

SUMMARY
The c-Fes protein-tyrosine kinase modulates cellular signaling pathways governing differentiation, the innate immune response, and vasculogenesis. Here we report the identification of Type I and II kinase inhibitors with potent activity against c-Fes both in vitro and in cell-based assays. One of the most potent inhibitors is the previously described anaplastic lymphoma kinase inhibitor, TAE684. The crystal structure of TAE684 in complex with the c-Fes SH2-kinase domain showed excellent shape complementarity with the ATP-binding pocket and a key role for the gatekeeper methionine in the inhibitory mechanism. TAE684 and two pyrazolopyrimidines with nanomolar potency against c-Fes in vitro were used to establish a novel role for this kinase in osteoclastogenesis, illustrating the value of these inhibitors as tool compounds to probe the diverse biological functions associated with this unique kinase.
INTRODUCTION
The c-fes/fps proto-oncogene encodes a 93 kDa protein-tyrosine kinase (c-Fes), and together with the homologous kinase Fer, defines a structurally unique kinase family [reviewed in (Greer et al., 2011; Hellwig and Smithgall, 2011)]. Sequences of c-fes and fps were first isolated as part of oncogenic Gag-Fes/Fps chimeras found in several avian and feline retroviruses (Snyder and Theilen, 1969; Wang et al., 1981), leading to subsequent identification of the corresponding mammalian and avian cellular proto-oncogenes (Huang et al., 1985; Roebroek et al., 1985). Human c-fes, which maps to chromosome 15, is expressed in embryonic tissues derived from all three germ layers (Carè et al., 1994). In adults, c-Fes is present in a variety of cell lineages, including myeloid hematopoietic, vascular endothelial, neuronal and epithelial cells (Carè et al., 1994; Delfino et al., 2006; Haigh et al., 1996).
The structural organization of c-Fes is distinct from other nonreceptor tyrosine kinases such as c-Src and c-Abl (Greer et al., 2011; Hellwig and Smithgall, 2011). The unique N-terminal region features a Fes/CIP4 homology (FCH) domain, followed by two-coiled coil motifs, a central Src-homology 2 (SH2) domain and a C-terminal kinase domain. The FCH region and first coiled-coil motif comprise an F-BAR homology domain. Other F-BAR domain proteins have been implicated in the regulation of plasma membrane curvature through phosphoinositide binding and induction of membrane tubulation (Itoh and De Camilli, 2006). A recent study demonstrated the ability of the c-Fes F-BAR domain to bind phospholipids and induce membrane tubulation in vitro, suggesting that phosphoinositides may recruit c-Fes to cellular membranes and contribute to its activation by FcεRI/Lyn complexes in mast cells (McPherson et al., 2009).
c-Fes biological activity is tightly regulated in cells, with the kinase domain adopting a catalytically repressed state (Greer et al., 1988). Downregulation of kinase activity is maintained despite the absence of negative regulatory structural elements, such as an SH3 domain or a negative regulatory tail found in the Src-family of nonreceptor tyrosine kinases (Engen et al., 2008). Cellular c-Fes kinase activity is stimulated by the experimental addition of the amino-terminal myristylation signal from c-Src (Greer et al., 1994), replacement of residues of the c-Fes kinase domain with homologous v-Fps sequences (Kim and Feldman, 2002), introduction of a point mutation predicted to disrupt the structure of the first N-terminal coiled-coil domain (Cheng et al., 2001; Shaffer et al., 2009) or substitution of the SH2 domain with that of v-Src (Rogers et al., 2000). Interestingly, insertional mutagenesis of the v-Fps SH2 domain reduced kinase and transforming functions, providing the first evidence that the SH2 domain is a positive regulator of kinase activity (Sadowski et al., 1986). Subsequent studies indicated that the same is true for c-Fes (Hjermstad et al., 1993).
A recent crystal structure of a truncated form of c-Fes, consisting of the SH2 and kinase domains, revealed the molecular mechanisms behind the positive impact of the SH2 domain on kinase activity (Filippakopoulos et al., 2008). Packing and electrostatic interactions between the SH2 and the kinase domain stabilize an active conformation of the critical αC helix found in the kinase domain N-lobe. Crystallization with a synthetic substrate peptide established that substrate binding to the phosphotyrosine site of the SH2 domain stabilizes an ordered SH2 conformation and primes the kinase for catalysis through proper orientation of the αC helix. These structures suggested a model of coordinated c-Fes activation in which substrate binding to SH2 and subsequent autophosphorylation of the activation loop on Y713 stabilize a catalytically competent kinase domain conformation.
Several lines of evidence suggest a possible role for c-Fes in oncogenesis. Kinase-active mutants of c-Fes drive focus-forming activity and soft-agar colony formation in rodent fibroblast transformation assays (Cheng et al., 2001; Li and Smithgall, 1998). More recently, c-Fes was identified as a phosphorylation target of the constitutively active D816V mutant of c-Kit, a mutation commonly found in human malignancies (Voisset et al., 2007). siRNA targeting of endogenous c-Fes in TF-1 cells expressing c-Kit D816V significantly reduced proliferation and phosphorylation of STATs and p70 S6 kinase. Active c-Fes has been observed in acute myeloid leukemia (AML), and reduction of c-fes expression by RNAi demonstrated a requirement for c-Fes in AML cell survival (Voisset et al., 2010). Downregulation of c-Fes by siRNA treatment was also shown to reduce proliferation of two human renal carcinoma cell lines (Kanda et al., 2009).
Angiogenesis is a common hallmark of tumorigenesis (Hanahan and Weinberg, 2000). A role for c-Fes in angiogenesis was first suggested by the observation that membrane-targeted c-fes expression led to hypervascularization and hemangioma formation in transgenic mice (Greer et al., 1994). Subsequently, c-Fes kinase activity was shown to contribute to FGF-2-induced chemotactic cell migration and tube formation by brain capillary endothelial cells (Kanda et al., 2000). Further studies confirmed that c-Fes is a common mediator of PI3-kinase activation by numerous angiogenic factors, including VEGF-A, Ang1 and Ang2 (Kanda et al., 2007).
Delineating a role for c-Fes in cancer is complicated by observations that c-Fes may also fulfill the role of a tumor suppressor. Large-scale sequencing of the tyrosine kinome in multiple colorectal tumor cell lines identified c-fes as a one of only a small number of consistently mutated genes (Bardelli et al., 2003). Subsequent work showed that none of the reported mutations stimulated c-Fes kinase activity, and several impaired kinase function, consistent with a tumor-suppressor role (Delfino et al., 2006; Sangrar et al., 2005). Expression of c-Fes is readily detected in normal colonic epithelium, but is frequently absent in matched tumor samples as well as in human colorectal cancer cell lines as a result of extensive promoter methylation (Delfino et al., 2006; Shaffer and Smithgall, 2009). In a mouse model of breast cancer, tumor onset was accelerated in homozygous-null c-fes mice, and this effect was rescued by a c-fes transgene (Sangrar et al., 2005). Taken together, these data point to a tumor suppressor function for c-Fes in some epithelial cancers.
Spearheaded by the clinical success of the Bcr-Abl inhibitor imatinib in chronic myelogenous leukemia, kinases have become the focus of major drug discovery efforts as targets for anti-cancer drug therapy (Zhang et al., 2009). As summarized above, mounting evidence points towards a role for c-Fes in human cancer through its involvement in cell proliferation, survival signaling, and angiogenesis, making it an attractive candidate for drug targeting (Kanda and Miyata, 2011). Selective small molecule inhibitors are urgently needed to clarify the roles of c-Fes as dominant oncogene vs. tumor suppressor depending upon the cellular context. Despite the intriguing biology associated with c-Fes, no inhibitors with a useful level of selectivity and cellular activity have been reported to date.
In this study, we report the discovery and characterization of potent c-Fes tyrosine kinase inhibitors with cellular activity. Using a recombinant c-Fes protein consisting of the SH2 and kinase domains, we first screened a kinase-biased small-molecule library using an in vitro kinase assay. ‘Hit’ compounds were then tested for their ability to inhibit c-Fes autophosphorylation and microtubule association in COS-7 cells and for their effect on rodent fibroblast transformation driven by constitutively active c-Fes mutants. Using these screens we identified both Type I and Type II c-Fes kinase inhibitors from diverse chemical classes, including diaminopyrimidines, pyrazolopyrimidines, pyrrolopyridines and pyrazines, with activity against c-Fes both in vitro and in vivo. Type I inhibitors bind to the ATP-binding site with the kinase assuming an ‘active’ conformation defined by the ‘DFG-motif’ of the activation loop adopting an ‘in’ conformation conducive to substrate binding. Type II inhibitors bind to the ‘inactive’ conformation with the ‘DFG-motif’ in an ‘out’ conformation blocking access to the substrate binding site (Liu and Gray, 2006).
Surprisingly, we discovered that TAE684, a compound previously identified as a potent and selective inhibitor of the anaplastic lymphoma kinase [Alk; (Galkin et al., 2007)], is also a potent inhibitor of c-Fes both in vitro and in vivo. We were able to obtain a crystal structure of the c-Fes SH2- kinase region in complex with TAE684 which will guide further modifications to enhance potency and selectivity. Finally, using several of these inhibitors as chemical probes, we were able to define a novel role for endogenous c-Fes activity in osteoclast differentiation from macrophages for the first time. Our findings represent an important first step towards the development of potent and selective inhibitors of c-Fes, which will have utility in the elucidation of the roles of this enigmatic kinase in normal cellular physiology and tumorigenesis.
RESULTS AND DISCUSSION
Identification of c-Fes inhibitors by FRET-based chemical library screening
The Z′-Lyte fluorescence resonance energy transfer (FRET)-based assay platform for high-throughput assessment of kinase activity has been successfully used in a chemical library screen to identify inhibitors of HIV Nef-induced Hck tyrosine kinase activity (Emert-Sedlak et al., 2009; Rodems et al., 2002). Here we used this assay and a recombinant catalytically active fragment of c-Fes, consisting of the SH2 and kinase domains (SH2-KD) for which the crystal structure is known (Filippakopoulos et al., 2008), to screen a kinase-biased library of small molecules. A total of 586 compounds were initially screened for inhibition of SH2-KD at a concentration of 1 μM (Figure S1). We found that 19 compounds inhibited Fes SH2-KD kinase activity by 90% or more, while an additional 13 compounds inhibited kinase activity by 80–90%. Of the inhibitors identified in the primary screen, 21 compounds representing eight diverse chemical scaffolds were chosen for further analysis. The inhibitory activities of these compounds were verified in dose-response experiments and IC50 values were determined by curve fitting (Figure S2). The IC50 values for all 21 compounds were in the sub-micromolar range (Table 1), with the lowest values observed for the pyrazolopyrimidine WZ-4-49-8 (IC50 67 nM) and the diaminopyrimidine TAE684 (IC50 118 nM). In addition to these type I inhibitors, two putative type II inhibitors, HG-7-27-01 (IC50 224 nM) and HG-7-92-01 (IC50 292 nM), were also discovered in this compound set. Structures and concentration-response curves for these four inhibitors are presented in Figure 1. Chemical syntheses and characterization data for these four compounds are presented in the Supplemental Experimental Procedures section.
Table 1. IC50 values of c-Fes inhibitors determined in vitro using a recombinant SH2-kinase protein and the Z′-Lyte assays.
Concentration response curves were generated using the recombinant c-Fes SH2-kinase protein and the Z′-Lyte assay as described under Experimental Procedures. Compounds are organized by chemical scaffold and predicted mode of inhibition (Type vs. Type II). See also Figure S2.
| Compound | Type (predicted) | IC50 (nM) |
|---|---|---|
| Diaminopyrimidines | ||
| TAE684 | I | 118 |
| Triaminopyrimidines | ||
| XMD5-130 | I | 308 |
| XMD6-3 | I | 275 |
| HG-5-91-01 | I | 275 |
| HG-5-145-01 | I | 537 |
| HG-5-150-01 | I | 252 |
| HG-5-56-01 | I | 654 |
| Pyrazolopyrimidines | ||
| WZ-4-49-1 | I | 214 |
| WZ-4-49-8 | I | 67 |
| WZ-4-49-9 | I | 302 |
| WZ-4-49-10 | I | 146 |
| Diaminopurines | ||
| WZ-4-24-1 | I | 168 |
| WZ-4-24-2 | I | 365 |
| WZ-4-24-3 | I | 363 |
| WZ-4-24-7 | I | 149 |
| Thiazolepyridines | ||
| HG-7-127-01 | II | 222 |
| Pyrrolopyridines | ||
| HG-7-92-01 | II | 298 |
| Pyrazines | ||
| HG-7-27-01 | II | 224 |
| Pyrimidine carbamates | ||
| WH-4-199-1 | II | 256 |
| WH-4-199-2 | II | 422 |
| WH-4-124-2 | II | 535 |
Figure 1. Inhibition of recombinant c-Fes SH2-KD using an in vitro kinase assay and FRET-peptide substrate.

The assay was performed as described under Experimental Procedures using the substrate peptide, Tyr2. Concentration-response curves for selected inhibitors of different chemical classes and the structure of these compounds are shown. See also Figure S1.
Kinome-wide selectivity of lead compounds
The kinome-wide selectivity for each of the four lead compounds was assessed using KINOMEscan™ screening technology, a high-throughput method for screening kinase inhibitors against a panel of either 353 kinases (for TAE684 and HG-7-92-01) or 442 kinases (for WZ-4-49-8 and HG-7-27-01). A kinome interaction map was constructed from the resulting data for each compound (Figure S3A). This approach revealed that the diaminopyrimidine TAE684, a type I kinase inhibitor, possessed a broad selectivity profile with a selectivity score of 0.38 (Karaman et al., 2008). In contrast, the pyrazolopyrimidine type I inhibitor WZ-4-49-8, which possesses an ortho-ethoxy group, displayed an exceptional selectivity profile with a selectivity score of only 0.027. The two type II inhibitors HG-7-92-01 and HG-7-27-01 displayed intermediate selectivity profiles (0.18 and 0.16, respectively). Selectivity profiling was intentionally performed at relatively high inhibitor concentrations (10 μM) to identify the full spectrum of possible targets. A complete listing of all of the kinases profiled in the screen and the relative selectivity profiles of each compound are shown in Supplemental Table S1. While this approach provides a broad measure of kinase selectivity, it is important to note that it does not necessarily translate to inhibition of kinase activity in biochemical or cellular assays. For example, TAE684 and WZ-4-49-8 scored as strongly active for Erk2 in the KINOMEscan™ assay (displacement scores of 0.3 and 0.7, respectively, Table S1). However, neither compound showed notable inhibition of Erk2 activity in kinase assays, with IC50 values for Erk2 inhibition at least 100-fold higher than those observed for the inhibition of Fes (Figure S3B,C). Further experiments will be required to determine whether any other kinases identified in this in vitro displacement assay represent true alternative targets in cells, especially at concentrations where c-Fes is inhibited.
Three-dimensional structure of the c-Fes SH2-KD region in complex with TAE684
The c-Fes SH2-kinase protein used in the primary screen was crystallized in complex with TAE684 and the resulting X-ray crystal structure was refined to 1.84 Å (data collection and refinement statistics are listed under Supplemental Table S2). In the crystal structure, the regulatory αC helix assumed an active conformation as indicated by the canonical salt bridge between the conserved αC glutamate (E607) and the active site lysine (K590). However, the activation segment was largely unstructured as expected for unphosphorylated, inactive c-Fes in the absence of SH2 ligands (Filippakopoulos et al., 2008). The inhibitor was very well defined by electron density (Figure 2A) and showed good shape complementarity with the c-Fes ATP binding pocket. The inhibitor pyrimidine and aniline amines formed two hydrogen bonds with the hinge backbone of V639 (Figure 2B). In addition, the 5-chloro substituent of TAE684 packed against the gatekeeper methionine (M636), an interaction that was also observed between the related inhibitor WZ-4002 and the T790M gatekeeper mutant of the EGF receptor tyrosine kinase (Zhou et al., 2009). Binding was further stabilized by hydrophobic interactions with residues A588, L638, V620, L690, V575 and I567 (Figure 2C). The piperazine moiety of TAE684 extended over helix αD and was shielded from the solvent channel by a symmetry related protein molecule.
Figure 2. X-ray crystal structure of the c-Fes SH2-kinase region bound to the diaminopyrimidine inhibitor, TAE684.
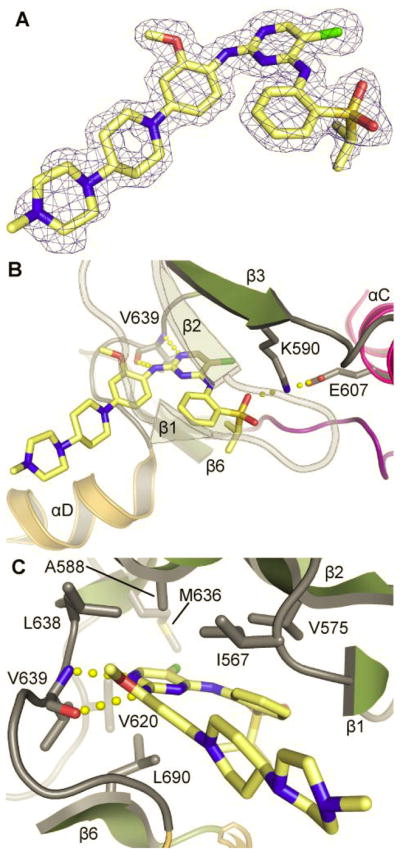
A) 2Fc-Fo map of TAE684 bound to the c-Fes active site. The map was contoured at 2σ. B) Binding mode of TAE864 in the c-Fes co-crystal structure is stabilized by hydrogen bonds with the hinge backbone V639 and the active site K590. C) Detailed view of the TAE684 c-Fes interaction. See also Figure S4.
Superimposition of the c-Fes/TAE684 co-crystal structure with that of Alk (Bossi et al., 2010) revealed an additional polar interaction of TAE684 with E1210 located in helix αD which is not present in c-Fes (Figure S4A,B). However, the electron density for the piperidine-piperazine group was not well defined in the Alk complex suggesting that this moiety is flexible. In c-Fes, the piperidine-piperazine group forms water-mediated hydrogen bonds with residues G641 and G642 located C-terminal to the hinge region (Figure S4C). Additional water-mediated hydrogen bonds were also observed between TAE684 and the active site lysine (K590), the P-loop residues F572 and N571, and D701 (Supplemental Figure S4D).
Analysis of inhibitors in a cell-based assay for c-Fes autophosphorylation and microtubule localization
We next examined whether the inhibitors identified in vitro also displayed activity against full-length c-Fes in a cell-based assay. The N-terminal region of c-Fes, which is not part of the crystal structure, contains two coiled-coil homology domains that have been implicated in the regulation of c-Fes kinase activity in cells (Hellwig and Smithgall, 2011). A leucine-to-proline point mutation in the first coiled-coil domain (L145P), which has been predicted to disrupt the coiled-coil structure, strongly activates c-Fes in vivo and results in fibroblast transformation (Cheng et al., 2001). When a GFP fusion of this active c-Fes mutant is expressed in COS-7 cells, autophosphorylation of the c-Fes kinase domain activation loop on Y713 can be readily detected by immunofluorescence along with redistribution of the protein to the prominent microtubule scaffold present in this cell line (Laurent et al., 2004). Microtubule association results from c-Fes-mediated phosphorylation of tubulin, followed by association via the c-Fes SH2 domain. Association of active c-Fes with microtubules is in striking contrast to the diffuse cytoplasmic distribution of wild-type c-Fes, which is downregulated despite the high-level over-expression achievable in this cell line (Figure 3A).
Figure 3. Inhibitors of c-Fes kinase activity block of c-Fes-L145P autophosphorylation and microtubule localization in vivo.

A) Confocal images of COS-7 cells expressing wild-type GFP-c-Fes (Fes-WT) or the active GFP-c-Fes-L145P mutant (Fes-L145P) stained with anti-Fes pY713 specific antibody (Texas Red, pFes). Wild-type c-Fes shows diffuse cytoplasmic distribution (upper panel) and kinase activity is repressed, as evidenced by lack of pFes immunofluorescence signal (lower panel). In contrast, GFP-c-Fes-L145P is autophosphorylated at Y713 and strongly localizes to microtubules. Representative images of cells expressing GFP-c-Fes-L145P treated with the inhibitors TAE684 and HG-7-27-01 are also shown. B) COS-7 cells expressing GFP-c-Fes-L145P were treated with the kinase inhibitors shown for 20 h and the percentage of GFP-expressing cells that stained positive for c-Fes pY713 was determined. Results with cells expressing wild-type GFP-c-Fes (WT) and GFP-c-Fes-L145P (LP) in the absence of inhibitors are also shown (black bars; left). For each condition, all cells in a minimum of ten 40X fields were scored, and the average of three independent experiments is shown ± SD. Data points that could not be determined due to cytotoxic effects of the compound are indicated.
Using the COS-7 expression system, we tested all 21 lead compounds from the in vitro screen for their ability to inhibit c-Fes autophosphorylation and association with microtubules in vivo. COS-7 cells were transiently transfected with the GFP-Fes-L145P fusion protein, followed by 24-hour incubation with each compound at concentrations of 1, 3 and 10 μM. Treated cells were fixed and immunostained for autophosphorylated c-Fes using a pY713-specific antibody (Takashima et al., 2003). As shown in Figure 3A, treatment with the compound TAE684 resulted in a dramatic loss of GFP-Fes localization from microtubules and concomitant loss of pY713 immunostaining. Additional experiments revealed that loss of autophosphorylation and cellular redistribution of GFP-Fes-L145P can be observed after as little as 1 hour of inhibitor treatment (data not shown). This observation suggests that inhibition of c-Fes kinase activity both reverses and prevents microtubule association.
Nine additional compounds also inhibited c-Fes-L145P autophosphorylation and microtubule association in at least a subset of cells. To quantify the effects of these inhibitors, the percentage of cells showing loss of c-Fes-L145P pY713 immunostaining was determined in three independent experiments, and the results are shown in Figure 3B. The strongest inhibition was observed with TAE684, with ~70% (at 1 μM) to ~90% (at 10 μM) of cells showing loss of c-Fes-L145P activity and microtubule association. These experiments identify TAE684 as a potent inhibitor of active c-Fes in a cellular context. The pyrazolopyrimidines WZ-4-49-1 and WZ-4-49-8 also showed strong effects in this system, with IC50 values in the low micromolar range. In contrast to these compounds, the predicted Type II inhibitor HG-7-27-01 reduced c-Fes-L145P autophosphorylation in only 10–15% of cells when tested at concentrations below the cytotoxicity threshold, despite its apparent potency in vitro. As described in the next section, this difference may be due to a preference of this compound for the downregulated conformation of the kinase domain.
Inhibition of tubulin phosphorylation by c-Fes in vitro
In addition to strong association with microtubules in vivo, purified c-Fes directly phosphorylates tubulin and catalyzes tubulin polymerization in vitro (Laurent et al., 2004). In support of the inhibitor-induced changes in c-Fes-L145P autophosphorylation and microtubule localization observed in COS-7 cells, we next performed immune-complex kinase assays using purified recombinant tubulin as substrate. Flag-tagged wild-type or L145P forms of c-Fes were expressed in COS-7 cells, and immunoprecipitates were incubated with tubulin in the presence of [γ32P]ATP over a range of inhibitor concentrations. For comparative purposes, tubulin phosphorylation assays were also performed with the recombinant SH2-KD form of c-Fes used in the initial inhibitor screen. TAE648 potently inhibited tubulin phosphorylation by both wild-type and L145P c-Fes, with average IC50 values of 15 nM and 30 nM, respectively (Figure 4 and Table 2). Interestingly, the IC50 value for inhibition of wild-type full-length c-Fes is ~3-fold lower than the IC50 for the SH2-KD protein in this assay (15 nM compared to 46 nM), suggesting that TAE684 may have increased affinity for full-length c-Fes.
Figure 4. Inhibition of tubulin phosphorylation by c-Fes, c-Fes-L145P and c-Fes SH2-KD in vitro.

A) c-Fes-Flag and c-Fes-L145P-Flag were expressed in 293T cells, and immunoprecipitates were subjected to in vitro kinase assays with [γ32P]ATP and purified tubulin as substrate in the presence of the inhibitors TAE684 or HG-7-27-01 as indicated. Similar assays were conducted using the recombinant purified c-Fes SH2-kinase domain protein (SH2-KD). Reaction products were separated by SDS-PAGE and transferred to PVDF membranes, followed by autoradiography (pFes, pTubulin). Equivalent levels of c-Fes protein in each assay were verified by immunoblotting (Fes) and tubulin substrate levels were confirmed by Coomassie staining (Tubulin). B) Concentration-response curves for TAE684 (left) and HG-7-27-01 (right) as quantified from at least three tubulin phosphorylation assays performed for each kinase. Average percent inhibition ± SEM for each inhibitor concentration is shown.
Table 2. IC50 values for TAE684 and HG-7-27-01 inhibition of tubulin phosphorylation by recombinant SH2-kinase vs. full-length c-Fes-Flag or c-Fes-L145P-Flag.
IC50 values as well as 95% confidence intervals (CI) from three independent determinations are shown. The concentration-response curves used to generate these values are shown in Figure 4B.
| TAE684 | HG-7-27-01 | |||
|---|---|---|---|---|
| Kinase | IC50 (nM) | 95% CI (nM) | IC50 (nM) | 95% CI (nM) |
| SH2-KD | 45.8 | 41.3 – 50.7 | 494.9 | 385.4 – 636.5 |
| Fes-Flag | 15.0 | 10.1 – 22.1 | 1297 | 517.2 – 3252 |
| Fes-L145P-Flag | 29.7 | 11.6 – 76.0 | 5243 | 3068 – 8961 |
For HG-7-27-01, which displayed only weak inhibition of c-Fes-L145P autophosphorylation in COS-7 cells (Figure 3), inhibition of tubulin phosphorylation by c-Fes-L145P-Flag was also weak in vitro, with an IC50 value of 5.2 μM (Figure 4 and Table 2). More potent inhibition was observed for wild-type c-Fes-Flag and SH2-KD, with average IC50 values of 1.3 and 0.5 μM, respectively. Inhibitor binding in the Type II mode typically requires the kinase domain to be in an inactive conformation, with the DFG-motif rotated to an outward orientation that allows for access to a hydrophobic pocket adjacent to the ATP-binding site (Liu and Gray, 2006). The observed differences in IC50 values for inhibition of wild-type vs. the L145P and truncated forms of c-Fes by HG-7-27-01 suggest a bias of this compound towards the inactive conformation of the kinase as expected for a type II inhibitor. In addition, these results provide indirect evidence that the unique Fes N-terminal region may influence the conformation of the inhibitor binding site in the kinase domain. Potent inhibition of tubulin phosphorylation by these c-Fes kinases was also observed for WZ-4-49-8 and HG-7-92-01 with IC50 values against wild-type c-Fes-Flag of 127 nM and 306 nM, respectively (Figure S3B,C and data not shown). No significant differences in potency against wild-type c-Fes over the L145P mutant were observed in this assay for these compounds. These results are consistent with the inhibition of c-Fes autophosphorylation and microtubule association in COS cells by these compounds (Figure 3).
c-Fes inhibitors selectively inhibit rodent fibroblasts transformed by active c-Fes mutants but not by the Src-family kinase, Hck
We next investigated whether the compounds that potently inhibited c-Fes activity in vitro and in the COS cell system could also suppress oncogenic transformation of cells by active forms of c-Fes. In previous work, we established that Rat-2 fibroblasts undergo rapid transformation upon stable expression of constitutively active mutants of c-Fes, including the N-terminal coiled-coil domain mutant L145P used in the COS cell assay (Cheng et al., 2001). Using a soft-agar colony-forming assay for anchorage-independent growth, we first tested the ability of TAE684 to inhibit Rat-2 transformation by two different active forms of c-Fes. Rat-2 cells expressing GFP fusions of c-Fes-L145P or an active c-Fes/v-Fps chimera (Kim et al., 2004) were grown in soft-agar in the presence of various inhibitor concentrations and the number of transformed colonies were counted two weeks later. As shown in Figure 5, TAE684 potently inhibited soft-agar colony formation by Rat-2 cells expressing either of the transforming variants of c-Fes by more than 50% at 100 nM. Growth of control Rat-2 cells in monolayer culture was only slightly reduced at this concentration of TAE684, strongly supporting a direct effect of the inhibitor on c-Fes-mediated transformation (Figure S5). Complete inhibition of c-Fes-mediated soft-agar colony formation was observed with TAE684 at 400 nM (c-Fes/v-Fps) and 600 nM (c-Fes-L145P).
Figure 5. Inhibition of soft-agar colony formation by rodent fibroblasts transfected with transforming variants of c-Fes.

Rat-2 fibroblasts were transformed with a constitutively active c-Fes-L145P coiled-coil mutant, a c-Fes/v-Fps chimeric kinase, a tail-activated mutant of the Src-family kinase Hck (Hck-YF) or the active Hck mutant plus a T338M gatekeeper mutation (Hck-TMYF). Fibroblast transformation was determined in a soft-agar colony formation assay in the presence of 0.1% DMSO (control) and increasing concentrations of TAE684 as shown. A) Scanned images of representative plates showing stained soft-agar colonies. B) The number of transformed colonies was determined from the scanned images using Quantity One® software (BioRad). Soft-agar colony growth is expressed as the mean percent of colonies observed relative to the untreated control in triplicate assays ± SD. C) Transformed Rat-2 cells were grown in monolayer culture and treated with the indicated concentrations of TAE684 for 16 h. Cell lysates were prepared and autophosphorylation of the transforming kinases were assessed by immunoblotting with phosphopspecific antibodies. D) The data shown in C were quantified and IC50 values were determined by curve fitting. See also Figure S5.
To rule out a role for Src-family kinases in the inhibitory effect of TAE684 on Rat-2 fibroblast transformation, we also performed soft-agar colony assays using cells transformed by an active mutant of the Src-family kinase, Hck (Briggs and Smithgall, 1999; Lerner and Smithgall, 2002). In contrast to the strong inhibition of c-Fes-mediated transformation by TAE684, only minimal inhibition of Hck-mediated soft-agar colony formation was observed with 200 nM TAE684 (Figure 5). Complete loss of soft-agar colony growth was observed at 1 μM TAE684 with both the c-Fes and Hck transformants. However, growth of control Rat-2 cells expressing GFP alone was reduced by 50% at 1 μM TAE684 after 72 hrs and completely abolished with a concentration of 3 μM of this compound. Together, these results indicate that the effects of TAE684 on Hck-transformed fibroblasts are largely due to non-specific suppression of cell growth as concentrations approach 1 μM or higher.
One important structural difference between the c-Fes and c-Src kinase families is the identity of the amino acid that occupies the “gatekeeper” position adjacent to the ATP binding site in the kinase domain. This residue impacts the access of some small molecule inhibitors to a hydrophobic cavity that is an important determinant of inhibitor specificity. In Hck and all other members of the Src-kinase family, threonine occupies the gatekeeper position, while a bulkier methionine residue is present in this position in c-Fes. In the X-ray crystal structure of the c-Fes kinase domain, the gatekeeper methionine (M636) comes in very close contact with the chloro group of TAE684 (Figure 2). To evaluate whether this key inhibitor specificity determinant impacts the sensitivity of Hck to TAE684, we created an active form of Hck in which the gatekeeper threonine was replaced by methionine. Remarkably, this single amino acid substitution dramatically enhanced the sensitivity of Hck-transformed fibroblasts to inhibition by TAE684 in a manner very similar to cells transformed with the c-Fes mutants (Figure 5). These results suggest that the gatekeeper residue is a critical specificity determinant for TAE684.
To correlate inhibition of transforming activity with effects on kinase activity, we next treated monolayer cultures of transformed Rat-2 cells with a range of TAE684 concentrations and assayed the autophosphorylation status of each kinase by immunoblotting the cell lysates with phosphospecific antibodies against the activation loop phosphotyrosine residues (Figure 5C,D). In agreement with the selectivity of TAE684 observed in the soft agar colony assays, both c-Fes and the Hck-TMYF gatekeeper mutant were sensitive to TAE684 treatment (IC50 values of 88 nM and 685 nM, respectively), while Hck-YF autophosphorylation remained largely unaffected (IC50 > 10 μM).
Rat-2 fibroblast transformation assays were also performed with WZ-4-49-8 as well as HG-7-92-01 (Figure S5), both of which demonstrated strong inhibition of c-Fes in vitro. WZ-4-49-8 potently inhibited c-Fes-L145P-mediated soft-agar colony growth and showed remarkable selectivity for c-Fes-L145P over several active SFKs tested. This pyrazolopyrimidine inhibited c-Fes-L145P-mediated soft-agar colony formation by more than 80% at 100 nM, while cells transformed with active Hck showed no reduction in soft-agar growth at this concentration. Interestingly, Rat-2 cells transformed by either of two active SFK mutants carrying a threonine to methionine gatekeeper mutation (Hck-TMYF and Yes-TMYF) were inhibited to 61% and 25% of control values at 300 nM, respectively. These results support an important role for the gatekeeper residue in determining inhibitor specificity as described above for TAE684. HG-7-92-01 was much less potent than the other two compounds in the transformation assay, producing only 50% inhibition of c-Fes-L145P-mediated colony forming activity at 1 μM. A similar degree of inhibition was also observed for fibroblasts transformed with activated Hck as well as the Hck-TM mutant, indicating a relative lack of specificity for c-Fes in this assay.
c-Fes Inhibitors Reveal a New Role for c-Fes Kinase Activity in Differentiation of Macrophages to Osteoclasts
Having identified a group of structurally distinct inhibitors for c-Fes kinase activity, we next investigated the utility of these compounds as chemical probes for endogenous c-Fes function. For these experiments we turned to macrophages, one of the cell lineages where c-Fes is most strongly expressed. Macrophages in turn give rise to osteoclasts, the multinucleated cells critical for resorption of bone (Boyle et al., 2003). This process is regulated in part by the coordinated action of macrophage colony-stimulating factor (M-CSF) and RANK ligand (RANKL), where M-CSF promotes macrophage proliferation and survival while RANKL induces differentiation. Given the strong expression of c-Fes in macrophages and its role myeloid differentiation (Craig, 2012), we were interested to see if c-Fes may contribute to induction of the osteoclast phenotype. To test this hypothesis, we employed both bone-marrow derived macrophages (BMM) as well as the RAW 264.7 macrophage cell line. As shown in Figure 6, treatment of BMM with M-CSF and RANKL induced strong induction of osteoclast differentiation, assessed as the formation of multi-nucleated cells that stained positive for tartrate-resistant acid phosphatase (TRAP). Remarkably, the c-Fes inhibitors TAE684 and WZ-4-49-8 (as well as the WZ-4-49-8 analog WZ-4-49-1) dramatically inhibited differentiation in this system, with IC50 values in the submicromolar range. Very similar results were observed using the same inhibitors in RAW 264.7 cells, where osteoclast differentiation was driven by RANKL in combination with vascular endothelial growth factor instead of M-CSF (Figure 6B). Importantly, none of the c-Fes inhibitors produced toxic effects on primary macrophages or RAW 264.7 cells when cultured in the absence of the differentiation inducers for eight days (data not shown).
Figure 6. c-Fes inhibitors suppress osteoclast differentiation from bone marrow-derived macrophages (BMM) and RAW 264.7 cells and reduce RANKL-induced upregulation of the osteoclast marker Cathepsin K.

Mouse BMM (A) and RAW 264.7 mouse macrophages (B) were cultured with 100 ng/ml RANKL and M-CSF (BMM) or VEGF-A (RAW 264.7 cells) to induce osteoclast formation in the presence of the indicated c-Fes inhibitors at 0.5 and 2.0 μM. Control cultures without inhibitors were supplemented with the DMSO carrier solvent (0.1%). Cells were fixed and stained for tartrate-resistant acid phosphatase (TRAP) activity. Representative fields of DMSO and inhibitor-treated cells are shown on the left. TRAP-positive multinuclear cells were counted from three independent cultures, and the mean values are plotted on the right ± SD. Three independent experiments produced comparable results. All inhibitor-treated cultures exhibited significantly fewer differentiated cells relative to the DMSO-treated control (p < 0.05 in each case). In additional experiments, RAW264.7 cells transfected with c-Fes specific siRNAs were grown in the presence of VEGF-A and RANKL for 6 days (C, left panel) or in the absence of cytokines for 24 h (C, right panel). RNA was isolated and levels of the osteoclast marker Cathepsin K were analyzed by real-time quantitative RT-PCR. Mean expression values ± SEM of triplicate samples are plotted relative to control cells. Quantitative RT-PCR was used to measure Cathepsin K mRNA expression in cell populations stimulated for 6 days with RANKL in the presence of DMSO or the indicated concentrations of c-Fes inhibitors (Mean ± SEM, Figure 6D). See also Figure S6.
As an independent validation of a role for c-Fes in osteoclast differentiation indicated by the inhibitors, we targeted endogenous c-Fes by transient siRNA transfection of RAW 264.7 cells (Figure S6A–C). Transfection with three murine c-Fes-specific siRNAs reduced c-Fes mRNA and protein expression by ~50% (Figure S6A,B), a level comparable to published targeting efficiencies by transient siRNAs in RAW264.7 cells (Wang and Grainger, 2010). Targeting of c-Fes had no effect on the mRNA levels of the closely related Fer kinase (Figure S6A). Transfection with c-Fes specific siRNAs suppressed RANKL-driven formation of TRAP-positive polykaryons when compared to mock-transfected cells and cells transfected with nonspecific control siRNA (Figure S6C). In addition, mRNA analyses of siRNA transfected cells by quantitative real-time RT-PCR revealed that knockdown of c-Fes reduced the expression of the osteoclast marker Cathepsin K in the cytokine-stimulated cell population, while basal expression levels of Cathepsin K in unstimulated RAW 264.7 remained unchanged (Figure 6C). This reduction in RANKL-stimulated upregulation of Cathepsin K mRNA levels is reproduced by treatment with c-Fes inhibitor compounds at submicromolar concentrations (Figure 6D). Taken together, these results suggest that c-Fes activity is required for the differentiation of osteoclasts from macrophages, and identify c-Fes as a possible therapeutic target for the treatment of bone loss associated with cancer metastasis and aging (Roodman, 2004).
Because the kinome-selectivity profile identified Erk kinases as possible alternative targets for TAE684 and WZ-4-49-8, we performed a control osteoclast differentiation assay from RAW 264.7 macrophages in the presence of the Erk inhibitor FR180204 (Figure S6C,D). Treatment with 1 μM or 10 μM FR180204 did not influence osteoclast differentiation, ruling out the Erk pathway as a target for TAE684 and WZ-4-49-8. These results are consistent with the very low potency of TAE684 and WZ-4-49-8 towards Erk in an in vitro kinase assay (Figure S3).
SIGNIFICANCE
In this report, we describe the first inhibitor discovery campaign directed at the c-Fes protein- tyrosine kinase. Identified in the context of several transforming retroviruses almost thirty years ago, c-Fes kinase activity has been implicated in diverse physiological processes, including innate immune receptor signaling, myeloid differentiation, and vasculogenesis. c-Fes has also been implicated in tumorigenesis, where it may act as a dominant oncogene or tumor suppressor depending upon the cellular context. Despite the importance of c-Fes to normal cellular function and disease, no useful inhibitors of c-Fes kinase activity have been described. Through a combination of in vitro and cell-based focused library screens, we identified eight distinct chemotypes with potent activity against c-Fes. One of the most potent compounds is TAE684, a diaminopyrimidine previously developed as an inhibitor of the Alk kinase associated with anaplastic lymphoma. The X-ray crystal structure of TAE684 bound to the c-Fes SH2-kinase region reveals the basis of the observed specificity, including a role for the gatekeeper position in the c-Fes kinase domain. The importance of the c-Fes gatekeeper methionine for TAE684 binding was confirmed by replacement of the threonine residue found in this position in several members of the Src kinase family. Substitution of Src family kinase gatekeeper residues with methionine dramatically enhanced their sensitivity to inhibition by TAE684 in a cell-based assay. In addition to TAE684, the novel pyrazolopyrimidines WZ-4-49-1 and WZ-4-49-8 also showed remarkable potency and selectivity for c-Fes. Using these three inhibitors as chemical probes, we discovered a novel role for endogenous c-Fes function in osteoclast formation from cultured macrophages, a physiological site of c-Fes expression. This observation identifies c-Fes as a possible therapeutic target in osteoporosis as well as osteolytic bone metastasis often associated with advanced cancers. In summary, our study provides a strong rationale for future development of c-Fes inhibitors with enhanced potency and specificity. Such compounds will represent important tools for elucidating the complex roles of c-Fes in innate immunity, differentiation, and cancer etiology.
EXPERIMENTAL PROCEDURES
Chemical Library Screen
The chemical library screen for c-Fes inhibitors was performed using the Z′-Lyte kinase assay system (Invitrogen/Life Technologies). This FRET-based assay utilizes a synthetic substrate peptide (Tyr2) labeled with the fluorophores coumarin and fluorescein on opposite peptide termini (Rodems et al., 2002). In a two-step reaction process, the substrate peptide is first incubated with the kinase to allow for phosphorylation of a single tyrosine residue. In the second step, site-specific proteolytic cleavage of nonphosphorylated but not of phosphorylated peptide occurs. The coumarin and fluorescein fluorophores constitute a FRET-pair and peptide cleavage results in loss of the FRET signal. Kinase activity is monitored by measuring the emission ratio after excitation of the donor fluorophore:
High kinase activity results in a low emission ratio, while inhibition of kinase activity results in a high emission ratio.
Reactions were carried out in a 384-well plate format in a volume of 10 μL according to the manufacturer’s protocols. Initial titration of kinase input verified linear reaction conditions for 25 ng of recombinant c-Fes SH2-KD with a 1 h reaction time at room temperature in the presence of 50 μM ATP. The chemical library screen was performed at 1 μM final compound concentration. IC50 values of “hit” compounds, defined as those inhibiting Fes activity by at least 80% in the primary screen, were determined using 10-point serial dilutions of compounds ranging from 0.5 nM to 10 μM. Results were graphed and IC50 values determined by curve fitting using Prism (Graph Pad).
Tubulin Phosphorylation Assay
COS-7 cells (5 × 105) were seeded onto 60 mm culture dishes and grown at 37 °C for 24 h. The cells were transfected with pCDNA3.1 vectors carrying c-Fes-Flag or c-Fes-L145P-Flag and grown for an additional 20 h. Cell extracts were prepared by sonication in 600 μL of Fes lysis buffer [50 mM Tris-HCl, pH 7.4, 1 mM EDTA, 50 mM NaCl, 1 mM MgCl2, 0.1% Triton X-100, protease inhibitor cocktail (Calbiochem), 2 mM Na3VO4, 20 mM NaF]. Kinases were immunoprecipitated using 2 μg of anti-Flag antibody (sc-807, Santa Cruz). The immunoprecipitates were washed twice in RIPA buffer, twice in kinase assay buffer (50 mM Hepes, pH 7.4, 10 mM MgCl2) and resuspended in kinase assay buffer to give a final volume of 100 μL. For reactions using recombinant c-Fes SH2-KD, 25 ng of kinase were diluted in 10 μL kinase assay buffer.
For kinase reactions, 10 μL of each immunoprecipitate was incubated with 2 μg of bovine tubulin (Cytoskeleton), 10 μCi of [γ32P]ATP, and inhibitor in kinase assay buffer in a total volume of 20 μL. The final DMSO concentration in all reactions was 1%. Following incubation for 10 min at 30 °C, reactions were quenched by addition of SDS-PAGE sample buffer and incubation at 95 °C for 5 min. The reaction products were analyzed by SDS gel electrophoresis followed by transfer to PVDF membranes and autoradiography. Consistent input of Fes and tubulin was verified by western blotting and Coomassie staining of the membrane, respectively. Relative tubulin phosphorylation was quantified by image analysis of the autoradiographs using ImageJ. IC50 values were determined by plotting phosphotubulin levels relative to the DMSO control against the inhibitor concentration followed by curve fitting using Prism.
Cell-based Assay for Microtubule Association
Association of active c-Fes with microtubules can be readily monitored by expression of GFP-fusions of active c-Fes mutants in transfected COS-7 cells (Laurent et al., 2004). To determine the effect of inhibitor compounds on c-Fes autophosphorylation and subcellular localization, COS-7 cells were grown in 48-well plates. Inhibitors were added 24 h later in 0.2% DMSO. Immediately following addition of the inhibitors, the cells were transfected with expression plasmids encoding GFP-Fes-L145P or the wild-type GFP-Fes control using Fugene 6 transfection reagent (Roche). Cells were fixed and immunostained 24 h later with a pY713-specific antibody (Takashima et al., 2003) and a secondary antibody conjugated to Texas Red (Southern Biotech). Fluorescence and subcellular localization were evaluated using a Nikon TE300 inverted microscope equipped with a SPOT CCD high-resolution digital camera. For the images in Figure 3A, COS-7 cells were grown, treated with inhibitors and subsequently fixed and immunostained on glass cover slips. Cover slips were mounted on slides and images were taken using an Olympus Fluoview FV1000 confocal microscope. Image acquisition settings were unchanged between treated and control slides.
Rat-2 Fibroblast Transformation Assay
The construction of GFP-fusions of c-Fes-L145P and the c-Fes/v-Fps chimera in the retroviral expression vector pSRαMSVtkneo, the production of recombinant retroviruses and the retroviral infection of Rat-2 fibroblasts are described in detail elsewhere (Cheng et al., 2001). Stably transfected Rat-2 cells were maintained in DMEM supplemented with 10% FBS and 400 μg/ml G418. For the soft agar colony formation assays, 35 mm culture dishes were prepared with bottom layers of 1 mL of 0.5% Seaplaque agarose in growth medium. This cell-free layer also contained test compounds at twice the final assay concentration and 0.2% DMSO. Bottom layers were solidified at 4 °C. Retrovirally transduced Rat-2 cells were trypsinized to a single-cell suspension and resuspended at a concentration of 10,000 cells/ml in medium containing 0.33% Seaplaque agarose. Each cell suspension (1 ml) was layered onto the drug-containing bottom layer and allowed to solidify at 4 °C, after which the plates were incubated at 37 °C for 13 d. Soft agar colonies were stained for 1 h at 37 °C with thiazolyl blue tetrazolium bromide (MTT) in growth medium (1 mg/ml; 1 ml/plate). Colony counts were obtained from scanned images of the plates with QuantityOne software (BioRad). A sensitivity setting of 5 and averaging setting of 5 was applied to all counts. For the immunoblot analyses in Figure 5C and D, cells were seeded in 12-well plates at 50,000 cells/well and grown for 48 h. Inhibitors were added at the indicated concentrations and cells were cultured for an additional 16 h. Extracts were prepared using RIPA buffer containing 1X protease inhibitors (Calbiochem), 1 mM Na3VO4 and 1 mM NaF. Anti-Fes pY713, anti-Src pY418 (Invitrogen), anti-Hck sc-72 and anti-Fes sc-7670 (Santa Cruz) were used for immunodetection. IC50 values were determined using Prism.
Isolation and culture of bone marrow monocytes/macrophages (BMM)
Six-week-old male ddY mice were purchased from Kyudo Co. Ltd., Tosu, Japan. All animal work was performed at the Nagasaki University Graduate School of Biomedical Sciences with Institutional Animal Care and Use Committee approval in accordance with institutional guidelines. Hematopoietic cells were isolated by the perfusion of femurs and tibias with α-MEM (Sigma-Aldrich Japan) containing 10% FBS (Invitrogen) and mononuclear cells were enriched by Histopaque-1077 density gradient centrifugation (Sigma-Aldrich Japan, Tokyo, Japan). Mononuclear cells were cultured in α-MEM containing 10% FBS overnight, and nonadherent cells were collected by centrifugation and seeded into 6 cm dishes in α-MEM containing 10% FBS and 20 ng/ml recombinant human M-CSF (R&D Systems). Seven days later, adherent cells (BMM) were harvested with trypsin and seeded into 48-well plates for osteoclast differentiation in growth medium containing 20 ng/ml M-CSF at a density of 2 × 104 cells/cm2.
Osteoclast differentiation assay
BMMs were cultured in the presence of recombinant human soluble RANK ligand (RANKL; PeproTech, Rocky Hill, NJ) and M-CSF for eight days. RAW 264.7, obtained from the American Type Culture Collection, were cultured for 6 days in α-MEM containing 10% FBS, 100 ng/mL VEGF-A (PeproTech) and 100 ng/mL RANKL. Cultures were fixed with 10% buffered formalin, followed by an ethanol:acetone (1:1) wash. Tartrate-resistant acid phosphatase (TRAP) positive cells were visualized using the TRAP staining kit (Sigma-Aldrich) and the number of TRAP-positive multinucleated cells (at least 3 nuclei per cell) was counted under the microscope. For each experiment, cells in three separate wells were counted and the mean osteoclast number/well ± SD was calculated. Statistical analysis was performed using Mann-Whitney’s U-test, and differences between samples were considered significant when the P-value was < 0.05.
Chemical Synthesis
Synthetic routes to compounds TAE684, WZ-4-49-8, HG-7-92-01, and HG-7-27-01 are provided in the Supplemental Experimental Procedures.
X-ray Crystallography
The recombinant c-Fes SH2-kinase domain protein was expressed in E. coli and purified as described previously (Filippakopoulos et al., 2008). Crystallization was conducted using the sitting drop vapor diffusion method at 4 °C. Aliquots of the purified proteins were dispensed for crystallization using a Mosquito crystallization robot (TTP Labtech, Royston UK). Coarse screens were typically set up on Greiner 3-well plates using three different drop ratios of precipitant to protein per condition (2:1, 1:1 and 1:2; 150 nl final volume). c-Fes crystals with TAE684 (1 mM final concentration) were grown by mixing 100 nl of the protein (6.0 mg/ml) with a 50 nl of reservoir solution containing 0.1 M SPG pH 8.0 and 30% PEG 1000. Crystals were flash frozen in liquid nitrogen. X-Ray data were collected in-house on an FR-E Superbright source using an RAXIS IV plate detector at 1.542 Å. Indexing and integration were carried out using MOSFLM (Leslie and Powell, 2007) and scaling was performed with SCALA (Evans, 2007). Initial phases were calculated by molecular replacement with PHASER (McCoy et al., 2005) using the known model of c-Fes SH2-kinase (PDB: 3BKB) (Filippakopoulos et al., 2008). Initial models were built by ARP/wARP (Perrakis et al., 1999) and building was completed manually with COOT (Emsley and Cowtan, 2004). Refinement was carried out in REFMAC5 (Murshudov et al., 1997). Thermal motions were analyzed using TLSMD (Painter and Merritt, 2006) and hydrogen atoms were included in late refinement cycles. Crystallographic data and refinement statistics can be found in Supplemental Table S2. The crystal coordinates and structure factor file will be deposited in the RCSB Protein Data Bank prior to publication.
Supplementary Material
Highlights.
Discovery of eight chemotypes with potent inhibitory activity against the c-Fes tyrosine kinase using in vitro and cell-based assay systems
Unexpected identification of c-Fes as a target for the anaplastic lymphoma kinase inhibitor, TAE684
Solution of a c-Fes SH2-kinase crystal structure in complex with TAE684
Use of TAE684 and other inhibitors reveals a novel role for endogenous c-Fes in osteoclast formation from macrophages
Acknowledgments
This work was supported by National Institutes of Health Grants CA123756 (to T.S.) and GM079575 and CA130876 (to N.S.G). The Structural Genomics Consortium is a registered charity (number 1097737) that receives funds from the Canadian Institutes for Health Research, the Canadian Foundation for Innovation, Genome Canada through the Ontario Genomics Institute, GlaxoSmithKline, the Karolinska Institute, the Knut and Alice Wallenberg Foundation, the Ontario Innovation Trust, the Ontario Ministry for Research and Innovation, Merck & Co., Inc., the Novartis Research Foundation, the Swedish Agency for Innovation Systems, the Swedish Foundation for Strategic Research and the Wellcome Trust. We thank Dr. Ritsuko Masuyama, Nagasaki University Graduate School of Biomedical Sciences, for expert assistance with the isolation and culture of mouse bone marrow macrophages.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bardelli A, Parsons DW, Silliman N, Ptak J, Szabo S, Saha S, Markowitz S, Willson JK, Parmigiani G, Kinzler KW, et al. Mutational analysis of the tyrosine kinome in colorectal cancers. Science. 2003;300:949. doi: 10.1126/science.1082596. [DOI] [PubMed] [Google Scholar]
- Bossi RT, Saccardo MB, Ardini E, Menichincheri M, Rusconi L, Magnaghi P, Orsini P, Avanzi N, Borgia AL, Nesi M, et al. Crystal structures of anaplastic lymphoma kinase in complex with ATP competitive inhibitors. Biochemistry. 2010;49:6813–6825. doi: 10.1021/bi1005514. [DOI] [PubMed] [Google Scholar]
- Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337–342. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- Briggs SD, Smithgall TE. SH2-kinase linker mutations release Hck tyrosine kinase and transforming activities in Rat-2 fibroblasts. J Biol Chem. 1999;274:26579–26583. doi: 10.1074/jbc.274.37.26579. [DOI] [PubMed] [Google Scholar]
- Carè A, Mattia G, Montesoro E, Parolini I, Russo G, Colombo MP, Peschle C. c-fes expression in ontogenetic development and hematopoietic differentiation. Oncogene. 1994;9:739–747. [PubMed] [Google Scholar]
- Cheng HY, Schiavone AP, Smithgall TE. A point mutation in the N-terminal coiled-coil domain releases c-Fes tyrosine kinase activity and survival signaling in myeloid leukemia cells. Mol Cell Biol. 2001;21:6170–6180. doi: 10.1128/MCB.21.18.6170-6180.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig AW. FES/FER kinase signaling in hematopoietic cells and leukemias. Front Biosci. 2012;17:861–875. doi: 10.2741/3961. [DOI] [PubMed] [Google Scholar]
- Delfino FJ, Stevenson H, Smithgall TE. A growth-suppressive function for the c-fes protein-tyrosine kinase in colorectal cancer. J Biol Chem. 2006;281:8829–8835. doi: 10.1074/jbc.M507331200. [DOI] [PubMed] [Google Scholar]
- Emert-Sedlak L, Kodama T, Lerner EC, Dai W, Foster C, Day BW, Lazo JS, Smithgall TE. Chemical library screens targeting an HIV-1 accessory factor/host cell kinase complex identify novel antiretroviral compounds. ACS Chem Biol. 2009;4:939–947. doi: 10.1021/cb900195c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Engen JR, Wales TE, Hochrein JM, Meyn MA, 3rd, Banu Ozkan S, Bahar I, Smithgall TE. Structure and dynamic regulation of Src-family kinases. Cell Mol Life Sci. 2008;65:3058–3073. doi: 10.1007/s00018-008-8122-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. SCALA - scale together multiple observations of reflections. Cambridge: MRC Laboratory of Molecular Biology; 2007. [Google Scholar]
- Filippakopoulos P, Kofler M, Hantschel O, Gish GD, Grebien F, Salah E, Neudecker P, Kay LE, Turk BE, Superti-Furga G, et al. Structural coupling of SH2-kinase domains links Fes and Abl substrate recognition and kinase activation. Cell. 2008;134:793–803. doi: 10.1016/j.cell.2008.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galkin AV, Melnick JS, Kim S, Hood TL, Li N, Li L, Xia G, Steensma R, Chopiuk G, Jiang J, et al. Identification of NVP-TAE684, a potent, selective, and efficacious inhibitor of NPM-ALK. Proc Natl Acad Sci U S A. 2007;104:270–275. doi: 10.1073/pnas.0609412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer P, Haigh J, Mbamalu G, Khoo W, Bernstein A, Pawson T. The Fps/Fes protein-tyrosine kinase promotes angiogenesis in transgenic mice. Mol Cell Biol. 1994;14:6755–6763. doi: 10.1128/mcb.14.10.6755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer PA, Kanda S, Smithgall TE. The contrasting oncogenic and tumor suppressor roles of FES. Front Biosci. 2011 doi: 10.2741/280. in press. [DOI] [PubMed] [Google Scholar]
- Greer PA, Meckling-Hansen K, Pawson T. The human c-fps/fes gene product expressed ectopically in rat fibroblasts is nontransforming and has restrained protein-tyrosine kinase activity. Mol Cell Biol. 1988;8:578–587. doi: 10.1128/mcb.8.2.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigh J, McVeigh J, Greer P. The fps/fes tyrosine kinase is expressed in myeloid, vascular endothelial, epithelial, and neuronal cells and is localized in the trans-golgi network. Cell Growth Differ. 1996;7:931–944. [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The Hallmarks of Cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hellwig S, Smithgall TE. Structure and regulation of the c-Fes protein-tyrosine kinase. Front Biosci. 2011;17:3146–3155. doi: 10.2741/3902. [DOI] [PubMed] [Google Scholar]
- Hjermstad SJ, Peters KL, Briggs SD, Glazer RI, Smithgall TE. Regulation of the human c-fes protein tyrosine kinase (p93c-fes) by its src homology 2 domain and major autophosphorylation site (Tyr-713) Oncogene. 1993;8:2283–2292. [PubMed] [Google Scholar]
- Huang CC, Hammond C, Bishop JM. Nucleotide sequence and topography of chicken c-fps. Genesis of a retroviral oncogene encoding a tyrosine-specific protein kinase. J Mol Biol. 1985;181:175–186. doi: 10.1016/0022-2836(85)90083-x. [DOI] [PubMed] [Google Scholar]
- Itoh T, De Camilli P. BAR, F-BAR (EFC) and ENTH/ANTH domains in the regulation of membrane-cytosol interfaces and membrane curvature. Biochim Biophys Acta. 2006;1761:897–912. doi: 10.1016/j.bbalip.2006.06.015. [DOI] [PubMed] [Google Scholar]
- Kanda S, Lerner EC, Tsuda S, Shono T, Kanetake H, Smithgall TE. The nonreceptor protein-tyrosine kinase c-Fes is involved in fibroblast growth factor-2-induced chemotaxis of murine brain capillary endothelial cells. J Biol Chem. 2000;275:10105–10111. doi: 10.1074/jbc.275.14.10105. [DOI] [PubMed] [Google Scholar]
- Kanda S, Miyata Y. The c-Fes protein tyrosine kinase as a potential anti-angiogenic target in cancer. Front Biosci. 2011;16:1024–1035. doi: 10.2741/3732. [DOI] [PubMed] [Google Scholar]
- Kanda S, Miyata Y, Kanetake H, Smithgall TE. Non-receptor protein-tyrosine kinases as molecular targets for antiangiogenic therapy (Review) Int J Mol Med. 2007;20:113–121. [PubMed] [Google Scholar]
- Kanda S, Miyata Y, Kanetake H, Smithgall TE. Downregulation of the c-Fes protein-tyrosine kinase inhibits the proliferation of human renal carcinoma cells. Int J Oncol. 2009;34:89–96. [PMC free article] [PubMed] [Google Scholar]
- Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- Kim J, Feldman RA. Activated Fes protein tyrosine kinase induces terminal macrophage differentiation of myeloid progenitors (U937 cells) and activation of the transcription factor PU.1. Mol Cell Biol. 2002;22:1903–1918. doi: 10.1128/MCB.22.6.1903-1918.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Ogata Y, Ali H, Feldman RA. The Fes tyrosine kinase: a signal transducer that regulates myeloid-specific gene expression through transcriptional activation. Blood Cells Mol Dis. 2004;32:302–308. doi: 10.1016/j.bcmd.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Laurent CE, Delfino FJ, Cheng HY, Smithgall TE. The human c-Fes tyrosine kinase binds tubulin and microtubules through separate domains and promotes microtubule assembly. Mol Cell Biol. 2004;24:9351–9358. doi: 10.1128/MCB.24.21.9351-9358.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner EC, Smithgall TE. SH3-dependent stimulation of Src-family kinase autophosphorylation without tail release from the SH2 domain in vivo. Nat Struct Biol. 2002;9:365–369. doi: 10.1038/nsb782. [DOI] [PubMed] [Google Scholar]
- Leslie AGW, Powell H. MOSFLM. Cambridge: MRC Laboratory of Molecular Biology; 2007. [Google Scholar]
- Li J, Smithgall TE. Fibroblast transformation by Fps/Fes tyrosine kinases requires Ras, Rac, and Cdc42 and induces extracellular signal-regulated and c-Jun N-terminal kinase activation. J Biol Chem. 1998;273:13828–13834. doi: 10.1074/jbc.273.22.13828. [DOI] [PubMed] [Google Scholar]
- Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol. 2006;2:358–364. doi: 10.1038/nchembio799. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- McPherson VA, Everingham S, Karisch R, Smith JA, Udell CM, Zheng J, Jia Z, Craig AW. Contributions of F-BAR and SH2 domains of Fes protein tyrosine kinase for coupling to the FcepsilonRI pathway in mast cells. Mol Cell Biol. 2009;29:389–401. doi: 10.1128/MCB.00904-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr. 2006;62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- Rodems SM, Hamman BD, Lin C, Zhao J, Shah S, Heidary D, Makings L, Stack JH, Pollok BA. A FRET-based assay platform for ultra-high density drug screening of protein kinases and phosphatases. Assay Drug Dev Technol. 2002;1:9–19. doi: 10.1089/154065802761001266. [DOI] [PubMed] [Google Scholar]
- Roebroek AJ, Schalken JA, Verbeek JS, Van den Ouweland AM, Onnekink C, Bloemers HP, Van de Ven WJ. The structure of the human c-fes/fps proto-oncogene. Embo J. 1985;4:2897–2903. doi: 10.1002/j.1460-2075.1985.tb04020.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers JA, Cheng HY, Smithgall TE. Src homology 2 domain substitution modulates the kinase and transforming activities of the Fes protein-tyrosine kinase. Cell Growth Differ. 2000;11:581–592. [PubMed] [Google Scholar]
- Roodman GD. Mechanisms of bone metastasis. N Engl J Med. 2004;350:1655–1664. doi: 10.1056/NEJMra030831. [DOI] [PubMed] [Google Scholar]
- Sadowski I, Stone JC, Pawson T. A noncatalytic domain conserved among cytoplasmic protein-tyrosine kinases modifies the kinase function and transforming activity of Fujinami sarcoma virus P130gag-fps. Mol Cell Biol. 1986;6:4396–4408. doi: 10.1128/mcb.6.12.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangrar W, Zirgnibl RA, Gao Y, Muller WJ, Jia Z, Greer PA. An identity crisis for fps/fes: oncogene or tumor suppressor? Cancer Res. 2005;65:3518–3522. doi: 10.1158/0008-5472.CAN-04-3468. [DOI] [PubMed] [Google Scholar]
- Shaffer JM, Hellwig S, Smithgall TE. Bimolecular fluorescence complementation demonstrates that the c-Fes protein-tyrosine kinase forms constitutive oligomers in living cells. Biochemistry. 2009;48:4780–4788. doi: 10.1021/bi900238f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer JM, Smithgall TE. Promoter methylation blocks FES protein-tyrosine kinase gene expression in colorectal cancer. Genes Chromosomes Cancer. 2009;48:272–284. doi: 10.1002/gcc.20638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder SP, Theilen GH. Transmissible feline fibrosarcoma. Nature. 1969;221:1074–1075. doi: 10.1038/2211074a0. [DOI] [PubMed] [Google Scholar]
- Takashima Y, Delfino FJ, Engen JR, Superti-Furga G, Smithgall TE. Regulation of c-Fes tyrosine kinase activity by coiled-coil and SH2 domains: analysis with Saccharomyces cerevisiae. Biochemistry. 2003;42:3567–3574. doi: 10.1021/bi0272499. [DOI] [PubMed] [Google Scholar]
- Voisset E, Lopez S, Chaix A, Georges C, Hanssens K, Prebet T, Dubreuil P, De Sepulveda P. FES kinases are required for oncogenic FLT3 signaling. Leukemia. 2010;24:721–728. doi: 10.1038/leu.2009.301. [DOI] [PubMed] [Google Scholar]
- Voisset E, Lopez S, Dubreuil P, De Sepulveda P. The tyrosine kinase FES is an essential effector of KITD816V proliferation signal. Blood. 2007;110:2593–2599. doi: 10.1182/blood-2007-02-076471. [DOI] [PubMed] [Google Scholar]
- Wang LH, Feldman R, Shibuya M, Hanafusa H, Notter MF, Balduzzi PC. Genetic structure, transforming sequence, and gene product of avian sarcoma virus UR1. J Virol. 1981;40:258–267. doi: 10.1128/jvi.40.1.258-267.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Grainger DW. siRNA knock-down of RANK signaling to control osteoclast-mediated bone resorption. Pharm Res. 2010;27:1273–1284. doi: 10.1007/s11095-010-0099-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, Cortot AB, Chirieac L, Iacob RE, Padera R, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462:1070–1074. doi: 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.