in cancer cells, associated with activation of AMPK signaling pathway 19-20. However, the caveat and limitation to the use of EGCG in cancer prevention and therapy is its low bioavailability due to its instability under neutral or alkaline conditions (i.e. physiologic pH) and to biologically inactivating processes that include methylation 21-22. An octa-acetate derivative of EGCG (pro-EGCG, Figure 1A) has been found to function as the pro-drug of EGCG with improved bioavailability 23.
Figure 1.
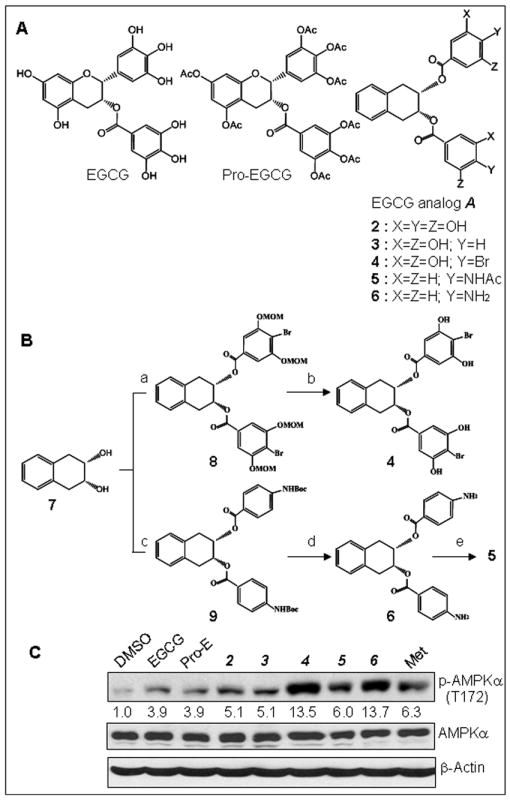
Chemical structures of EGCG analogs, their chemical synthetic scheme and their effect on AMPK activation. (A) Chemical structures of natural EGCG, synthetic pro-EGCG and general structure A of EGCG analogs. (B) Brief summary of a synthetic scheme of EGCG analogs 4, 5 and 6 including steps a - e. The step a: 4-bromo-3,5-dimethoxymethoxybenzoic acid/diisopropylethylamine/ dicyclohexylcarbodiimide in DCM; b: p-toluenesulfonic acid/MeOH; c: N-boc-p-aminobenzoic acid/diisopropylethylamine/ dicyclohexylcarbodiimide in DCM; d: TFA in DCM; e: Ac2O/pyridine. (C) EGCG analogs 4 and 6 are more potent AMPK activators when compared with natural EGCG and metformin. MDA-MB-231 cells were treated with 20 μM of natural EGCG or synthetic EGCG analogs or 10 mM of metformin for 3 h, followed by Western blot analysis. The numbers underneath the Western results of p-AMPKα indicate normalized phosphor-AMPKα / β-actin ratios.
We have previously shown that (+)-EGCG, the synthetic enantiomer of natural (-)-EGCG, is equally potent in the inhibition of the chymotrypsin like activity of proteasome.24 This suggested that the β5 active site binds equally well to (+)- and (-)-EGCG and is thus pseudo-symmetric. On this basis, we synthesized simple, symmetrical analogs 2 and 3 of general structure A (Figure 1A) and they were found to be inhibitors of proteasome as well.25 Presumably, they bind to the same β5 active site and the gallate function in 2 resembles the gallate ester of EGCG 26. Furthermore, the 4’-deoxy analog 3 can also mimic EGCG with the additional advantage that 3 is not a substrate of human catechol-O-methyltransferase (COMT) and can not be biomethylated as natural EGCG. The compound 3 exhibits higher cytotoxicity than natural EGCG in human breast cancer cells which harbor high catechol-O-methyltransferase activity 27. We also found that p-aminobenzoate 28 and fluorobenzoate 29 could replace the gallate group of EGCG to induce cancer cell apoptosis.
We have thus synthesized a focused library of EGCG analogs of general structure A (Figure 1A) with the substituents X, Y and Z being H, OH, OAc, NH2, NHAc, alkyl and halogens (Figure 1B). By screening this library of about 58 different compounds (U. S. Patent Application 2011/0152210 A1, June 23, 2011),30 we found that compared with natural product EGCG and metformin, analogs 4 and 6 were AMPK activators with greater potency. Activation of AMPK by these EGCG analogs resulted in inhibition of cell proliferation, suppression of tumorsphere formation and decrease of cancer stem cell population in human breast cancer cells, which is associated with down-regulation of mTOR pathway and up-regulation p21 protein. The results also showed that these EGCG analogs were able to enhance efficacy of anti-cancer drugs in human breast cancer cells, associated with activation of AMPK pathway. Therefore, it would be an attractive approach to develop novel AMPK activators as anti-cancer agents from structure-modified natural compound, which possess unique properties of higher efficacy, lower toxicity and targeting cancer stem cells as well.
2. Chemistry
2.1. General
Compounds 2 and 3 were synthesized as previously described 25. The library of compounds of general structure A (Figure 1A) were synthesized as reported.30 Generally, the starting materials and reagents, purchased from commercial suppliers, were used without further purification. Anhydrous methylene chloride was distilled under nitrogen from CaH2. Anhydrous DMF was distilled under vacuum from CaH2. Reaction flasks were flame-dried under a stream of N2. All moisture-sensitive reactions were conducted under a nitrogen atmosphere. Flash chromatography was carried out using silica-gel 60 (70-230 mesh). The melting points were uncorrected. 1H-NMR and 13C NMR (300 MHz) spectra were measured with TMS as an internal standard when CDCl3, CD3OD and acetone-d6 were used as solvent. High-resolution (ESI) MS spectra were recorded using a QTOF-2 Micromass spectrometer to analyze the compounds.
2.2. Synthesis of EGCG analog 4
2.2.1. 4-Bromo-3,5-dimethoxymethoxybenzoic acid
A solution of 4-bromo- 3, 5-dihydroxylbenzoic acid (500mg, 2.1 mmol) in dry DCM at 0 °C was added with diisopropylethylamine (2.49gm, 18.9 mmol) dropwise, followed by methoxymethyl chloride (1.5gm, 18.9 mmol) dropwise and stirred for 48h at room temperature. After completion of the reaction as indicated by TLC, the reaction mixture was quenched with saturated NH4Cl sol (10ml) and extracted twice with DCM. Removal of the solvent gave the intermediate ether ester. To this ether ester in MeOH (80ml) was added 15% NaOH in MeOH (80ml) and the whole was heated at 70 °C for 3h. The pH of the reaction was adjusted to 5-6 by addition of 6N HCl at 0 °C followed by filtration. The MOM protected benzoic acid was obtained as white solid, m.p. 172 °C, (400mg, 60% yield): 1H NMR (400MHz, CDCl3) δ: 7.54 (s, 2H), 5.32 (s, 4H), 3.53 (s, 6H); 13C NMR (400 MHz, d4- CH3OH) δ: 167.3, 154.8, 130.7, 109.6, 108.5, 94.8, 55.2; ESI MS m/z: 318(M-1).
2.2.2. 2,3-Naphthalene-1,2,3,4-tetrahydro-2,3-bis(4-bromo-3,5-dimethoxymethoxybenzoate), (2R,3S)rel (8)
The acid (466 mg, 1.4 mmol) obtained above and dicyclohexylcarbodiimide (296 mg, 1.4 mmol) were taken in dry DCM and stirred for 1h at room temperature (rt). Then 4-dimethylaminopyridine (14mg, 0.08mmol) and the cis-diol 7 (100mg, 0.6 mmol), was added and stirred at rt. After 24h the formed ppt was filtered off, and purified by column chromatography (3:7, Ethyl Acetate:Hexane) to give the product as clear white solid, m.p. 138 °C, (400 mg, 85% yield): 1H NMR (400MHz, CDCl3) δ: 7.41 (s, 4H), 7.22-7.16 (m, 4H), 5.71-5.65 (m, 2H), 5.2 (s, 8H), 3.42 (s, 12H), 3.38-3.22 (m, 4H); 13C NMR (400MHz, CDCl3) δ: 165.0, 154.8, 132.0, 130.1, 129.0, 126.5, 110.2, 109.8, 95.1, 70.6, 56.4, 32.0; ESI MS m/z: 793 (M+ Na); HRMS (ESI) m/z Calc for (M+23) C32 H34 Br2 O12 Na, 791.0309, Found 791.0338.
2.2.3. 2,3-Naphthalene-1,2,3,4-tetrahydro-2,3-bis(4-bromo-3,5-dihydroxybenzoate), (2R,3S)rel (4)
Para-Toulenesulfonic acid (35mg, 0.18mmol) was added to a solution of the diester 8 (350mg, 0.45mmol) obtained above in MeOH and refluxed for 3h. After completion of the reaction, the reaction mixture was neutralized by solid NaHCO3, filtered and dried over NaSO4. Purification by column chromatography gave the product 4 as white solid, m.p. 164 °C, (180mg, 65% yield): 1H NMR (400 MHz, CDCl3) δ: 9.13 (brs, 4H), 7.20 (s, 4H), 7.11 (s, 4H), 5.75-5.6 (m, 2H), 3.4-3.2 (m, 4H),; 13C NMR (400 MHz, d3- CH3Cl) δ: 205.4, 164.8, 155.3, 132.4 130.2, 129.0, 126.4, 107.8, 70.2, 31.6; ESI MS m/z: 594 (M); HRMS (ESI) m/z, Calc for (M+23) C24 H18 Br2 O8 Na, 614.9260, Found 614.9280.
2.3. Synthesis of compounds 5 and 6
2.3.1. 2,3-Naphthalene-1,2,3,4-tetrahydro-2,3-bis(N-t-butoxycarbonyl-4-aminobenzoate), (2R,3S)rel (9)
The N-boc-p-aminobenzoic acid (45 mg, 0.21 mmol) and dicyclohexylcarbodiimide (42mg, 0.21 mmol) were taken in dry DCM and stirred for 2h at rt. 4-Dimethylaminopyridine (3mg, 0.03mmol) and the cis-diol 7 (20mg, 0.12mmol) were added and stirred at rt. After 24h the formed precipitate was filtered off, and purified by column chromatography (1.5:8.5, Ethyl Acetate:Hexane) to give compound 9 as white solid, m.p. 110 °C, (45 mg, 60%yield): 1H NMR (400 MHz, CDCl3) δ: 8.78 (brs, 2H), 7.90 (d, 4H), 7.63 (d, 4H), 7.2 (s, 4H), 5.8-5.65 (m, 2H), 3.4-3.35 (m, 4H), 1.47(s, 18H),; 13C NMR (400MHz, d6-Acetone) δ: 205.3, 165.0, 152.4, 144.2, 132.7, 130.5, 129.0, 126.3, 123.7, 117.2, 117.1, 79.8, 69.8, 31.8, 27.5; ESI MS m/z: 625 (M + Na). HRMS (ESI) m/z, Calc for (M+23) C34 H38 N2 O8 Na, 625.2520, Found 625.2543.
2.3.2. 2,3-Naphthalene-1,2,3,4-tetrahydro-2,3-bis(4-aminobenzoate), (2R,3S)rel (6)
Compound 9 (130mg, 0.2mmol) was dissolved in DCM and a little excess of trifluoroacetic acid (246mg, 2mmol) was added and stirred at rt for overnight. The excess trifluoroacetic acid was removed and the crude product was purified by column chromatography to give compound 6 as white solid, m.p. 94 °C, (80mg, 92% yield): 1H NMR (300 MHz, d6-Acetone) δ : 7.69 (d, 4H), 7.17 (m, 4H), 6.62 (d, 4H), 5.62 (t, 2H), 5.42 (brs, 4H), 3.29-3.36 (m, 4H),; 13C NMR (400MHz, d6-Acetone) δ: 205.3, 165.4, 132.9, 131.3, 128.9, 126.1, 117.6, 112.8, 69.2, 32.0; ESI MS m/z: 425 (M + Na); HRMS (ESI) m/z, Calc for (M+23) C24 H22 N2 O4 Na, 425.1471, Found 425.1471.
2.3.3. 2,3-Naphthalene-1,2,3,4-tetrahydro-2,3-bis(4-acetamidobenzoate), (2R,3S)rel (5)
Compound 6 (40mg, 0.09mmol), acetic anhydride (0.5ml) and pyridine (0.5ml) were stirred at rt for 24h. After completion of the reaction, ethyl acetate was added and stirred for 5 min then 1N HCL (1ml) was added and stirred for another 5 min. The solution was washed with CuSO4 solution (2×10ml), water (2×10ml), brine (2×10ml), dried over Na2SO4 and purified by column chromatography to give compound 5 as white solid, m. p. 128 °C, (40mg, 82% yield): 1H NMR (300 MHz, d2-DCM) δ: 7.90 (d, 6H). 7.55 (d, 4H), 7.18 (s, 4H), 5.5.72-5,62 (m, 2H), 3.34-3.24 (m, 4H), 2.14 (s, 6H),; 13C NMR (300 MHz, d2-DCM) δ: 169.7, 165.5, 142.9, 132.4, 130.6, 129.0, 126.3, 124.9, 118.7, 70.0, 31.9, 24.0; ESI MS m/z: 509 (M + Na); HRMS (ESI) m/z, Calc for (M+23) C28 H26 N2 O6 Na, 509.1683, Found 509.1702.
3. Materials and methods for biological studies
3.1. Materials
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), dimethyl sulfoxide (DMSO) and metformin were purchased from Sigma–Aldrich (St. Louis, MO, USA). Antibodies against phosphor-AMPKα (T172), AMPKα, phosphor-Raptor, were purchased from Cell Signaling Technology, Inc. (Danvers, MA). Antibodies against β-actin and p21 were obtained from Santa Cruz Biotechnologies (Santa Cruz, CA, USA). Mouse monoclonal antibody against human PARP was purchased from BIOMOL International LP (Plymouth Meeting, PA, USA). Antibodies CD44-FITC and CD24-PE were purchased from BD Biosciences (San Jose, CA, USA).
3.2. Cell culture, protein extraction, and Western blot assay
Human breast cancer MDA-MB-231 cells were grown in DMEM/F12 (1:1) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin and maintained at 37°C in a humidified incubator with an atmosphere of 5% CO2. A whole cell extract was prepared as previously described32. For Western blot analysis, the cell extracts were separated by SDS–PAGE and transferred to a nitrocellulose membrane, followed by visualization using the enhanced chemiluminescence (ECL) reagent (Amersham Biosciences, Piscataway, NJ, USA).
3.3. Cell viability/proliferation assay
The MTT assay was used to determine the effects of various compounds on proliferation of MDA-MB-231 breast cancer cells. Cells were plated onto a 96-well plate and grown to 70-80% confluency followed by adding indicated EGCG analogs or metformin for 24 h incubation at 37°C. Inhibition of cell proliferation was measured as previously described 33. All samples were assayed in triplicate in three independent experiments, and the mean value for each experiment was calculated. The results are displayed as mean (± standard deviation) and are expressed as percentage of the control, which was considered to be 100%.
3.4. Mammosphere formation assay
Mammosphere formation assay was performed to assess the capacity of cancer stem cell self-renewal. Single cell suspensions of MDA-MB-231 cells were thoroughly suspended and plated on ultra low adherent wells of 6-well plates (Corning, Lowell, MA) at 1,000 cells/well in 1.5 ml of sphere formation medium (1:1 DMEM/F12 medium supplemented with 50 units/ml penicillin, 50 mg/ml streptomycin, B-27, and N-2). One milliliter of sphere formation medium was added every 3–4 days. After 7 days of incubation with different concentrations of EGCG analogs 4, 6 or metformin, the formed spheres were collected by centrifugation at 300 g for 5 min and counted with an inverted phase-contrast Zeiss Axiovert 25 microscope.
3.5. Flow cytometry
MDA-MB-231 cells were cultured on low adherent plates in sphere formation medium for 5 days and the formed colonies were washed with PBS, and incubated with accutase solution (Sigma) at 37°C for 5 min. The cells were then seeded on p100 dishes and treated with different concentrations of compounds 4 or 6 for 48 hours. The treated cells were detached by using 0.02% EDTA in phosphate-buffered saline (PBS), counted and washed in 0.1% BSA in PBS, and followed by staining with anti-CD24-PE and anti-CD44-FITC (BD Pharmingen, San Diego, CA, USA), or respective isotype controls for 30 min in the dark at 4°C. After washing steps, the labeled cells were analyzed by flow cytometry using a FACS FACscan (Becton Dickinson, CA, USA).
4. Results
4.1. EGCG analogs 4 and 6 significantly activate AMPK in human breast cancer cells
In order to discover AMPK activators from natural resource, we screened a series of EGCG analogs (see Figure 1A) in breast cancer cells. Treatment of human breast cancer MDA-MB-231 cells with 20 μM of the EGCG analogs was performed. Metformin and natural product EGCG were used as controls. After treatment for 3 hours, the cell lysates of the treated cells were analyzed by Western blot to measure levels of phosphor-AMPKα (T172), an active form of AMPK protein (Figure 1C). As reported, metformin and EGCG can activate AMPK signaling pathway 17-19, 34. Our results (Figure 1C) showed that pro-EGCG, the pro-drug of EGCG, was as effective as EGCG in activating the AMPK signaling pathway. Among our synthetic EGCG analogs, 4 and 6 not only significantly activated AMPK but also with greater potency compared with metformin, even at lower concentrations (20 μM vs. 10 mM) (Figure 1C). The compounds 4 and 6 were also more potent AMPK activator than natural product EGCG (Figure 1C).
4.2. EGCG analogs 4 and 6 inhibit proliferation of breast cancer cells in a dose-dependent manner associated with activation of AMPK and induction of p21 protein
It has been reported that AMPK activation by an authentic AMPK activator AMP-mimetic 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR) results in cell cycle arrest and inhibition of cell proliferation in hepatoma HepG2 cells 35. In order to determine whether EGCG analogs 4 and 6 could play a similar role as AICAR in suppression of cell proliferation, we treated human breast cancer MDA-MB-231 cells with different concentrations of compounds 4 and 6, followed by Western-blot analysis and cell proliferation assay. The cells treated with metformin and natural product of EGCG were served as controls. The results showed that both EGCG analogs 4 and 6 could inhibit cell proliferation in a dose-dependent manner and their inhibitory effects were more potent than EGCG and metformin even when analogs 4 and 6 were used at much lower concentrations compared with metformin treatment (Figure 2A). Results in Western blot showed that compounds 4 and 6 activated AMPK in a dose-dependent manner as well, as measured by increased levels of phosphor-AMPKα and phosphor-Raptor, one of the direct downstream substrate proteins of AMPK (Figure 2B). Our data also showed that activation of AMPK by 4 and 6 could suppress mTOR pathway measured by decreased phosphor-p70-S6K (Figure 2B), demonstrating the functionality of these EGCG analogs as AMPK activators.
Figure 2.
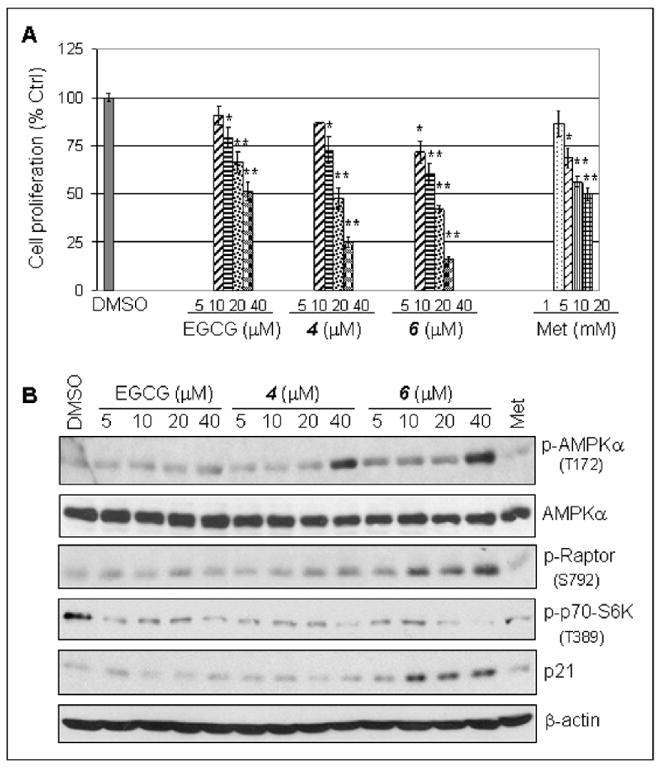
EGCG analogs 4 and 6 inhibited breast cancer cell proliferation through activation of AMPK and upregulation of p21. (A) EGCG analogs 4 and 6 inhibited breast cancer cell proliferation. MDA-MB-231 cells were treated with indicated concentrations of 4, 6 or metformin for 24 h, followed by a MTT assay. *, P < 0.1; **, P < 0.01. Columns, mean of three experiments; bars, SD. (B) EGCG analogs 4 and 6 activated AMPK at a dose-dependent manner, measured by elevated level of phosphor-AMPKα and its downstream proteins of phosphor-Raptor. Treatment with 4 and 6 also showed increased level of p21. MDA-MB-231 cells were treated with indicated concentrations of 4 or 6 for 3 h, followed by Western blot analysis with the indicated antibodies. The numbers underneath the Western results of p-AMPKα indicated normalized phosphor-AMPKα / β-actin ratios. The numbers underneath the Western results of p-AMPKα indicate normalized phosphor-AMPKα / β-actin ratios.
The cell proliferation inhibitory results in the figure 2 were supported by others’ reports, in which AMPK activation was able to inhibit cell proliferation and induce cell cycle arrest in various cancer cells in vitro and in vivo through increasing p53-p21 axis 11, 35. Inhibition of breast cancer cell proliferation by treatment with compounds 4 and 6 was indeed associated with increased levels of p21 protein (Figure 2B).
4.3. Compounds 4 and 6 significantly inhibit mammosphere formation
Tumor stem cells have the characteristic of forming tumor sphere. An experiment of mammosphere formation is a useful tool to identify a human mammary stem/progenitor-cell population and measure stem cell-like behavior. To examine whether compounds 4 and 6 can target cancer stem or stem-like cells and inhibit mammosphere formation, we conducted a mammosphere formation assay. Metformin and EGCG were used as controls. The results showed that treatment of MDA-MB-231 cells with 10 or 20 μM of 4 or 6 for 7 days resulted in inhibition of mammosphere formation by 45.1% and 66.7% or 52.2% and 73.3%, respectively (Figure 3). As comparisons, treatment with 10 or 20 μM of EGCG only inhibited mammosphere formation by 20.1% and 51.3%, and treatment with 5 and 10 mM of metformin inhibited mammosphere formation by 41.4% and 67.6%, respectively (Figure 3). Therefore, EGCG analogs 4 or 6 are much more potent than EGCG and metformin in terms of inhibition of mammosphere formation.
Figure 3.
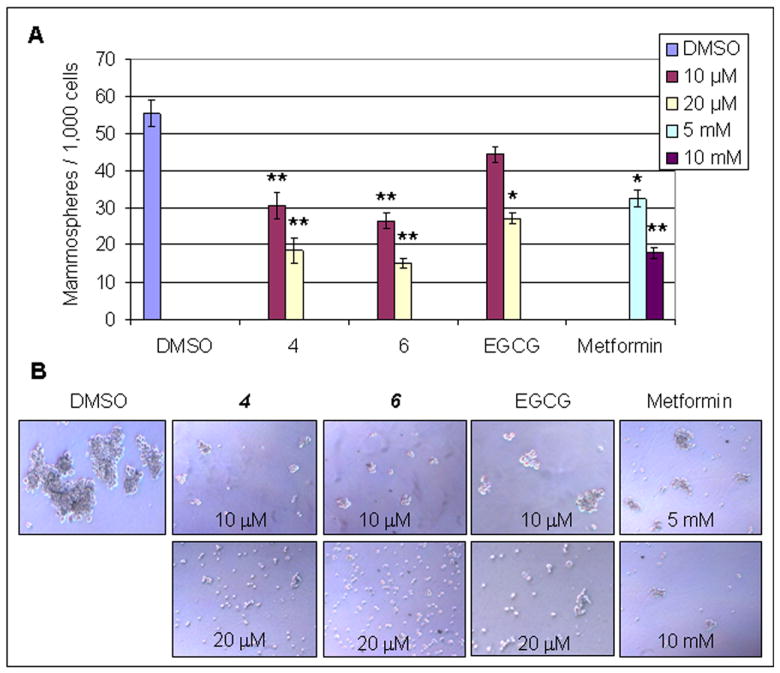
EGCG analogs 4 and 6 inhibited Mammosphere formation. MDA-MB-231 cells were seeded in low attached 6-well plates (1,000 cells/well) and treated with indicated concentrations of 4 and 6 for 7 days, followed by calculating numbers of mammosphere (A) and taking photos of mammosphere morphology (B). Metformin and EGCG were served as controls. *, p<0.1; **, p< 0.01. Columns, mean of three experiments; bars, SD.
4.4. Both EGCG analogs 4 and 6 significantly decreased the CD44+high/CD24-low population in breast cancer cells
Metformin could selectively target cancer stem cells and reduce CD44high/CD24low cell population in breast cancer cells through activation of AMPK signaling 18. To determine whether EGCG analogs 4 and 6 possess the similar property, we treated MDA-MB-231 cells with different concentrations of 4 and 6 for 48 hours. The treated cells were stained with specific antibodies against human CD44 (FITC), CD24 (PE) or their respective isotype controls, followed by washing, fixing and analysis by flow cytometry. The results showed that both EGCG analogs 4 and 6 could reduce CD44high/CD24low cell population. Treatment of MDA-MB-231 cells with 10 or 20 μM of 6 resulted in reduction of the CD44high/CD24low population by 43.3% and 71.7%, respectively (Figure 4). The same results were observed in the cells treated with compound 4, which was slightly less potent than 6 (Figure 4).
Figure 4.
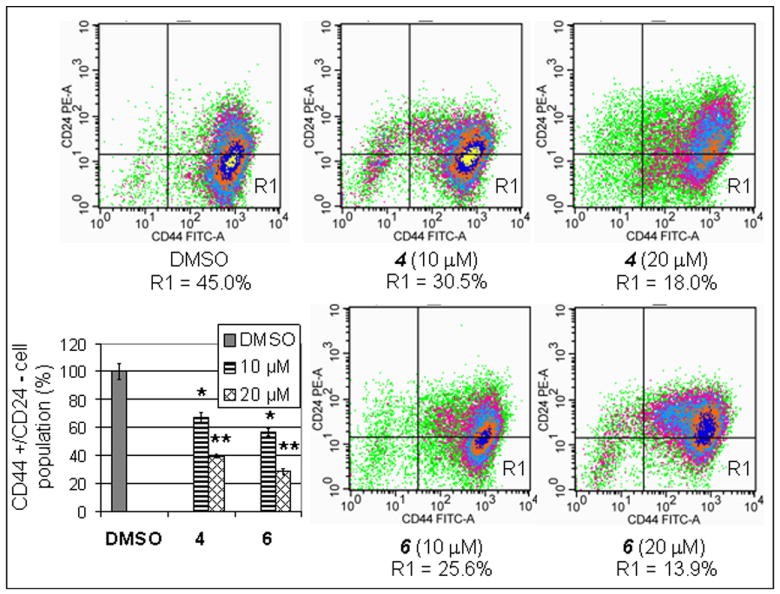
Treatment with EGCG analogs 4 and 6 effectively decreased cancer stem cell population in breast cancer cells. The MDA-MB-231 cells were treated with indicated concentrations of 4 and 6 for 48 hours, followed by flow cytometry analysis of the CD44-FITC and CD24-PE labeled cells. *, p<0.1; **, p< 0.01. Columns, mean of three experiments; bars, SD.
5. Discussion
Breast cancer remains a leading cause of morbidity and mortality in women living in Western countries. The major reason of breast cancer mortality is metastasis of the primary tumor. Experimental evidence from preclinical and clinical studies suggests that cancer stem cells may contribute to tumor progression and metastasis spread. However, most chemotherapeutical drugs can not target cancer stem cells. Furthermore, even though chemotherapy plays an important role as single modality for cancer treatment, limited effectiveness of anti-cancer drugs are often observed because induction of drug-resistance and severe side-effects37. In an attempt to improve conventional chemotherapies, development of more effective agents as an adjuvant therapy is critically important, especially agents that can target cancer stem cells.
AMPK signaling pathway recently becomes a “hot” field in cancer prevention and therapy. Hirsch H et al. reported that metformin selectively targets breast cancer stem cells and inhibited tumor growth in mouse models mediated by activation of AMPK pathway 18. It is becoming an attractive approach to discover more AMPK activators with greater potent and more specific properties. It has been reported that natural compounds genistein (rich in soy been), EGCG (abundant in green tea), and capsaicin (from hot pepper) are able to activate AMPK pathway 34 and anti-cancer effects of these compounds have been also reported previously 38-40. Disadvantages of natural compounds as anti-cancer agents include poor bioavailability, short half-life and weak potency 41. In order to increase bioavailability and stability, we synthesized a pro-drug of EGCG (termed pro-EGCG, Figure 1A), in which all −OH group were protected with acetate groups which can be cleaved by cellular esterases to release EGCG in vivo. Through testing pro-EGCG in vitro and in vivo, we reported that compared with natural EGCG, Pro-EGCG increased the bioavailability, stability, and proteasome inhibitory and anticancer activities in human breast cancer cells and breast cancer xenografts 23.
Recently we synthesized a series of EGCG analogs based on the general chemical structure A (Figure 1A) to mimic natural EGCG in their proteasome inhibition activity. By testing these synthetic compounds we found that EGCG analogs 4 and 6 could inhibit breast cancer proliferation, suppress mammosphere formation and decrease cancer stem cell population. These effects of EGCG analogs 4 and 6 appeared to be associated with their AMPK activating property. Furthermore these two synthetic compounds showed greater potency when compared with EGCG and metformin in terms of activation of AMPK, inhibition of cell proliferation and mammosphere formation (Figures 2, 3).
The AMPK signaling pathway is composed of a series of tumor suppressor genes including LKB1, p53, p21, Forkhead box O-class (FoxO), Raptor and tuberous sclerosis complex 1/2 (TSC1/2). Accumulating evidence indicates that AMPK activation strongly suppresses cell proliferation. These actions of AMPK activators appear to be mediated through multiple mechanisms including regulation of the cell cycle, inhibition of protein synthesis and down-regulation of PI3K-Akt pathway. Cell cycle regulation by AMPK activators is mediated by up-regulation of the p53–p21 axis as well as down-regulation of TSC2–mTOR 42. The cell cycle machinery is tightly controlled by multiple events that are positively regulated by cyclin-dependent kinases (CDKs) and negatively regulated by CDK inhibitors (CDKIs) 43, such as p21CIP and p27KIP, and tumor suppressor proteins, such as p53 44 and FoxO transcription factors 45-46. Most of AMPK activating stimuli, such as glucose deprivation and cellular stresses, can activate p53 through phosphorylation of p53 at the serine (Ser) 15 47. The phosphorylated p53 induces cell-growth arrest via the transcriptional regulation of p53 response genes such as p21CIP and p53-regulated apoptosis-inducing protein 1 (p53AIP1) 48. In order to determine mechanisms of inhibitory cell proliferation by EGCG analogs 4 and 6, we measured the protein level of p21 in MDA-MB-231 cells treated with increasing doses of EGCG analogs 4 and 6. The results revealed that treatment with the compounds 4 and 6 resulted in elevated protein level of p21, as well as phosphor-AMPKα (Thr 172) (Figures 2 and 3).
AMPK activation in breast cancer cells treated with EGCG analogs 4 and 6 resulted in increased protein levels of phosphor-Raptor, a mTOR binding partner and inhibitor, and decreased levels of phosphor-p70S6K, a down-stream protein of mTOR (Figure 2). These data demonstrate that the compounds 4 and 6 are able to down-regulate mTOR pathway mediated by activation of AMPK.
Breast cancer tissue contains a rare population of multi-potent cells with the capacity to self-renew, termed cancer stem cells. These breast cancer stem cells could be enriched and measured by a colony formation assay in three-dimensional cultures, referred to as mammosphere formations. Klopp A et al. reported that cancer stem cells can promote mammosphere formation 49. In the current study, we examined whether exposure of breast cancer cells to EGCG analogs 4 and 6 could block their mammosphere formation. The results showed that mammosphere formation was decreased in a dose-dependent manner in MDA-MB-231 cells treated with the compounds 4 and 6. The inhibitory effect of these synthetic compounds was more potent than the treatment with metformin control (Figure 3).
To further verify whether EGCG analogs 4 and 6 can target cancer stem cells, we selected cell surface protein CD44+ high/CD24- low as breast cancer stem cell-related biomarkers. The CD44 antigen is a cell-surface glycoprotein and a receptor for hyaluronic acid. The CD44 protein belongs to cell surface adhesion molecules involved in a wide variety of cellular functions including cell-cell and cell-matrix interactions 50. CD44 expression is positively associated with stem cell-like characteristics and CD24 is related to differentiation of epithelial cells 51. Breast cancer stem cells, which are rich in CD44+ high/CD24- low cells, were first identified by Al-Hajj et al. 52. This pioneer study showed that this distinct population of cells had the exclusive ability to form tumors in mice. Later, these human breast cancer cells were further demonstrated to contain a defined subpopulation of CD44+ high/CD24- low cells, which possess the property of tumorigenesis 53. As we expected, EGCG analogs 4 and 6 indeed targeted cancer stem cells and significantly decreased the CD44+ high/CD24- low cell population in MDA-MB-231 cells in a dose-dependent manner (Figure 4).
Another candidate marker of cancer stem cells is aldehyde dehydrogenase 1 (ALDH1) 54, which is responsible for the oxidation of intracellular aldehydes to carboxylic acids 55. Increased ALDH1 activity has been found in stem cell populations in both normal and malignant cells 51, 56-57. The ALDH1+ cell population has a small overlap with the CD44+ high/CD24- low cells. Ginestier et al. reported that the overlap of both cell populations represented approximately 1% or less of the total cancer cell population. However, these small populations of cells appeared to harbor highly tumorigenic capability and could generate tumors from as few as 20 cells 54. It will be worthwhile for us to further determine effects of the EGCG analogs on both populations of ALDH1+ and CD44+ high/CD24- low cells and verify the finding in animal tumor models in the near future.
Our previous studies have indicated that EGCG is able to induce tumor cell apoptosis and growth arrest in G1 phase of the cell cycle via inhibition of the chymotrypsin-like activity of the proteasome 40. However, the relationship between proteasome inhibition and AMPK activation mediated by EGCG analog remain unclear. At the present time, there is only limited information on the structure activity relationship among these synthetic compounds.
6. Conclusions
The present study demonstrates that two EGCG analogs 4 and 6 of our library exhibit inhibitory properties in MDA-MB-231 human breast cancer cells through inhibition of cell proliferation and mammosphere formation, suppression of CD44+ high/CD24- low cell populations and activation of AMPK signaling pathway. The activation of AMPK appears to be a key element in the various molecular events presented, and the involved mechanisms were summarized in Figure 5. It is currently unclear how these EGCG analogs activate AMPK, whether any other responsible molecular targets are involved, and what the relationship among these potential targets would be, since besides AMPK pathway, the synthetic EGCG analogs might have multiple molecular targets in cancer cells. Future investigation on this matter including in vivo study may provide new insight into the questions and related mechanisms.
Figure 5.
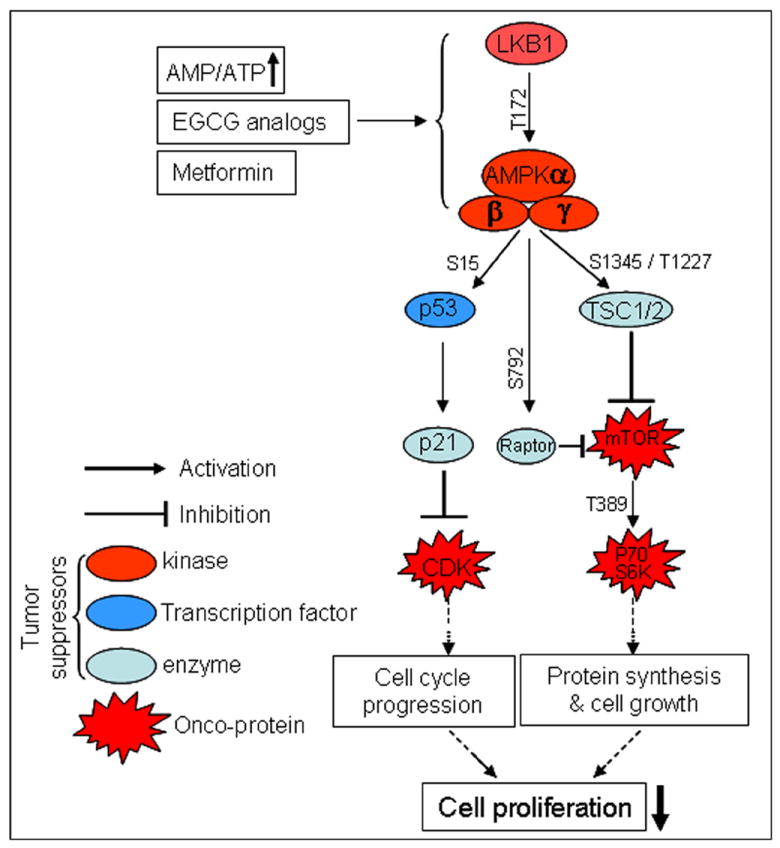
Schematic model for inhibition of cell proliferation by EGCG analogs.
Acknowledgments
This work was partially supported by a pilot fund from Karmanos Cancer Institute and grants from the National Cancer Institute (1R01CA120009, 3R01CA120009-04S1 and 5R01CA127258-05, to QPD).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hardie DG, Carling D, Carlson M. Annu Rev Biochem. 1998;67:821. doi: 10.1146/annurev.biochem.67.1.821. [DOI] [PubMed] [Google Scholar]
- 2.Wang W, Yang X, Lopez de Silanes I, Carling D, Gorospe M. J Biol Chem. 2003;278:27016. doi: 10.1074/jbc.M300318200. [DOI] [PubMed] [Google Scholar]
- 3.Xiao B, Heath R, Saiu P, Leiper FC, Leone P, Jing C, Walker PA, Haire L, Eccleston JF, Davis CT, Martin SR, Carling D, Gamblin SJ. Nature. 2007;449:496. doi: 10.1038/nature06161. [DOI] [PubMed] [Google Scholar]
- 4.Mungai PT, Waypa GB, Jairaman A, Prakriya M, Dokic D, Ball MK, Schumacker PT. Mol Cell Biol. 2011;31:3531. doi: 10.1128/MCB.05124-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hattori Y, Nakano Y, Hattori S, Tomizawa A, Inukai K, Kasai K. FEBS Lett. 2008;582:1719. doi: 10.1016/j.febslet.2008.04.037. [DOI] [PubMed] [Google Scholar]
- 6.Minokoshi Y, Kim YB, Peroni OD, Fryer LG, Muller C, Carling D, Kahn BB. Nature. 2002;415:339. doi: 10.1038/415339a. [DOI] [PubMed] [Google Scholar]
- 7.Fu A, Ng AC, Depatie C, Wijesekara N, He Y, Wang GS, Bardeesy N, Scott FW, Touyz RM, Wheeler MB, Screaton RA. Cell Metab. 2009;10:285. doi: 10.1016/j.cmet.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 8.Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. Proc Natl Acad Sci U S A. 2004;101:3329. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abbott MJ, Edelman AM, Turcotte LP. Am J Physiol Regul Integr Comp Physiol. 2009;297:R1724. doi: 10.1152/ajpregu.00179.2009. [DOI] [PubMed] [Google Scholar]
- 10.Zou MH, Hou XY, Shi CM, Kirkpatick S, Liu F, Goldman MH, Cohen RA. J Biol Chem. 2003;278:34003. doi: 10.1074/jbc.M300215200. [DOI] [PubMed] [Google Scholar]
- 11.Rattan R, Giri S, Singh AK, Singh I. J Biol Chem. 2005;280:39582. doi: 10.1074/jbc.M507443200. [DOI] [PubMed] [Google Scholar]
- 12.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. Mol Cell. 2008;30:214. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petroulakis E, Mamane Y, Le Bacquer O, Shahbazian D, Sonenberg N. Br J Cancer. 2006;94:195. doi: 10.1038/sj.bjc.6602902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. BMJ. 2005;330:1304. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alimova IN, Liu B, Fan Z, Edgerton SM, Dillon T, Lind SE, Thor AD. Cell Cycle. 2009;8:909. doi: 10.4161/cc.8.6.7933. [DOI] [PubMed] [Google Scholar]
- 16.Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Cancer Res. 2006;66:10269. doi: 10.1158/0008-5472.CAN-06-1500. [DOI] [PubMed] [Google Scholar]
- 17.Liu B, Fan Z, Edgerton SM, Deng XS, Alimova IN, Lind SE, Thor AD. Cell Cycle. 2009;8:2031. doi: 10.4161/cc.8.13.8814. [DOI] [PubMed] [Google Scholar]
- 18.Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Cancer Res. 2009;69:7507. doi: 10.1158/0008-5472.CAN-09-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Collins QF, Liu HY, Pi J, Liu Z, Quon MJ, Cao W. J Biol Chem. 2007;282:30143. doi: 10.1074/jbc.M702390200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hwang JT, Ha J, Park IJ, Lee SK, Baik HW, Kim YM, Park OJ. Cancer Lett. 2007;247:115. doi: 10.1016/j.canlet.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 21.Chen Z, Zhu QY, Tsang D, Huang Y. J Agric Food Chem. 2001;49:477. doi: 10.1021/jf000877h. [DOI] [PubMed] [Google Scholar]
- 22.Lu H, Meng X, Yang CS. Drug Metab Dispos. 2003;31:572. doi: 10.1124/dmd.31.5.572. [DOI] [PubMed] [Google Scholar]
- 23.Landis-Piwowar KR, Huo C, Chen D, Milacic V, Shi G, Chan TH, Dou QP. Cancer Res. 2007;67:4303. doi: 10.1158/0008-5472.CAN-06-4699. [DOI] [PubMed] [Google Scholar]
- 24.Smith DM, Daniel KG, Wang Z, Guida WC, Chan TH, Dou QP. Proteins: Structure, Function, and Bioinformatics. 2004;54:58. doi: 10.1002/prot.10504. [DOI] [PubMed] [Google Scholar]
- 25.Huo C, Yang H, Cui QC, Dou QP, Chan TH. Bioorg Med Chem. 2010;18:1252. doi: 10.1016/j.bmc.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kazi A, Wang Z, Kumar N, Falsetti SC, Chan TH, Dou QP. Anticancer Res. 2004;24:943. [PubMed] [Google Scholar]
- 27.Huo C, Yang H, Cui QC, Dou QP, Chan TH. Bioorg Med Chem. 2010;18:1252. doi: 10.1016/j.bmc.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Osanai K, Landis-Piwowar KR, Dou QP, Chan TH. Bioorg Med Chem. 2007;15:5076. doi: 10.1016/j.bmc.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chan TH, Dou QP. 0152210 A1. US patent application. 2011 Jun 23;
- 31.Yang H, Sun DK, Chen D, Cui QC, Gu YY, Jiang T, Chen W, Wan SB, Dou QP. Cancer Lett. 2010;292:48. doi: 10.1016/j.canlet.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen D, Cui QC, Yang H, Dou QP. Cancer Res. 2006;66:10425. doi: 10.1158/0008-5472.CAN-06-2126. [DOI] [PubMed] [Google Scholar]
- 33.Chen D, Landis-Piwowar KR, Chen MS, Dou QP. Breast Cancer Res. 2007;9:R80. doi: 10.1186/bcr1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hwang JT, Park IJ, Shin JI, Lee YK, Lee SK, Baik HW, Ha J, Park OJ. Biochem Biophys Res Commun. 2005;338:694. doi: 10.1016/j.bbrc.2005.09.195. [DOI] [PubMed] [Google Scholar]
- 35.Imamura K, Ogura T, Kishimoto A, Kaminishi M, Esumi H. Biochem Biophys Res Commun. 2001;287:562. doi: 10.1006/bbrc.2001.5627. [DOI] [PubMed] [Google Scholar]
- 36.Eckhoff L, Nielsen M, Moeller S, Knoop A. Acta Oncol. 2011;50:1075. doi: 10.3109/0284186X.2011.602111. [DOI] [PubMed] [Google Scholar]
- 37.Jhanwar YS, Divgi C. J Nucl Med. 2005;46(Suppl 1):141S. [PubMed] [Google Scholar]
- 38.Dou D, Ahmad A, Yang H, Sarkar FH. Nutr Cancer. 2011;63:272. doi: 10.1080/01635581.2011.523497. [DOI] [PubMed] [Google Scholar]
- 39.Kazi A, Daniel KG, Smith DM, Kumar NB, Dou QP. Biochem Pharmacol. 2003;66:965. doi: 10.1016/s0006-2952(03)00414-3. [DOI] [PubMed] [Google Scholar]
- 40.Nam S, Smith DM, Dou QP. J Biol Chem. 2001;276:13322. doi: 10.1074/jbc.M004209200. [DOI] [PubMed] [Google Scholar]
- 41.Harvey AL. Drug Discov Today. 2008;13:894. doi: 10.1016/j.drudis.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 42.Motoshima H, Goldstein BJ, Igata M, Araki E. J Physiol. 2006;574:63. doi: 10.1113/jphysiol.2006.108324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reis T, Edgar BA. Cell. 2004;117:253. doi: 10.1016/s0092-8674(04)00247-8. [DOI] [PubMed] [Google Scholar]
- 44.Grana X, Reddy EP. Oncogene. 1995;11:211. [PubMed] [Google Scholar]
- 45.Ho KK, Myatt SS, Lam EW. Oncogene. 2008;27:2300. doi: 10.1038/onc.2008.23. [DOI] [PubMed] [Google Scholar]
- 46.Huang H, Tindall DJ. Future Oncol. 2006;2:83. doi: 10.2217/14796694.2.1.83. [DOI] [PubMed] [Google Scholar]
- 47.Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. Mol Cell. 2005;18:283. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 48.Bode AM, Dong Z. Nat Rev Cancer. 2004;4:793. doi: 10.1038/nrc1455. [DOI] [PubMed] [Google Scholar]
- 49.Klopp AH, Lacerda L, Gupta A, Debeb BG, Solley T, Li L, Spaeth E, Xu W, Zhang X, Lewis MT, Reuben JM, Krishnamurthy S, Ferrari M, Gaspar R, Buchholz TA, Cristofanilli M, Marini F, Andreeff M, Woodward WA. PLoS One. 2010;5:e12180. doi: 10.1371/journal.pone.0012180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goodison S, Urquidi V, Tarin D. Mol Pathol. 1999;52:189. doi: 10.1136/mp.52.4.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ricardo S, Vieira AF, Gerhard R, Leitao D, Pinto R, Cameselle-Teijeiro JF, Milanezi F, Schmitt F, Paredes J. J Clin Pathol. 2011;64:937. doi: 10.1136/jcp.2011.090456. [DOI] [PubMed] [Google Scholar]
- 52.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Proc Natl Acad Sci U S A. 2003;100:3983. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fillmore C, Kuperwasser C. Breast Cancer Res. 2007;9:303. doi: 10.1186/bcr1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, Wicha MS, Dontu G. Cell Stem Cell. 2007;1:555. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sophos NA, Vasiliou V. Chem Biol Interact. 2003:143–144. 5. doi: 10.1016/s0009-2797(02)00163-1. [DOI] [PubMed] [Google Scholar]
- 56.Hess DA, Wirthlin L, Craft TP, Herrbrich PE, Hohm SA, Lahey R, Eades WC, Creer MH, Nolta JA. Blood. 2006;107:2162. doi: 10.1182/blood-2005-06-2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pearce DJ, Taussig D, Simpson C, Allen K, Rohatiner AZ, Lister TA, Bonnet D. Stem Cells. 2005;23:752. doi: 10.1634/stemcells.2004-0292. [DOI] [PubMed] [Google Scholar]
- 58.Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, Rimm DL, Wong H, Rodriguez A, Herschkowitz JI, Fan C, Zhang X, He X, Pavlick A, Gutierrez MC, Renshaw L, Larionov AA, Faratian D, Hilsenbeck SG, Perou CM, Lewis MT, Rosen JM, Chang JC. Proc Natl Acad Sci U S A. 2009;106:13820. doi: 10.1073/pnas.0905718106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kumar A, Gao H, Xu J, Reuben J, Yu D, Mehta K. PLoS One. 2011;6:e20701. doi: 10.1371/journal.pone.0020701. [DOI] [PMC free article] [PubMed] [Google Scholar]