

Abstract
A Rh-catalyzed 1,3-acyloxy migration of propargyl ester followed by intramolecular [4 + 2] cycloaddition of vinylallene and unactivated alkyne was developed. This tandem reaction provides access to bicyclic compounds containing a highly functionalized isotoluene or cyclohexenone structural motif, while only aromatic compounds were observed in related transition metal-catalyzed cycloadditions.
The Diels–Alder cycloaddition is arguably the most powerful reaction for the construction of substituted six-membered rings from various 2π and 4π reactants.1 However, unactivated 2π and 4π systems either require very harsh conditions or fail to undergo cycloadditions. In many cases, transition metal catalysts facilitated chemical transformations that were inaccessible under thermal conditions and they have proven to be of great value in cycloaddition reactions.2 Vinylallenes are unique 4π-components that have been used in [4 + 2] cycloadditions with activated alkenes and alkynes under thermal conditions.3,4 The cycloaddition of vinylallenes with unactivated alkynes required transition metal catalysts such as platinum5 or rhodium complexes.6 In both cases, polysubstituted benzene derivatives were obtained.
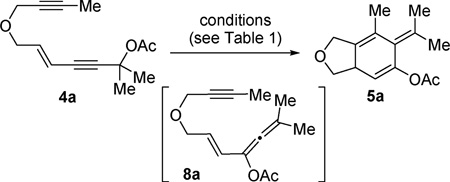
We recently found that [Rh(CO)2Cl]2 could catalyze 1,3-acyloxy migration of propargyl ester 1 to form allene 3,7 which was previously realized mainly by π-acidic metal-based catalysts such as silver, copper, platinum, and gold in various tandem transformations.8,9 We envisioned that the combination of this novel reactivity of Rh(i) catalyst and its ability to promote cycloadditions might allow the tandem transformation from enyne 4 to product 6: a net 1,3-acyloxy migration of propargyl ester 4, a subsequent [4 + 2] cycloaddition of the resulting vinylallene with a tethered alkyne, and a final isomerization of isotoluene 5 to polysubstituted aromatic compound 6.10 In a related report, Liang and co-workers developed a PtCl2-catalyzed tandem 1,3-acyloxy migration followed by [4 + 2] cycloaddition of vinylallene and alkyne to form similar aromatic products.5 The unactivated 2π alkyne partners, however, were limited to terminal alkynes in the PtCl2-catalyzed reaction. We thought that the scope of this tandem process could be expanded to include internal alkynes using rhodium catalyst via a different mechanism. Surprisingly, isotoluene derivative 5 was observed as the major product from rhodium-catalyzed isomerization of enyne 4. A stable bicyclic compound 7 containing a cyclohexenone substructure was isolated upon hydrolysis (Scheme 1).
Scheme 1.

1,3-Acyloxy migration and [4 + 2] cycloaddition of vinylallene and alkyne.
When we treated compound 4a11 with a catalytic amount of [Rh(CO)2Cl]2, an isotoluene derivative 5a was isolated in 88% yield (Table 1, entry 1). The expected aromatic compound was not detected at all. When the reaction time was extended to approximately 1 h, compound 5 was almost completely decomposed into unidentified byproducts. Other Rh(i) catalysts did not promote the tandem acyloxy migration and cycloaddition process (entries 2–4), suggesting that the electron-withdrawing CO ligand was critical. Certain gold and platinum catalysts facilitated the 1,3-acyloxy migration;9 no cycloaddition product, however, was observed in any case (entries 5–10).
Table 1.
Screening of catalysts and conditions for 1,3-acyloxy migration followed by [4 + 2] cycloaddition
 | ||
|---|---|---|
| Entry | Conditions | Results |
| 1 | [Rh(CO)2Cl]2 (5 mol%), DCE, 80 °C, 15 min | 88%a |
| 2 | [Rh(COD)Cl]2 (5 mol%), DCE, 80 °C, 15 min | NR |
| 3 | Rh(PPh3)3Cl (5 mol%), DCE, 80 °C, 15 min | NR |
| 4 | [Rh(COD)]BF4 (5mol%), DCE, 80 °C, 15min | Complex mixture |
| 5 | AuCl (5 mol%), DCE, 80 °C, 15 min | Complex mixture |
| 6 | AuCl3 (5 mol%), DCE, 80 °C, 15 min | Complex mixture |
| 7 | Au(PPh3)Cl (5 mol%), DCE, 80 °C, 15 min | 4a/8a = 2 : 1b |
| 8 | Au(PPh3)Cl (5 mol%), AgOTf (5 mol%), DCE, 80 °C, 10 min | Complex mixture |
| 9 | PtCl2 (5 mol%), DCE, 80 °C, 15 min | 4a/8a = 6 : 1b |
| 10 | PtCl2 (10 mol%), CO (1 atm), DCE, 80 °C, 24 h | Complex mixture |
Isolated yield.
No cycloaddition product was observed. The ratio was determined by 1H NMR of the crude product.
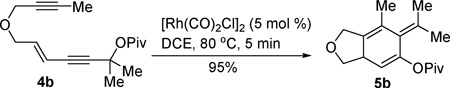
The reaction worked equally well when the acetate was changed to pivolate (eqn (1)). Product 5b could be isolated in high yield when the reaction was run for 5min. Isotoluene derivatives 5a and 5b were stable enough to be isolated and characterized by NMR. We tried to expand the scope of this tandem reaction, yet we found that most of the isotoluene derivatives could not be stored for long time. Many of them were decomposed into unidentified byproducts after a few days of storage.
 |
(1) |
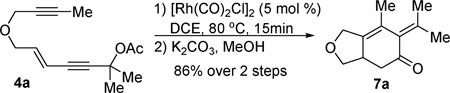
We then decided to convert the isotoluene derivative into a more stable compound. The enol ester in product 5a is essentially a masked ketone. The crude product from the cycloaddition reaction was treated with a basic solution of potassium carbonate in methanol (eqn (2)).11 Cyclohexenone 7a was obtained in 86% yield after two steps.
 |
(2) |
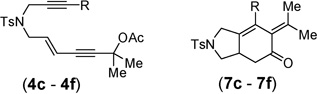
With the above two-step protocol in hand, we then examined the scope of the 1,3-acyloxy migration and [4 + 2] cycloaddition cascade. Terminal and various internal alkynes could participate in the [4 + 2] cycloaddition as 2π components (Table 2, entries 1–4). The X-ray structure of 7f (CCDC 846678) further confirmed our structural assignment.11 A substrate with a methyl substituent on the olefin led to the formation of product 7g with an angular methyl group in the bicyclic system (entry 5). Both 5–6 and 6–6 fused carbocycles could be prepared efficiently (entries 6–8). The gem-dimethyl group of the propargyl ester could be replaced by a cyclohexane ring (entry 9).
Table 2.
Synthesis of cyclohexenone derivatives via Rh-catalyzed 1,3-acyloxy migration, [4 + 2] cycloaddition, and hydrolysisa
| Entry | Substrate | Product | Yield |
|---|---|---|---|
 |
|||
| 1 | R = H, 4c | 7c | 95% |
| 2 | R = Me, 4d | 7d | 95% |
| 3 | R = Ph, 4e | 7e | 92% |
| 4 | R = 4-BrC6H4, 4f | 7f | 87% |
| 5 | 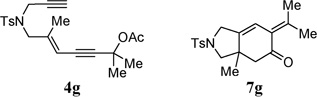 |
90% | |
| 6 | 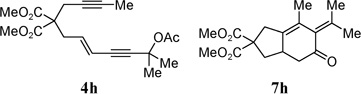 |
86% | |
| 7 | 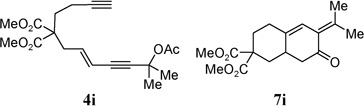 |
95% | |
| 8 | 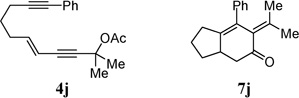 |
91% | |
| 9 | 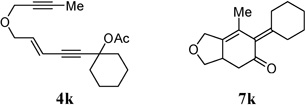 |
86% | |
 |
|||
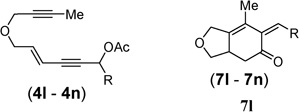
| 10b,d | R = t-Bu, 4l | 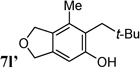 |
(Z/E > 20 : 1)cZ-7l 61% 7l′: 30% |
| 11b | R = i-Pr, 4m | 7m | (Z/E = 3.7 : 1)cZ-7m 58% |
| 12b | R = CH2OTBS, 4n | 7n | (Z/E = 3 : 1)cZ-7n 52% |
Condition A: (1) [Rh(CO)2Cl]2 (5 mol%), DCE, 80 °C, 5–20 min; (2) K2CO3, MeOH.
Condition B: (1) [Rh(CO)2Cl]2 (5 mol%), [(CF3)2CHO]3P (20 mol%), DCE, 80 °C, 15–20 min; (2) K2CO3, MeOH.
The ratio was determined by 1H NMR of the crude product.
The substrate was heated at 80 °C for 45 min under condition B.
A stereoselectivity issue arose for the exo-cyclic olefins when secondary propargyl esters were employed (entries 10–12). Although a high Z/E selectivity was observed for substrate 4l with a tert-butyl group (entry 10), moderate selectivities were obtained for other substrates (entries 11 and 12). Phenol 7l′ derived from hydrolysis after isomerization was isolated in 30% yield. The (Z)-isomers of products 7m and 7n were obtained in over 50%yields. Small amounts of phenol products were also observed in these two cases. In addition, the rate of reaction dropped significantly for all secondary esters. Nonetheless, the addition of phosphite ligand [(CF3)2CHO)]3P improved both the rate and the yield.
To further probe the mechanism, we decided to try the [4 + 2] cycloaddition of independently prepared vinylallene with alkyne. When we treated ester 4b with AgSbF6 catalyst,8 an inseparable mixture of vinylallene 8b and enyne 4b was obtained after 1 h (Scheme 2). Desired product 5b was isolated in 78% yield from the above mixture in the presence of [Rh(CO)2Cl]2 catalyst, suggesting that the acyloxy substituted vinylallene was a plausible intermediate for the tandem reaction. No desired product was observed when this mixture was heated for 2 h in the absence of any catalyst.
Scheme 2.

Preparation of vinylallene and its involvement in [4 + 2] cycloaddition.
We proposed that η4-vinylallene complex 9 could be generated from enyne 4 via a Rh-catalyzed net 1,3-acyloxy migration of propargyl ester (Scheme 3). An oxidative cyclization of intermediate 9 would then yield alkylidene metallacyclopentene 10.12 Insertion of a tethered alkyne into metallacycle 10 followed by reductive elimination of metallacycloheptatriene 11 would then produce product 5 with a isotoluene motif, which might be stabilized by the acyloxy substituent because the isomerized aromatic products were obtained in previous transition metal catalyzed [4 + 2] cycloadditions of vinylallenes and unactivated alkynes.5,6
Scheme 3.

Proposed mechanism for the tandem 1,3-acyloxy migration and subsequent [4 + 2] cycloaddition.
In conclusion, we have developed a Rh-catalyzed tandem 1,3-acyloxy migration and subsequent intramolecular [4 + 2] cycloaddition of vinylallenes and unactivated alkynes. The resulting product with an isotoluene substructure could be isolated and characterized. Upon hydrolysis, more stable cyclohexenones formed. The acyloxy group in the propargyl ester starting material eased the preparation of reactive allene functionality, stabilized the isotoluene moiety, and also differentiated the three olefins in the product for further functionalization. The Rh(i) catalyst in the reaction first served as a π-acid to promote the net 1,3-acyloxy migration of propargyl esters. The same catalyst then underwent oxidative addition, migratory insertion, and reductive elimination to afford the [4 + 2] cycloaddition product. The combination of these properties may provide myriad opportunities for the design of new reactions.
Supplementary Material
Acknowledgments
We thank the Chinese Scholarship Council (to S.H.), NIH (R01 GM088285) and the University of Wisconsin for financial support and a Young Investigator Award (to W.T.) from Amgen.
Footnotes
Electronic supplementary information (ESI) available: 1H NMR, 13C NMR, HRMS, and IR data and copies of NMR spectra for all starting materials and products. CCDC 846678. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2cc17406e
Notes and references
- 1.For selected reviews, see: Danishefsky S. Acc. Chem. Res. 1981;14:400. Winkler JD. Chem. Rev. 1996;96:167. doi: 10.1021/cr950029z. Corey EJ. Angew. Chem., Int. Ed. 2002;41:1650. doi: 10.1002/1521-3773(20020517)41:10<1650::aid-anie1650>3.0.co;2-b. Nicolaou KC, Snyder SA, Montagnon T, Vassilikogiannakis G. Angew. Chem., Int. Ed. 2002;41:1668. doi: 10.1002/1521-3773(20020517)41:10<1668::aid-anie1668>3.0.co;2-z. Takao K, Munakata R, Tadano K. Chem. Rev. 2005;105:4779. doi: 10.1021/cr040632u.
- 2.For selected reviews on transition metal catalyzed cycloadditions, see: Lautens M, Klute W, Tam W. Chem. Rev. 1996;96:49. doi: 10.1021/cr950016l. Fruhauf HW. Chem. Rev. 1997;97:523. doi: 10.1021/cr941164z. Aubert C, Buisine O, Malacria M. Chem. Rev. 2002;102:813. doi: 10.1021/cr980054f. Evans PA. Modern Rhodium-Catalyzed Organic Reactions. Wiley-VCH: Weinheim; 2005. Michelet V, Toullec PY, Genet JP. Angew. Chem., Int. Ed. 2008;47:4268. doi: 10.1002/anie.200701589. Yu Z, Wang Y, Wang Y. Chem.–Asian J. 2010;5:1072. doi: 10.1002/asia.200900712. Inglesby PA, Evans PA. Chem. Soc. Rev. 2010;39:2791. doi: 10.1039/b913110h. Aubert C, Fensterbank L, Garcia P, Malacria M, Simonneau A. Chem. Rev. 2011;111:1954. doi: 10.1021/cr100376w. Inagaki F, Kitagaki S, Mukai C. Synlett. 2011:594.
- 3.For reviews on Diels–Alder cycloaddition of vinylallenes, see: Okamura WH, Curtin ML. Synlett. 1990:1. Murakami M, Matsuda T. In: Modern Allene Chemistry. Krause N, Hashmi ASK, editors. vol. 2. Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim; 2004. p. 727.
- 4.For selected examples on Diels–Alder cycloaddition of vinylallenes, see: Angelov CM, Mondeshka DM, Tancheva TN. J. Chem. Soc., Chem. Commun. 1985:647. Reich HJ, Eisenhart EK. J. Org. Chem. 1984;49:5282. Reich HJ, Eisenhart EK, Olson RE, Kelly MJ. J. Am. Chem. Soc. 1986;108:7791. doi: 10.1021/ja00284a051. Gibbs RA, Okamura WH. J. Am. Chem. Soc. 1988;110:4062. Reich HJ, Eisenhart EK, Whipple WL, Kelly MJ. J. Am. Chem. Soc. 1988;110:6432. Gibbs RA, Bartels K, Lee RWK, Okamura WH. J. Am. Chem. Soc. 1989;111:3717. Robinson JM, Sakai T, Okano K, Kitawaki T, Danheiser RL. J. Am. Chem. Soc. 2010;132:11039. doi: 10.1021/ja1053829. Choi S, Hwang H, Lee PH. Eur. J. Org. Chem. 2011:1351.
- 5.Lu L, Liu X, Shu X, Yang K, Ji K, Liang Y. J. Org. Chem. 2009;74:474. doi: 10.1021/jo802043z. [DOI] [PubMed] [Google Scholar]
- 6.Murakami M, Ubukata M, Itami K, Ito Y. Angew. Chem., Int. Ed. 1998;37:2248. doi: 10.1002/(SICI)1521-3773(19980904)37:16<2248::AID-ANIE2248>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 7.(a) Shu D, Li X, Zhang M, Robichaux PJ, Tang W. Angew. Chem., Int. Ed. 2011;50:1346. doi: 10.1002/anie.201006881. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li X, Zhang M, Shu D, Robichaux PJ, Huang S, Tang W. Angew. Chem., Int. Ed. 2011;50:10421. doi: 10.1002/anie.201104861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For pioneering work on metal-catalyzed 1,3-acyloxy migration of propargyl esters, see: Saucy G, Marbet R, Lindlar H, Isler O. Helv. Chim. Acta. 1959;42:1945.
- 9.For a leading review on π-acidic metal catalyzed 1,3-acyloxy migration of propargyl esters, see: Wang S, Zhang G, Zhang L. Synlett. 2010:692.
- 10.For leading references on the stability and reactivity of isotoluenes, see: Gajewski JJ, Gortva AM. J. Am. Chem. Soc. 1982;104:334. Gajewski JJ, Gortva AM. J. Org. Chem. 1989;54:373.
- 11.See ESI† for details.
- 12.Murakami M, Itami K, Ito Y. Organometallics. 1999;18:1326. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.