

Abstract
Both exogenous and endogenous cannabinoids can allosterically modulate glycine receptors (GlyRs). However, little is known about the molecular basis of cannabinoid-GlyR interactions. Here we report that sustained incubation with the endocannabinoid anandamide (AEA) substantially increased the amplitude of glycine-activated current in both rat cultured spinal neurons and in HEK-293 cells expressing human α1, rat α2 and α3 GlyRs. While the α1 and α3 subunits were highly sensitive to AEA-induced potentiation, the α2 subunit was relatively insensitive to AEA. Switching a serine at 296 and 307 in the TM3 (transmembrane domain 3) of the α1 and α3 subunits with an alanine (A) at the equivalent position in the α2 subunit converted the α1/α3 AEA-sensitive receptors to sensitivity resembling that of α2. The S296 residue is also critical for exogenous cannabinoid-induced potentiation of IGly. The magnitude of AEA potentiation decreased with removal of either the hydroxyl or oxygen groups on AEA. While desoxy-AEA was significantly less efficacious in potentiating IGly, desoxy-AEA inhibited potentiation produced by both Δ9-tetrahydrocannabinol (THC), a major psychoactive component of marijuana, and AEA. Similarly, didesoxy-THC, a modified THC with removal of both hydroxyl/oxygen groups, did not affect IGly when applied alone but inhibited the potentiation of IGly induced by AEA and THC. These findings suggest that exogenous and endogenous cannabinoids potentiate GlyRs via a hydrogen bonding-like interaction. Such a specific interaction likely stems from a common molecular basis involving the S296 residue in the TM3 of the α1 and α3 subunits.
Introduction
Emerging evidence has suggested that glycine receptors (GlyRs) are an important target for actions of exogenous and endogenous cannabinoids in the CNS (Zhang and Xiong, 2009). A number of recent studies have shown that cannabinoids can potentiate GlyR-mediated responses in various neurons and in cells expressing recombinant GlyRs via CB1 and CB2 independent mechanisms (Hejazi et al., 2006; Yang et al., 2008; Ahrens et al., 2009; Delaney et al., 2010; Xiong et al., 2011; Yevenes and Zeilhofer, 2011a). In addition to modulating GlyRs, both exogenous and endogenous cannabinoids have been shown to allosterically modulate all other members of the Cys-loop ligand-gated ion channels (LGICs) including serotonin type 3A (5-HT3A), neuronal nicotinic acetylcholine (nACh) and GABAA receptors (Fan, 1995; Barann et al., 2002; Oz et al., 2004; Hejazi et al., 2006; Xiong et al., 2008; Sigel et al., 2011). There is strong evidence to suggest that some of the cannabinoid-induced behavioral effects are independent of CB1 receptors. The endocannabinoid anandamide (AEA) stimulates GTPγS binding in brain membranes isolated from mice lacking CB1 receptors, and this effect is not altered by CB1 and CB2 antagonists (Di Marzo et al., 2000). Δ9-tetrahydrocannabinol (THC), the principle psychoactive component of marijuana, and AEA-induced analgesic effects in the tail flick test (for THC) and hot-plate test (for AEA) remain intact in mice with depleted CB1 receptors (CB1−/−) (Zimmer et al., 1999; Di Marzo et al., 2000) or both CB1 and CB2 receptors (Rácz et al., 2008). A recent study from our laboratory has suggested that exogenous and synthetic cannabinoid potentiation of GlyRs contributes to psychoactive and nonpsychoactive cannabinoid-induced analgesic effect in the tail flick reflex in mice (Xiong et al., 2011).
The GlyRs are involved in several physiological and pathological processes including neuromotor activity, antinociception, muscle relaxation, anxiety and reward mechanisms (Lynch, 2009). The GlyRs are formed as pentameric homomeric chloride channels of α1, α2, α3 and α4 subunits or as αβ heteromeric functional channels (Lynch, 2004). Each GlyR subunit is composed of an extracellular domain, four transmembrane domains (TMs) and a large cytoplasmic domain between TM3 and TM4. There is strong evidence showing that distinct sites located in these domains mediate functional modulation of GlyRs by different allosteric modulators (Lobo and Harris, 2005; Harris et al., 2008; Foadi et al., 2010; Xiong et al., 2011; Yevenes and Zeilhofer, 2011a). A recent study has revealed a serine at position 296 in the TM3 of GlyR as a distinct site to critically regulate THC-induced potentiation of GlyR (Xiong et al., 2011). However, little is known about the molecular mechanisms underlying endocannabinoid AEA potentiation of GlyRs. Here we report that the α1, α2 and α3 GlyR subunits are differentially sensitive to the AEA-induced potentiation of IGly. The S296 residue already implicated in THC potentiation of α1 GlyR function contributes to the differential sensitivity of different GlyR subunits to sustained AEA application. We have also provided evidence to suggest that exogenous and endogenous cannabinoids interact with GlyRs through a common molecular basis.
Materials and Methods
Cultured spinal neurons
Animals were treated and handled according to NIH guidelines. Postnatal day 0 rats of either sex were killed by cervical dislocation. The spinal cords were removed from three to five rats. The tissue, which was chopped into small pieces, was incubated with 5 ml of papain solution (Worthington, 20 U/ml) at 37°C for 40 min. The tissue was washed and triturated through a 9 inch glass Pasteur pipette with the tip fire polished to an opening of 0.7–0.9 mm diameter. The cell suspension was centrifuged at 120 × g for 5 min and the supernatants were discarded. Spinal neurons were resuspended and plated, at a concentration of 300,000 cells/ml, into 35 mm tissue culture dishes coated with poly-d-lysine (0.1 mg/ml). The neuronal feeding medium consisted of 90% minimum essential medium, 10% heat-inactivated fetal bovine serum and a mixture of nutrient supplements (Invitrogen). New medium was added every 3 d. Cells were cultured for at least 10 d and washed with normal external solution (see below) for 30 min before starting the electrophysiological experiments.
HEK-293 cell transfection and electrophysiological recording
HEK-293 cells were cultured as described previously (Hu et al., 2006). The plasmid cDNAs coding for the wild-type and mutant GlyR subunits and human 5-HT3A receptors (5-HT3ARs) were transfected using the SuperFect Transfection Kit (Qiagen). Electrophysiological recordings were performed 2 d after transfection. HEK-293 cells were treated with 0.25% (w/v) Trypsin and 0.53 mm EDTA 2 h before recording. The HEK-293 cells were lifted and continuously superfused with a solution containing (in mm): 140 NaCl, 5 KCl, 1.8 CaCl2, 1.2 MgCl2, 5 glucose, and 10 HEPES (pH 7.4 with NaOH; ∼340 mOsm with sucrose). Patch pipettes (3–5 MΩ) were filled with the intracellular solution that contained the following (in mm): 120 CsCl, 4 MgCl2, 10 EGTA, 10 HEPES, 0.5 Na-GTP, and 2 Mg-ATP (pH 7.2 with CsOH, ∼280 mOsm). Membrane currents were recorded in the whole-cell configuration using an Axopatch 200B amplifier (Axon) at 20−22°C. Cells were held at −60 mV unless otherwise indicated. Data were acquired using pClamp 9.2 software (Molecular Devices). Data were filtered at 1 kHz and digitized at 2 kHz. Bath solutions were applied through 3 barrel square glass tubing (Warner Instrument) with a tip diameter of ∼700 μm. Drugs were applied using a Warner fast-step stepper-motor driven system. The solution exchange time constants were ∼4 ms for an open pipette tip and 4–12 ms for whole-cell recording.
Site-directed mutagenesis
Point-mutations of the human α1 GlyR, rat α2 and α3 GlyR subunits, and human 5-HT3AR were introduced using a QuikChange Site-Directed Mutagenesis Kit (Stratagene). The authenticity of the DNA sequence through the mutation sites was confirmed by double-stranded DNA sequencing using a CEQ 8000 Genetic Analysis System (Beckman Coulter, Inc).
Molecular modeling and simulations
α1 GlyR and 5-HT3AR were modeled with SWISS-MODEL (Guex and Peitsch, 1997; Schwede et al., 2003; Arnold et al., 2006), using a homologous structure (PDB code: 3ehz, chain A) as a template. Five α1 GlyR or 5-HT3AR chains were superimposed on the five chains of the nicotinic acetylcholine receptor (PDB code: 2bg9) to create a pentamer conformation. CHARMM (Brooks et al., 1983, 2009) package was used to perform minimization and simulation. For efficient simulation, the N-terminal extracellular domains (1–252) were removed. THC and AEA molecules were placed in the membrane region near TM3. The C-terminal transmembrane helix bound together with the AEA molecules was minimized and simulated for 2 ns with the self-guided Langevin dynamics method (Wu and Brooks, 2003, 2011) to search for stable conformations.
Drugs
Most chemicals including glycine (Gly) were from Sigma. Solutions were prepared on the day of the experiment. Agonists and other compounds were diluted either directly in the bath solution or dissolved in ethanol before further dilution. The maximal concentration of ethanol in the bath solution was <8 mm, which, when applied alone, did not affect either IGly or AEA-induced potentiation of IGly. The maximal concentration of THC and AEA used was 30 μm because the compounds were difficult to keep in solution at concentrations >30 μm.
Chemical synthesis
Desoxyanandamide.
LiAlH4 of 0.14 ml at 1 m in THF, 0.14 ml) was added dropwise to 5 ml of anandamide (25 mg, 0.072 mmol) in anhydrous THF under Argon at 0°C. The resulting solution was warmed up to room temperature for 1 h and heated to reflux overnight. The reaction mixture was cooled to 0°C again and a solution of Rochelle salt was added cautiously. After being stirred for 1 h, the mixture was extracted with CH2Cl2 (3 × 20 ml). The combined extracts were washed with brine and dried over anhydrous Na2SO4. After filtration and concentration, the crude product was purified by flash chromatography (CHCl3:MeOH:NH4OH = 90:9:1) to afford desoxyanandamide (desoxy-AEA) (6.1 mg, 25.4%) as clear oil. ESI-MS 334.3 (M+ + 1); HRMS (ES+) calculated for C22H40NO, 334.3110; found, 334.3100.
Dehydroxylanandamide.
Arachidonic acid of 5 ml (31 mg, 0.1 mmol) in CH2Cl2 was added to a solution containing ethylamine in THF (2 m, 0.1 ml), Et3N (0.3 mmol, 42 μl) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (0.2 mmol, 38 mg) successively under argon at 0°C. The reaction mixture was stirred at room temperature overnight and then diluted with CH2Cl2 (20 ml). The diluted mixture was washed successively with 2 m HCl (10 ml) and water. The product was saturated with NaHCO3 and brine, and it was then dried over anhydrous Na2OS4. After filtration and concentration, the crude product was purified by flash chromatography (CHCl3:MeOH:NH4OH = 98:1.9:0.1) to give dehydroxylanandamide (dehydroxyl-AEA; 23.7 mg, 71.6%) as clear oil. 1H NMR (CDCl3, 400 MHz) δ 5.36 (m, 8H), 3.27 (m, 2H), 2.82 (m, 6H), 2.10 (m, 6H), 1.71 (m, 2H), 1.31 (m, 6H), 1.13 (t, J = 7.2 Hz, 3H), 0.88 (t, J = 6.4 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 172.6, 130.5, 129.2, 128.7, 128.6, 128.23, 128.20, 127.9, 127.5, 36.1, 34.3, 31.5, 29.3, 27.2, 26.7, 25.6, 25.5, 22.6, 14.9, 14.0; ESI-MS 332.3 (M+ + 1); HRMS (ES+) calculated for C22H38NO, 332.2953; found, 332.2960.
Didesoxy-THC.
Didesoxy-THC was synthesized as described previously (Xiong et al., 2011).
Data analysis
Statistical analysis of concentration–response data was performed using the nonlinear curve-fitting program (Prism 5.0). Data were fit using the Hill equation I/Imax = Bottom + (Top − Bottom)/(1 + 10((LogEC50 − Log[Agonist]) * Hill Slope) where I is the current amplitude activated by a given concentration of agonist ([Agonist]), Imax is the maximum response of the cell, and EC50 is the concentration eliciting a half-maximal response. Data were statistically compared by the unpaired t test, or ANOVA, as noted. Average values are expressed as mean ± SE.
Results
AEA potentiation of native and recombinant GlyRs
While AEA did not trigger detectable current even at high concentrations (>30 μm), AEA at 1 μm increased the magnitude of currents activated by a 2% maximal effective (EC2) concentration of Gly in cultured spinal neurons and in HEK-293 cells expressing human α1 GlyRs (Fig. 1B). AEA was always applied after stable IGly as basal current was achieved. When AEA was applied continuously with intermittent Gly applications every minute, the potentiation gradually increased over the first few minutes of sustained AEA exposure. The maximal magnitude of potentiation was reached after 5 min of sustained AEA application in both spinal neurons and in HEK-293 cells expressing the α1 homomeric and α1β1 heteromeric GlyRs (Fig. 1C). Consistent with a previous study showing that the α1 subunits were abundantly expressed in cultured spinal neurons (Tapia and Aguayo, 1998), the magnitudes of AEA potentiation of IGly appeared similar between spinal neurons and HEK-293 cells expressing the α1 subunits. For instance, the average percentage potentiation of IGly induced by 1 μm AEA was 86 ± 24% (n = 6) in spinal neurons and 97 ± 16% (n = 7) or 85 ± 12% (n = 6) in HEK-293 cells expressing the α1 or α1β1 subunits. These values were significantly higher than the initial values observed after 1 min of AEA exposure, 6 ± 6% (neurons) and 3 ± 1% (HEK-293 cells) (p < 0.05, one-way ANOVA followed by Dunnett's test comparing the last response to the response in the first minute).
Figure 1.
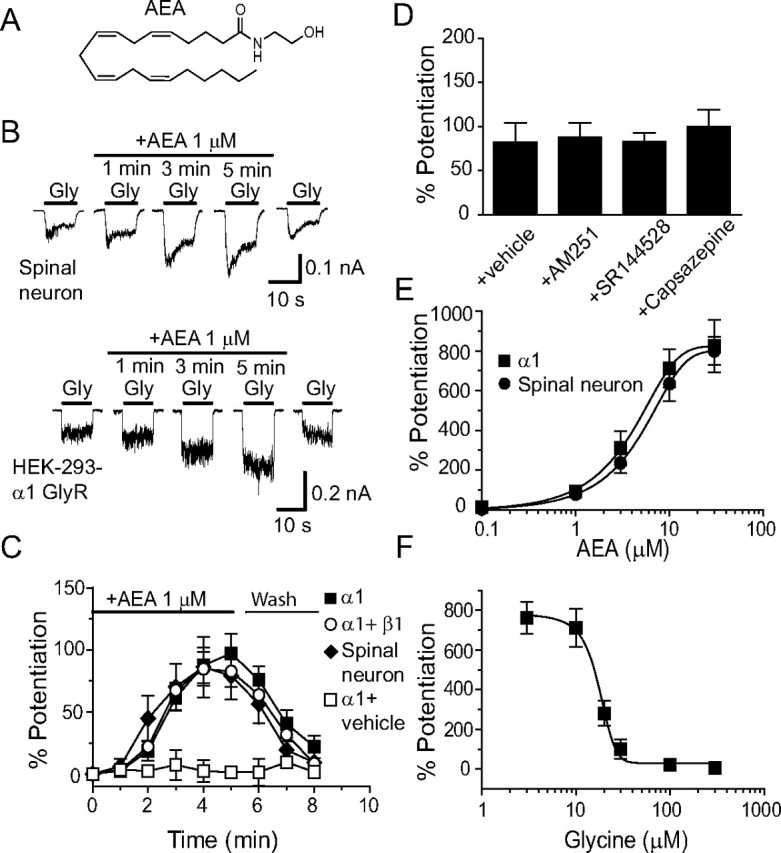
AEA potentiation of IGly in cultured spinal neurons and HEK-293 cells expressing α1 and β1 GlyRs. A, Chemical structure of AEA. B, Current traces showing IGly activated by EC2 concentrations of Gly (5–10 μm) before, during and after a 5 min continuous incubation with AEA in cultured spinal neurons (top) and in HEK-293 cells expressing the α1 GlyR subunits (bottom). C, Time courses of average percentage potentiation induced by 1 μm AEA during a 5 min period of continuous incubation. The solid bar indicates AEA application time in cultured neurons (solid diagonals) and in HEK-293 cells expressing α1 GlyRs (solid squares) and α1β1 GlyRs (open circles). Each point represents mean ± SE of at least 6 cells. D, Effects of AM251, SR144528 and capsazepine on AEA potentiation (n = 5–7). E, The concentration–response curve of the AEA-induced potentiation in cultured neurons (n = 6) and HEK-293 cells expressing the α1 subunit (n = 6). F, Agonist concentration dependence of AEA-induced potentiation of IGly in HEK-293 cells expressing the α1 subunits (n = 5). The error bars that are not visible are smaller than the size of symbols.
In addition to activating CB1 and CB2 receptors, AEA also activates vanilloid receptors in neurons (Pertwee, 2000; Smart et al., 2000; Ross, 2003). To determine whether AEA potentiates IGly through activating CB1, CB2 and vanilloid receptors, we tested the effects of specific antagonists of these receptors on AEA-induced potentiation of IGly in cultured spinal neurons (Fig. 1D). Selective antagonists of CB1 (AM251, 1 μm), CB2 (SR144528, 1 μm) and vanilloid (capsazepine, 2 μm) receptors did not significantly alter the AEA-induced potentiation of IGly in cultured spinal neurons (AM251, p = 0.84; SR144528, p = 0.97; Capsazepine, p = 0.56 compared with vehicle solution using one-way ANOVA followed by Dunnett's post hoc test), suggesting that AEA potentiation of native GlyRs does not depend on the CB1, CB2 and vanilloid receptors. AEA enhanced the peak amplitude of IGly in a concentration-dependent manner in spinal neurons and HEK-293 cells expressing the homomeric GlyR α1 subunit (Fig. 1E). The magnitudes of average percentage potentiation induced by 1, 3, 10, and 30 μm AEA were 91 ± 9%, 311 ± 86%, 712 ± 96%, and 824 ± 278%, respectively, in HEK-293 cells, and 79 ± 19%, 236 ± 52%, 636 ± 87% and 800 ± 71%, respectively, in spinal neurons. The EC50 values for AEA potentiation were 4.2 ± 1.95 μm in HEK-293 cells expressing α1 homomeric GlyRs and 5.5 ± 2.0 μm in spinal neurons. These values are not significantly different from one another (p = 0.85, unpaired t test, n = 5–7). AEA potentiation depended on the Gly concentration (Fig. 1F). With increasing Gly concentrations, the AEA potentiation was decreased. For example, the magnitude of average percentage potentiation induced by 10 μm AEA was 712 ± 96% in the presence of Gly at 10 μm in HEK-293 cells expressing the α1 subunit, while the magnitude of AEA potentiation was 102 ± 49% or 24 ± 6% when Gly concentrations were increased up to 30 and 100 μm.
S296: essential for AEA potentiation of the α1 and α3 subunit-containing GlyRs
Next we asked whether or not three distinct α subunits of GlyRs are differentially sensitive to AEA-induced potentiation. We first determined the EC50 values from the Gly concentration–response curves in HEK-293 cells transfected with α1 α2 or α3 subunits of GlyR. The EC50 values for Gly were 64 ± 13 μm for the α1 subunit, 128 ± 10 μm for the α2 subunit and 354 ± 29 μm for the α3 subunit. These values were significantly different from one another (p < 0.05, one-way ANOVA with Dunnett's post hoc test against α1, n = 6–11). We then examined the effect of AEA on IGly activated by EC2 concentrations of Gly in HEK-293 cells expressing each subtype of GlyRs. While the α1 and α3 GlyR subunits appeared to be equally sensitive to AEA-induced potentiation, the α2 GlyR subunits were less sensitive to AEA when expressed in HEK-293 cells (Fig. 2A). The magnitudes of average percentage potentiation induced by 10 μm AEA were 695 ± 76% and 730 ± 58% in cells expressing the α1 and α3 subunits. In contrast, the magnitude of AEA enhancement of receptors containing the α2 subunit (127 ± 37%) was significantly lower than that of receptors containing the α1 and α3 subunits (p < 0.001 compared with α1 or α3, one-way ANOVA with Dunnett's post hoc test, n = 6) (Fig. 2B).
Figure 2.
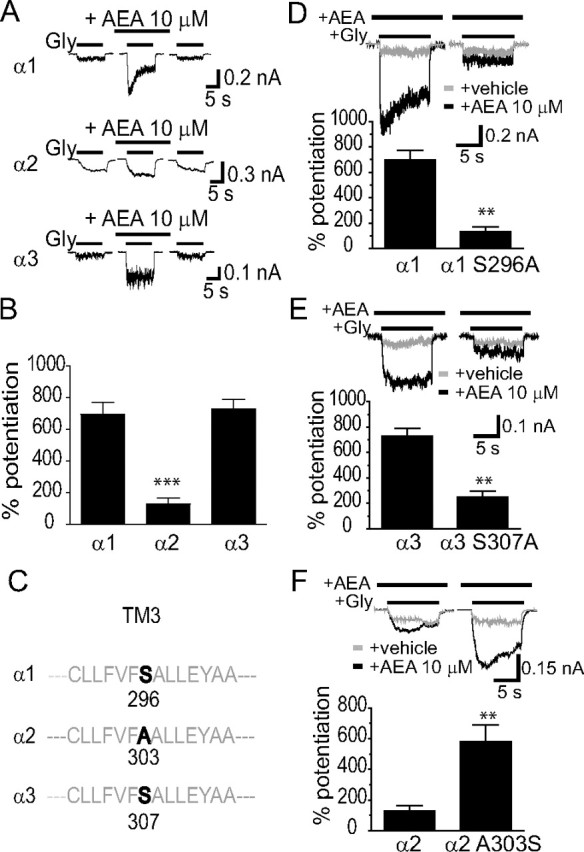
S296 is critical for AEA potentiation of α1 and α3 GlyRs. A, Differential AEA potentiation of IGly activated by EC2 concentrations of Gly (10 μm for the α1 subunit, 20 μm for the α2 subunit and 100 μm for the α3 subunit) in HEK-293 cells. The AEA (10 μm) was applied before agonist, and applied continuously for 5 min with intermittent 10 s Gly + AEA applications as shown in Figure 1B. B, Average potentiation by AEA of IGly in different HEK-293 cells expressing α1, α2 or α3 GlyRs (n = 6–7). ***p < 0.001, α2 vs α1 or α3, one-way ANOVA followed by Dunnett's post hoc test. C, Amino acid alignment of the TM3 region flanking S296 (α1) or equivalent residues in the α2 (A303) and α3 (S307) subunits. D, The effect of AEA (10 μm) on IGly activated by an EC2 concentration of Gly (10 μm) in cells expressing the S296A mutant and wild-type α1 GlyRs. E, The effects of AEA (10 μm) on IGly activated by the EC2 concentration of Gly (100 μm) in cells expressing the S307A mutant and wild-type α3 GlyRs. F, The effects of AEA (10 μm) on IGly activated by the EC2 concentration of Gly (20 μm) in cells expressing the A303S mutant and wild-type α2 GlyRs. **p < 0.01, unpaired t test.
A recent study from our laboratory has suggested that the S296 residue of the α1 and the S307 of the α3 GlyRs are critical for the THC-induced potentiation of IGly (Xiong et al., 2011). We next tested whether the S296/S307 residue is also critical for differential AEA sensitivity of GlyR subunits. The serine (S) at 296 and 307 in the TM3 is conserved between the α1 and α3 subunits, respectively. In contrast, the equivalent residue at 303 of the α2 subunit is Alanine (A), which is the only residue throughout the entire 4 TMs of the α2 subunit that differs from a corresponding residue identical between the α1 (S296) and α3 (S307) subunits (Fig. 2C). The S296A mutation in the α1 subunit significantly reduced the magnitude of AEA-induced potentiation from 695 ± 77% to 131 ± 41% (Fig. 2D, p < 0.01, n = 7, unpaired t test). Similarly, The S307A mutation in the α3 subunit also significantly reduced AEA potentiation from 731 ± 58% to 249 ± 48% (Fig. 2E, p < 0.01, n = 6, unpaired t test). Conversely, substitution of the corresponding residue, A303, of the α2 subunit with serine significantly increased the magnitude of AEA-induced potentiation from 127 ± 37% to 580 ± 110% (Fig. 2F, p < 0.01, n = 6, unpaired t test). The S296A mutation in the α1 subunit did not significantly affect the Gly EC50 value (Fig. 3A, 64 ± 13 μm vs 66 ± 7 μm, p = 0.67, n = 6, unpaired t test) and maximal current density (MCD) induced by a maximally efficacious Gly concentration (3 mm) (Fig. 3B, 912 ± 153 pA/pF vs 906 ± 147 pA/pF, p = 0.67, n = 6, unpaired t test). Similarly, neither the A303S mutation of α2 GlyR nor the S307A mutation of α3 GlyR significantly changed the Gly EC50 value and MCD (Fig. 3C–F). We also examined the inhibition of IGly by strychnine, a selective glycine receptor antagonist, in cells expressing the S296A mutant and WT α1 receptors. The S296A mutation in the α1 subunit did not significantly affect the strychnine (100 nm)-induced inhibiting effect on IGly activated by Gly at an EC50 concentration (78 ± 9% vs 81 ± 13%, n = 7, p > 0.2).
Figure 3.
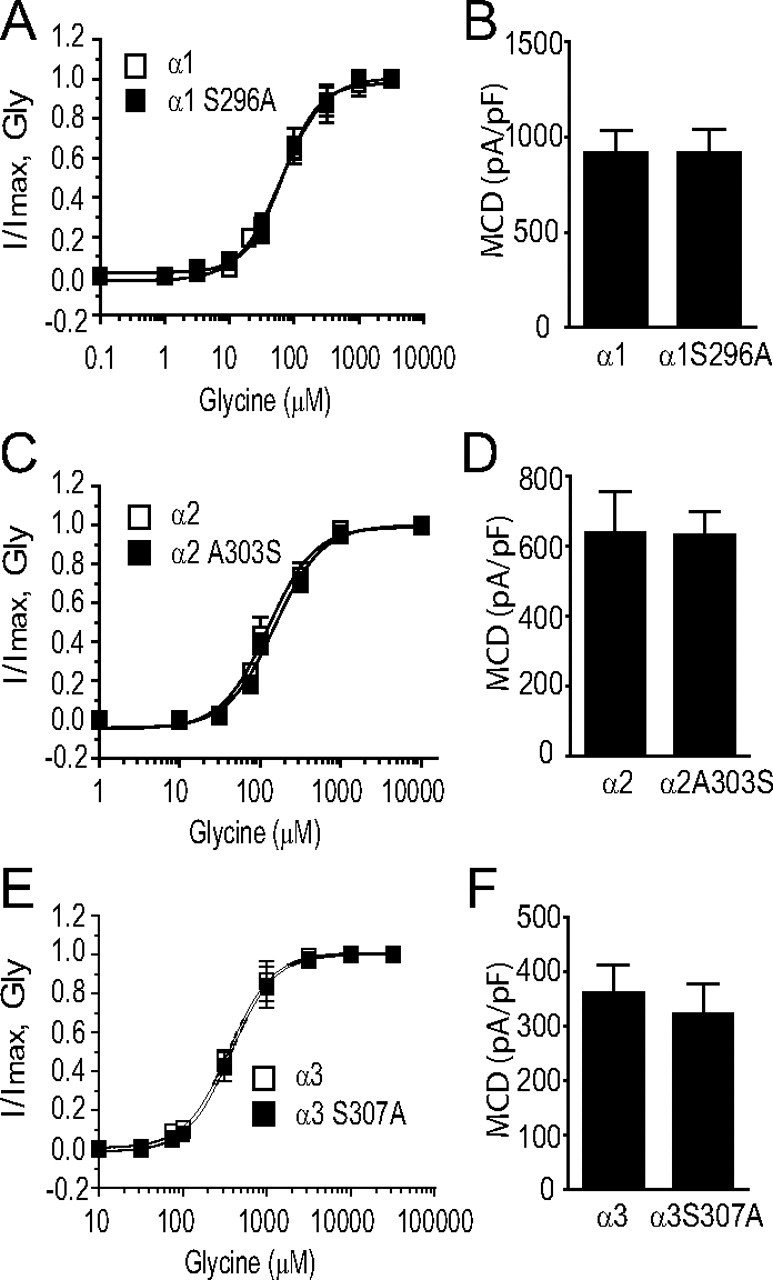
S296 mutation does not affect the basic function of GlyR. A, Gly concentration–response curves in HEK-293 cells expressing the S296A mutant and wild-type α1 GlyRs. B, The average MCD of IGly in HEK-293 cells expressing the S296A mutant and wild-type α1 GlyRs. C, Gly concentration–response curves in HEK-293 cells expressing the A303S mutant and wild-type α2 GlyRs. D, The average MCD of IGly in HEK-293 cells expressing the A303S mutant and wild-type α2 GlyRs. E, Gly concentration–response curves in HEK-293 cells expressing the S307A mutant and wild-type α3 GlyRs. F, The average MCD of IGly in HEK-293 cells expressing the S307A mutant and wild-type α3 GlyRs. The error bars that are not visible are smaller than the size of symbols.
S296 selectively contributes to sustained AEA application-induced potentiation
The maximal magnitude of AEA potentiation of GlyRs reported here was significantly higher than what we and others described in previous studies in which AEA was simultaneously coapplied with agonists (Hejazi et al., 2006; Ahrens et al., 2009; Yevenes and Zeilhofer, 2011a). One possibility that might account for this discrepancy is that distinct molecular processes are involved in AEA potentiation of GlyRs with and without sustained AEA incubation. To test this hypothesis, we first compared the potentiation of three GlyR subunits induced by sustained AEA application and simultaneous AEA application. Unlike the result obtained from sustained THC and AEA incubation (Fig. 4A), there was no significant difference in either simultaneous THC or simultaneous AEA-induced potentiation of the α1, α2 and α3 subunits expressed in HEK-293 cells (Fig. 4B, p > 0.2). Next we compared the role of the S296A mutation in both sustained and simultaneous AEA incubation-induced potentiation of the α1 subunit. While the S296A mutation significantly inhibited AEA potentiation after 5 min sustained incubation (695 ± 77% vs 131 ± 41%, p < 0.001, n = 5–6, unpaired t test), the S296A mutant and WT receptors did not significantly differ in their sensitivity to AEA (10 μm)-induced potentiation when simultaneously applied only with Gly (Fig. 4C,D, 108 ± 15% vs 99 ± 20%). A very recent study has suggested that the G254 residue in TM2 of the α1 GlyR regulates the receptor's sensitivity to the endocannabinoid 2-arachidonoylglycerol when these compounds are coapplied with agonist (Yevenes and Zeilhofer, 2011a). In view of this, we examined the effect of the G254A mutation (Fig. 4E) on the maximal potentiating effect on IGly induced by AEA after 5 min sustained incubation with intermittent application of Gly. The G254A mutation did not significantly alter the sustained AEA incubation-induced potentiation of IGly (Fig. 4F, 695 ± 77% vs 597 ± 77%, p = 0.40, n = 5, unpaired t test).
Figure 4.

Comparison between sustained and simultaneous application of AEA-induced potentiation of GlyRs. A, Potentiation of IGly induced by 5 min sustained incubation of THC (1 μm)/AEA (10 μm) in HEK-293 cells expressing α1, α2 and α3 GlyRs. B, Potentiation of IGly induced by simultaneous application of THC (1 μm)/AEA (10 μm) in HEK-293 cells expressing α1, α2 and α3 GlyRs. C, Current trace showing potentiation of IGly induced by simultaneous application of AEA or 5 min sustained incubation of 10 μm AEA in HEK-293 cells expressing the S296A mutant and wild-type α1 GlyRs. The solid bars on the top of traces indicate the time of drug application. D, Quantification of data in experiments shown in C. ***p < 0.001, unpaired t test. Note that the S296A mutation completely abolished the additional potentiation of IGly induced by sustained AEA incubation but not by simultaneously coapplied AEA. E, Amino acid alignment of the TM2 region flanking G254 (α1) or equivalent residues in the α2 and α3 subunits. F, The average percentage potentiation induced by sustained AEA application in HEK-293 cells expressing the G254A mutant and wild-type α1 GlyRs.
Chemical modification of AEA: critical role of hydroxyl/oxygen groups of AEA
Several phytocannabinoids such as THC and cannabidiol (CBD) can potentiate GlyRs (Hejazi et al., 2006; Yang et al., 2008; Ahrens et al., 2009). We next examined the influence of the S296A mutation on THC, CBD and HU210-induced potentiation of the α1 GlyR (Fig. 5A). The S296A mutation significantly reduced the potentiation of IGly induced by THC (1 μm, p < 0.001, n = 8, unpaired t test), CBD (1 μm, p < 0.05, n = 6, unpaired t test) and HU210 (1 μm, p < 0.001, n = 6, unpaired t test). Removal of both hydroxyl and oxygen groups from THC resulted in a new compound, didesoxy-THC, which substantially reduced THC-induced potentiation of the α1 GlyR (Xiong et al., 2011). While AEA and THC are structurally different, both compounds are potent agonists of CB1 receptors and likely bind to a similar molecular pocket of the receptor (Pertwee, 2008). We proposed that exogenous and endogenous cannabinoids interact with GlyRs via a similar molecular process. To test this hypothesis, we removed sequentially the hydroxyl or oxygen groups from AEA (Fig. 5B). The compounds with removal of hydroxyl (dehydroxyl-AEA) or oxygen group (desoxy-AEA) showed a reduced efficacy in potentiating IGly in receptors containing the α1 subunit (Fig. 5B). The reduction in the efficacy of AEA potentiation was more obvious with removal of the oxygen group from AEA (desoxy-AEA). To determine whether THC and AEA may share common interacting sites on the GlyRs, we cross-examined the effect of desoxy-AEA and didesoxy-THC on THC and AEA-induced potentiation of IGly in cells expressing the α1 subunits. Desoxy-AEA at 3 μm inhibited the magnitude of AEA and THC-induced potentiation in an apparently competitive manner (Fig. 5C,D). Both concentration–response curves of AEA and THC-induced potentiation were shifted in parallel to the right in the presence of desoxy-AEA. Similarly, didesoxy-THC inhibited AEA and THC-induced potentiation in a competitive manner that was also observed in desoxy-AEA (Fig. 5E,F).
Figure 5.
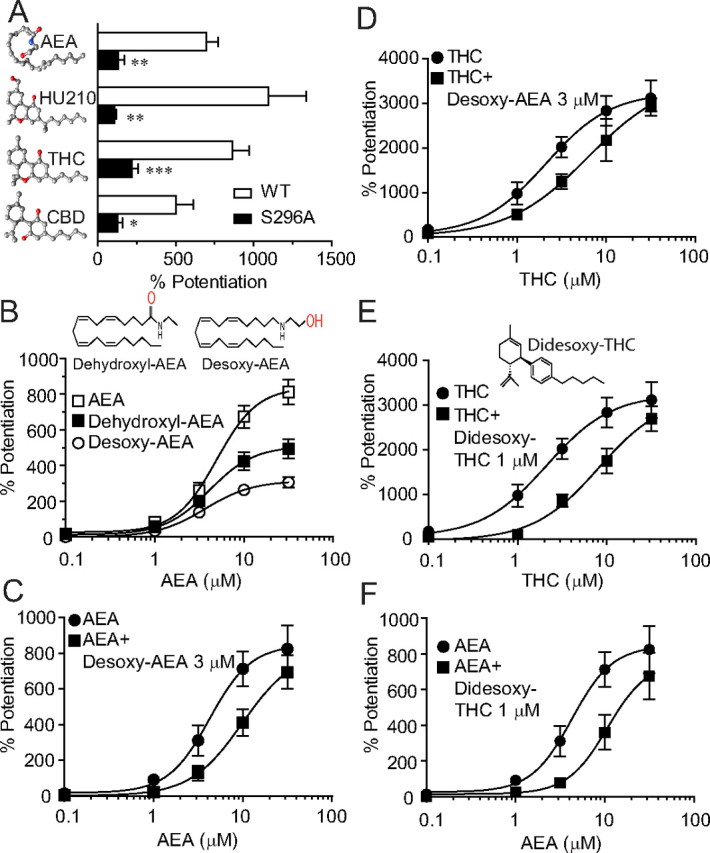
Desoxy-AEA shows reduced efficacy in potentiating IGly and antagonizes both AEA and THC-induced potentiation. A, The effects of AEA (10 μm), HU-210 (1 μm), THC (1 μm) and CBD (1 μm) on IGly activated by an EC2 concentration of Gly (10 μm) in cells expressing the S296A mutant and wild-type α1 GlyRs. n = 5–8, *p < 0.05, **p < 0.01, ***p < 0.001, one-way ANOVA followed by Dunnett's post hoc test against wild-type. B, The concentration–response curves for the AEA and modified AEA (dehydroxyl-AEA and desoxy-AEA)-induced potentiation in HEK-293 cells expressing the α1 GlyRs (n = 5–6). C, The concentration–response curves of the AEA potentiation without (solid circles) and with (solid squares) coapplication of desoxy-AEA. (n = 4–6). D, The concentration–response curves for THC potentiation without (solid circles) and with (solid squares) coapplication of desoxy-AEA. (n = 5–8). E, The concentration–response curves for THC potentiation without (solid circles) and with (solid squares) coapplication of didesoxy-THC (n = 5–8). F, The concentration–response curves for AEA potentiation without (solid circles) and with (solid squares) coapplication of didesoxy-THC (n = 5–7). The error bars that are not visible are smaller than the size of symbols.
S296 is not involved in AEA-induced inhibition of 5-HT3A receptors
There is strong evidence that AEA can inhibit serotonin-gated ion channels (5-HT3AR) through a direct interaction (Fan, 1995; Barann et al., 2002; Oz et al., 2002; Xiong et al., 2008). The 5-HT3AR and GlyR are both members of the Cys-loop LGIC superfamily and thus share a high level of amino acid sequence homology, especially within the transmembrane domains. For instance, the S296 residue is conserved between 5-HT3AR and GlyRs (Fig. 6A). In view of this, we explored the role of S296 in the sensitivity of 5-HT3ARs to AEA (Fig. 6A). Consistent with a previous observation (Xiong et al., 2008), incubation of AEA (0.1 μm) for 5 min produced gradually developing inhibition of 5-HT (30 μm)-activated currents in HEK-293 cells expressing the WT 5-HT3A receptors (Fig. 6B). In these cells, the AEA-induced inhibition of I5-HT reached a maximum after a continuous exposure to 0.1 μm AEA for 5 min. Even though AEA produced different types of modulation (inhibition vs potentiation) of 5-HT3ARs and GlyRs, the time courses of AEA modulation of both receptors appeared similar. However, the S296A mutation of the 5-HT3A receptors did not significantly alter the AEA-induced inhibition of 5-HT-activated current (Fig. 6C, p = 0.45, n = 6–10, Two-way ANOVA). This finding suggests that the S296A residue selectively regulates AEA modulation of GlyRs but not 5-HT3ARs. Although the S296 residue is conserved in the amino acid sequence of GlyR and 5-HT3AR, molecular modeling at the two dimensional level revealed that the orientation of S296 of GlyR differs from that of 5-HT3AR. For instance, S296 of GlyR is facing outside of the ion channel protein and lipid interface, a supposed docking site for AEA (Fig. 6D). In contrast, S296 of 5-HT3A receptor is hiding inside of the channel protein and away from lipid-protein interfaces (Fig. 6E). Both S296 residues do not overlap (Fig. 6F).
Figure 6.

The S296A mutation does not alter AEA-induced inhibition of 5-HT3A receptors. A, Amino acid alignment of the TM3 region flanking S296 (α1 GlyR) or equivalent residues in other ligand-gated ion channels. B, Representative current records for I5-HT activated by a maximally efficacious concentration (30 μm) of 5-HT before, during and after a continuous incubation with 0.1 μm AEA in HEK-293 cells expressing human 5-HT3ARs. C, Time course of the average AEA inhibition of I5-HT in HEK-293 cells expressing wild-type and S296A mutant 5-HT3A receptors. Each point represents mean ± SE of 5–7 cells. D–F, Comparison of the S296 orientation between α1 GlyR (gray ribbons) and 5-HT3AR (cyan ribbons) transmembrane domains. Side and top views of the transmembrane domains of GlyR (D), 5-HT3AR (E), and their superimposition (F). The sidechain oxygen atoms of S296 (shown as spheres) are colored in red for GlyR and in purple for 5-HT3AR.
Molecular docking of AEA and THC onto the α1 GlyR
Molecular modeling of AEA/THC and the TM1–4 of the GlyRα1 subunit revealed that the hydroxyl groups (in red, Fig. 7A,B) of AEA and THC likely interact with the GlyR in a location between TM3 and TM4 (Fig. 7C–F). The predicted interaction of AEA and THC with TM3 may occur via a hydrogen bond between the hydroxyl groups in AEA and the side chain of S296. AEA is well situated in the vicinity of TM3–4 of the α1 subunit in the monomer and pentamer. The hydrogen bonding interaction between AEA/THC and the side chain of S296 can be clearly seen by the close contact of their hydroxyl groups. With the exception of this hydrogen bond, the remaining interactions between AEA/THC and GlyR are likely to be via van der Waals forces.
Figure 7.

Molecular modeling of AEA and THC with the S296 residue in TM3 of the α1 subunit. A, Structure of AEA. B, Structure of THC. C, Side view of the monomeric α1 subunit bound with AEA. D, Side view of the monomeric α1 subunit bound with THC. E, Top view of the monomeric α1 subunit bound with AEA. F, Top view of the monomeric α1 subunit bound with THC. AEA and THC are in the pocket in proximity to S296 in the TM3 of the α1 GlyR subunit. The side chain and oxygen atoms of S296 are shown as van der Waals spheres and the atoms of AEA/THC are shown as ball-and-stick. All atoms are colored in red for oxygen, blue for nitrogen, and gray for carbon.
Discussion
The data presented in this study suggest that both exogenous and endogenous cannabinoids potentiate GlyRs based on a common molecular process. Like THC, AEA-induced potentiation of IGly developed gradually, and required sustained incubation. A similar finding was described in recent studies of the exogenous cannabinoid-induced potentiation of IGly in both spinal neurons and in cells expressing recombinant GlyRs (Xiong et al., 2011). This feature is also associated with cannabinoid modulation of a number of the Cys-loop LGICs. For instance, AEA inhibits 5-HT3ARs and nAChRs in a time-dependent manner, which required sustained incubation of AEA (Fan, 1995; Spivak et al., 2007). AEA differentially modulated different GlyR subunits in a manner similar to that of THC (Xiong et al., 2011). The amino acid residue serine at 296 was found to be critical for both AEA and THC-induced potentiating effect on IGly in cells expressing the α1 and α3 subunits. Conversion of the equivalent residue, alanine, in the α2 subunit to serine rescued the sensitivity of the GlyRs to AEA and THC potentiation. The hydroxyl/oxygen groups appeared to be important functional groups for both AEA and THC in potentiating IGly. Finally, deletion of these groups resulted in reduction in the efficacy of AEA and THC potentiation. Desoxy-AEA and didesoxy-THC competitively inhibited both AEA and THC-induced potentiation of GlyRs in a similar manner.
It is worth mentioning that different protocols for AEA application (sustained application vs simultaneous application with agonists) could cause several notable discrepancies between this study and previous reports. First, the maximal magnitude of AEA potentiation of the α1 GlyRs was estimated to be 80–100% by 1 μm AEA in previous studies when AEA was applied only simultaneously with Gly (Hejazi et al., 2006; Yang et al., 2008; Yevenes and Zeilhofer, 2011a). In the current study the maximal potentiation of the α1 GlyRs was nearly 700–800% when AEA was applied continuously for 5 min with intermittent supplication of Gly. Second, because of a substantial change in Emax of AEA potentiation and maximally efficacious concentration of AEA, the EC50 value of AEA potentiation of GlyRs increased from a range of 38–110 nm range described in previous studies to 4.2 μm observed in the current study. Third, AEA did not significantly alter IGly in HEK-293 cells expressing the α3 GlyRs in a previous study (Yang et al., 2008; Yevenes and Zeilhofer, 2011a), whereas AEA significantly potentiated the α3 GlyRs as reported in this study, and it is likely that the larger effect of sustained AEA application explains this difference. Finally, the data presented in this study suggest that two distinct molecular processes may be involved in the potentiating effect on IGly induced by sustained AEA application with intermittent application of Gly and simultaneous AEA application with Gly. The S296A mutation appeared to selectively contribute to the mechanism underlying sustained cannabinoid-induced potentiation of the α1 and α3 subunits. The molecular determinants for simultaneous AEA and THC-induced potentiation remain elusive. There is no differential sensitivity of GlyR subunits to simultaneous AEA and THC-induced potentiation.
The S296 residue appeared critical for increased sensitivity of the α1 and α3 GlyRs to the AEA-induced potentiation of IGly compared with the α2 subunit. Conversion of this amino acid residue to the corresponding alanine in the α2 subunit selectively altered the receptor's sensitivity to AEA potentiation without significantly changing the EC50 value for Gly or the expression level of receptor protein. The precise molecular mechanism by which S296 interacts with AEA remains to be determined. However, one prominent possibility is that S296 is involved in a hydrogen bonding interaction between AEA and GlyR protein. The most notable difference between the structures of alanine (A) and serine (S) is that one of the methylic hydrogens in the serine side chain is substituted by a hydroxyl group. This allows serine to form a hydrogen bond with potential ligands/modulators. Consistent with this, molecular docking of the GlyR-AEA system suggests a high probability binding mode, perhaps via a possible hydrogen bonding interaction between AEA and the hydroxyl group of the side chains of S296. This hypothesis fits well with a recent study showing that the polarity of the amino acid residue at 296 or the equivalent position in both α1 and α3 GlyRs is correlated with the magnitude of THC-induced potentiation of IGly (Xiong et al., 2011). Alternatively, the S296 residue may play a role in sensing or stabilizing the AEA-induced conformational change that occurs at protein-lipid interface since S296 is thought to be located in the membrane-embedded portion of TM3 of GlyR proteins. This hypothesis is favored by the slow onset of AEA modulation of GlyRs as well as other LGICs, suggesting that AEA may have difficulty in molding itself to the lipid face of receptor transmembrane domains. Consistent with this hypothesis, it has been suggested that the lipid bilayer plays a central role in determining the location and orientation of AEA with respect to membrane targeted proteins (Di Marzo et al., 1998). It should be noted, however, that other residues that differ between the α1/α3 and α2 subunits likely contribute to differential AEA potentiation of GlyRs. Both S296A and S307A mutations in the α1 and α3 subunits only partially inhibited the AEA potentiation.
While the expression level of the α2 subunit is significantly reduced after 1 week in cultured neurons, the α1 and α3 subunits are highly expressed in embryonic spinal cord neurons after a prolonged period (1 or 2 weeks) of in vitro growth (Aguayo et al., 2004). This expression pattern (high expression of the α1 and α3 subunits and low expression of the α2 subunits) in cultured spinal neurons fits well with what we found in this study. The sensitivity of GlyRs to AEA in native spinal neurons was similar to that of the α1 and α3 subunits expressed in HEK-293 cells. Subunit-specific modulation of GlyRs has been described for a number of allosteric modulators of GlyRs (Yevenes and Zeilhofer, 2011b). The α1 GlyRs are more sensitive to ethanol-induced potentiation compared with α2 and α3 GlyRs (Mascia et al., 1996; Perkins et al., 2008; Yevenes et al., 2010). Neurosteroids are also found to potentiate the GlyRs in a subunit-specific manner (Maksay et al., 2001). There is strong evidence that distinct molecular sites contribute to allosteric modulation of GlyRs by different substances (Lobo and Harris, 2005; Harris et al., 2008; Yevenes and Zeilhofer, 2011a). It is interesting to note that the S296 is not involved in AEA modulation of 5-HT3ARs even though the S296 residue is conserved between the α1GlyRs and 5-HT3ARs. Such a dramatic difference can be explained by the molecular modeling of the surrounding vicinity of S296 of the two receptors, which reveals different orientations of the S296 in its relationship with subunit interfaces and protein lipid interfaces.
AEA's binding affinity (Ki) for CB1 and CB2 receptors are 61–543 nm and 279–1940 nm (Pertwee, 2000). AEA at the concentration range (1 μm) used in this study will likely activate CB1 and CB2 receptors in cultured spinal neurons. These receptors are unlikely to contribute to the effect of AEA on IGly in this study because selective antagonists of CB1 and CB2 receptors did not prevent the AEA potentiation of IGly in spinal neurons. Direct modulation of GlyRs by AEA through CB1-independent mechanism was also reported in a previous study (Lozovaya et al., 2005). It must be noted that AEA was found to inhibit IGly in the presence of CB1/CB2 receptor antagonists in hippocampal neurons (Lozovaya et al., 2005). One possible explanation for the different effects of AEA on IGly could be that different GlyR subunits and signal transduction pathways that exist in different neurons could also contribute to the apparently discrepant effects. Alternatively, the apparent discrepancy may be partially caused by differences in experimental design. For example, different concentrations of Gly were used in the two studies. We and others observed that the magnitude of AEA-induced potentiation of IGly depended on Gly concentrations (Hejazi et al., 2006; Yang et al., 2008). The maximal AEA potentiation was observed when the lowest concentrations of Gly were applied. The extent of the AEA-induced potentiation of α1 GlyRs could be reduced substantially when the concentrations of Gly were >100 μm as used in the Lozovaya et al. (2005) study. Thus other factors, such as receptor desensitization, must be considered when trying to explain the inhibition observed by Lozovaya et al. (2005).
The α1 GlyR is expressed mainly in the spinal motor neurons (Legendre, 2001; Lynch, 2004). Humans and rodents carrying certain single amino acid polymorphisms/mutations on the α1 GlyRs at postsynaptic sites have severe deficiencies in neuromotor activity (Shiang et al., 1993, 1995). The α3 GlyRs are abundantly expressed in the adult spinal cord dorsal horn where these receptors critically regulate inflammatory pain sensation (Harvey et al., 2004). Some reports showed that AEA-induced antinociception can occur through a CB1-independent pathway (Adams et al., 1998; Smith et al., 1998; Vivian et al., 1998; Zimmer et al., 1999; Di Marzo et al., 2000). Future studies are awaited for exploring in vivo consequences of AEA potentiation of GlyRs.
Footnotes
This work was supported by funds from the intramural program of the National Institute on Alcohol Abuse and Alcoholism. We thank Drs. Kejun Cheng and Fuying Li for providing modified cannabinoids (didesoxy-THC, desoxy-AEA, and dehydroxyl-AEA). We also thank Dr. Zhifeng Zhou for technical assistance with DNA sequencing.
References
- Adams IB, Compton DR, Martin BR. Assessment of anandamide interaction with the cannabinoid brain receptor: SR 141716A antagonism studies in mice and autoradiographic analysis of receptor binding in rat brain. J Pharmacol Exp Ther. 1998;284:1209–1217. [PubMed] [Google Scholar]
- Aguayo LG, van Zundert B, Tapia JC, Carrasco MA, Alvarez FJ. Changes on the properties of glycine receptors during neuronal development. Brain Res Brain Res Rev. 2004;47:33–45. doi: 10.1016/j.brainresrev.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Ahrens J, Demir R, Leuwer M, de la Roche J, Krampfl K, Foadi N, Karst M, Haeseler G. The nonpsychotropic cannabinoid cannabidiol modulates and directly activates alpha-1 and alpha-1-Beta glycine receptor function. Pharmacology. 2009;83:217–222. doi: 10.1159/000201556. [DOI] [PubMed] [Google Scholar]
- Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- Barann M, Molderings G, Brüss M, Bönisch H, Urban BW, Göthert M. Direct inhibition by cannabinoids of human 5-HT3A receptors: probable involvement of an allosteric modulatory site. Br J Pharmacol. 2002;137:589–596. doi: 10.1038/sj.bjp.0704829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4:187. [Google Scholar]
- Brooks BR, Brooks CL, Mackerell AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, et al. CHARMM: The biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney AJ, Esmaeili A, Sedlak PL, Lynch JW, Sah P. Differential expression of glycine receptor subunits in the rat basolateral and central amygdala. Neurosci Lett. 2010;469:237–242. doi: 10.1016/j.neulet.2009.12.003. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Melck D, Bisogno T, De Petrocellis L. Endocannabinoids: endogenous cannabinoid receptor ligands with neuromodulatory action. Trends Neurosci. 1998;21:521–528. doi: 10.1016/s0166-2236(98)01283-1. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Breivogel CS, Tao Q, Bridgen DT, Razdan RK, Zimmer AM, Zimmer A, Martin BR. Levels, metabolism, and pharmacological activity of anandamide in CB(1) cannabinoid receptor knockout mice: evidence for non-CB(1), non-CB(2) receptor-mediated actions of anandamide in mouse brain. J Neurochem. 2000;75:2434–2444. doi: 10.1046/j.1471-4159.2000.0752434.x. [DOI] [PubMed] [Google Scholar]
- Fan P. Cannabinoid agonists inhibit the activation of 5-HT3 receptors in rat nodose ganglion neurons. J Neurophysiol. 1995;73:907–910. doi: 10.1152/jn.1995.73.2.907. [DOI] [PubMed] [Google Scholar]
- Foadi N, Leuwer M, Demir R, Dengler R, Buchholz V, de la Roche J, Karst M, Haeseler G, Ahrens J. Lack of positive allosteric modulation of mutated alpha(1)S267I glycine receptors by cannabinoids. Naunyn Schmiedebergs Arch Pharmacol. 2010;381:477–482. doi: 10.1007/s00210-010-0506-9. [DOI] [PubMed] [Google Scholar]
- Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- Harris RA, Trudell JR, Mihic SJ. Ethanol's molecular targets. Sci Signal. 2008;1:re7. doi: 10.1126/scisignal.128re7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey RJ, Depner UB, Wässle H, Ahmadi S, Heindl C, Reinold H, Smart TG, Harvey K, Schütz B, Abo-Salem OM, Zimmer A, Poisbeau P, Welzl H, Wolfer DP, Betz H, Zeilhofer HU, Müller U. GlyR alpha3: an essential target for spinal PGE2-mediated inflammatory pain sensitization. Science. 2004;304:884–887. doi: 10.1126/science.1094925. [DOI] [PubMed] [Google Scholar]
- Hejazi N, Zhou C, Oz M, Sun H, Ye JH, Zhang L. Delta9-tetrahydrocannabinol and endogenous cannabinoid anandamide directly potentiate the function of glycine receptors. Mol Pharmacol. 2006;69:991–997. doi: 10.1124/mol.105.019174. [DOI] [PubMed] [Google Scholar]
- Hu XQ, Sun H, Peoples RW, Hong R, Zhang L. An interaction involving an arginine residue in the cytoplasmic domain of the 5-HT3A receptor contributes to receptor desensitization mechanism. J Biol Chem. 2006;281:21781–21788. doi: 10.1074/jbc.M600676200. [DOI] [PubMed] [Google Scholar]
- Legendre P. The glycinergic inhibitory synapse. Cell Mol Life Sci. 2001;58:760–793. doi: 10.1007/PL00000899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo IA, Harris RA. Sites of alcohol and volatile anesthetic action on glycine receptors. Int Rev Neurobiol. 2005;65:53–87. doi: 10.1016/S0074-7742(04)65003-3. [DOI] [PubMed] [Google Scholar]
- Lozovaya N, Yatsenko N, Beketov A, Tsintsadze T, Burnashev N. Glycine receptors in CNS neurons as a target for nonretrograde action of cannabinoids. J Neurosci. 2005;25:7499–7506. doi: 10.1523/JNEUROSCI.0977-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch JW. Molecular structure and function of the glycine receptor chloride channel. Physiol Rev. 2004;84:1051–1095. doi: 10.1152/physrev.00042.2003. [DOI] [PubMed] [Google Scholar]
- Lynch JW. Native glycine receptor subtypes and their physiological roles. Neuropharmacology. 2009;56:303–309. doi: 10.1016/j.neuropharm.2008.07.034. [DOI] [PubMed] [Google Scholar]
- Maksay G, Laube B, Betz H. Subunit-specific modulation of glycine receptors by neurosteroids. Neuropharmacology. 2001;41:369–376. doi: 10.1016/s0028-3908(01)00071-5. [DOI] [PubMed] [Google Scholar]
- Mascia MP, Mihic SJ, Valenzuela CF, Schofield PR, Harris RA. A single amino acid determines differences in ethanol actions on strychnine-sensitive glycine receptors. Mol Pharmacol. 1996;50:402–406. [PubMed] [Google Scholar]
- Oz M, Zhang L, Morales M. Endogenous cannabinoid, anandamide, acts as a noncompetitive inhibitor on 5-HT3 receptor-mediated responses in Xenopus oocytes. Synapse. 2002;46:150–156. doi: 10.1002/syn.10121. [DOI] [PubMed] [Google Scholar]
- Oz M, Zhang L, Ravindran A, Morales M, Lupica CR. Differential effects of endogenous and synthetic cannabinoids on alpha7-nicotinic acetylcholine receptor-mediated responses in Xenopus oocytes. J Pharmacol Exp Ther. 2004;310:1152–1160. doi: 10.1124/jpet.104.067751. [DOI] [PubMed] [Google Scholar]
- Perkins DI, Trudell JR, Crawford DK, Alkana RL, Davies DL. Targets for ethanol action and antagonism in loop 2 of the extracellular domain of glycine receptors. J Neurochem. 2008;106:1337–1349. doi: 10.1111/j.1471-4159.2008.05476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG. Cannabinoid receptor ligands: clinical and neuropharmacological considerations, relevant to future drug discovery and development. Expert Opin Investig Drugs. 2000;9:1553–1571. doi: 10.1517/13543784.9.7.1553. [DOI] [PubMed] [Google Scholar]
- Pertwee RG. Ligands that target cannabinoid receptors in the brain: from THC to anandamide and beyond. Addict Biol. 2008;13:147–159. doi: 10.1111/j.1369-1600.2008.00108.x. [DOI] [PubMed] [Google Scholar]
- Rácz I, Bilkei-Gorzo A, Markert A, Stamer F, Göthert M, Zimmer A. Anandamide effects on 5-HT(3) receptors in vivo. Eur J Pharmacol. 2008;596:98–101. doi: 10.1016/j.ejphar.2008.08.012. [DOI] [PubMed] [Google Scholar]
- Ross RA. Anandamide and vanilloid TRPV1 receptors. Br J Pharmacol. 2003;140:790–801. doi: 10.1038/sj.bjp.0705467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiang R, Ryan SG, Zhu YZ, Hahn AF, O'Connell P, Wasmuth JJ. Mutations in the alpha 1 subunit of the inhibitory glycine receptor cause the dominant neurologic disorder, hyperekplexia. Nat Genet. 1993;5:351–358. doi: 10.1038/ng1293-351. [DOI] [PubMed] [Google Scholar]
- Shiang R, Ryan SG, Zhu YZ, Fielder TJ, Allen RJ, Fryer A, Yamashita S, O'Connell P, Wasmuth JJ. Mutational analysis of familial and sporadic hyperekplexia. Ann Neurol. 1995;38:85–91. doi: 10.1002/ana.410380115. [DOI] [PubMed] [Google Scholar]
- Sigel E, Baur R, Rácz I, Marazzi J, Smart TG, Zimmer A, Gertsch J. The major central endocannabinoid directly acts at GABA(A) receptors. Proc Natl Acad Sci U S A. 2011;108:18150–18155. doi: 10.1073/pnas.1113444108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart D, Gunthorpe MJ, Jerman JC, Nasir S, Gray J, Muir AI, Chambers JK, Randall AD, Davis JB. The endogenous lipid anandamide is a full agonist at the human vanilloid receptor (hVR1) Br J Pharmacol. 2000;129:227–230. doi: 10.1038/sj.bjp.0703050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith FL, Fujimori K, Lowe J, Welch SP. Characterization of delta9-tetrahydrocannabinol and anandamide antinociception in nonarthritic and arthritic rats. Pharmacol Biochem Behav. 1998;60:183–191. doi: 10.1016/s0091-3057(97)00583-2. [DOI] [PubMed] [Google Scholar]
- Spivak CE, Lupica CR, Oz M. The endocannabinoid anandamide inhibits the function of alpha4beta2 nicotinic acetylcholine receptors. Mol Pharmacol. 2007;72:1024–1032. doi: 10.1124/mol.107.036939. [DOI] [PubMed] [Google Scholar]
- Tapia JC, Aguayo LG. Changes in the properties of developing glycine receptors in cultured mouse spinal neurons. Synapse. 1998;28:185–194. doi: 10.1002/(SICI)1098-2396(199803)28:3<185::AID-SYN1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Vivian JA, Kishioka S, Butelman ER, Broadbear J, Lee KO, Woods JH. Analgesic, respiratory and heart rate effects of cannabinoid and opioid agonists in rhesus monkeys: antagonist effects of SR 141716A. J Pharmacol Exp Ther. 1998;286:697–703. [PubMed] [Google Scholar]
- Wu X, Brooks BR. Self-guided Langevin dynamics simulation method. Chem Phys Lett. 2003;381:512–518. [Google Scholar]
- Wu X, Brooks BR. Toward canonical ensemble distribution from self-guided Langevin dynamics simulation. J Chem Phys. 2011;134:134108. doi: 10.1063/1.3574397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W, Hosoi M, Koo BN, Zhang L. Anandamide inhibition of 5-HT3A receptors varies with receptor density and desensitization. Mol Pharmacol. 2008;73:314–322. doi: 10.1124/mol.107.039149. [DOI] [PubMed] [Google Scholar]
- Xiong W, Cheng K, Cui T, Godlewski G, Rice KC, Xu Y, Zhang L. Cannabinoid potentiation of glycine receptors contributes to cannabis-induced analgesia. Nat Chem Biol. 2011;7:296–303. doi: 10.1038/nchembio.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Aubrey KR, Alroy I, Harvey RJ, Vandenberg RJ, Lynch JW. Subunit-specific modulation of glycine receptors by cannabinoids and N-arachidonyl-glycine. Biochem Pharmacol. 2008;76:1014–1023. doi: 10.1016/j.bcp.2008.07.037. [DOI] [PubMed] [Google Scholar]
- Yevenes GE, Zeilhofer HU. Molecular sites for the positive allosteric modulation of glycine receptors by endocannabinoids. PLoS One. 2011a;6:e23886. doi: 10.1371/journal.pone.0023886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yevenes GE, Zeilhofer HU. Allosteric modulation of glycine receptors. Br J Pharmacol. 2011b;164:224–236. doi: 10.1111/j.1476-5381.2011.01471.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yevenes GE, Moraga-Cid G, Avila A, Guzmán L, Figueroa M, Peoples RW, Aguayo LG. Molecular requirements for ethanol differential allosteric modulation of glycine receptors based on selective Gbetagamma modulation. J Biol Chem. 2010;285:30203–30213. doi: 10.1074/jbc.M110.134676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Xiong W. Modulation of the Cys-loop ligand-gated ion channels by fatty acid and cannabinoids. Vitam Horm. 2009;81:315–335. doi: 10.1016/S0083-6729(09)81012-1. [DOI] [PubMed] [Google Scholar]
- Zimmer A, Zimmer AM, Hohmann AG, Herkenham M, Bonner TI. Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc Natl Acad Sci U S A. 1999;96:5780–5785. doi: 10.1073/pnas.96.10.5780. [DOI] [PMC free article] [PubMed] [Google Scholar]