

SUMMARY
The retinoblastoma protein pRb is essential for regulating many cellular activities through its binding and inhibition of E2F transcription activators, and pRb inactivation leads to many cancers. pRb activity can be perturbed by viral oncoproteins including human papillomavirus (HPV) that share an LxCxE motif. Since there are no treatments for existing HPV infection leading to nearly all cervical cancers and other cancers to a lesser extent, we screened for compounds that inhibit the ability of HPV-E7 to disrupt pRb/E2F complexes. This lead to the identification of thiadiazolidinedione compounds that bind to pRb with mid-high nanomolar dissociation constants, are competitive with the binding of viral oncoproteins containing an LxCxE motif and are selectively cytotoxic in HPV positive cells alone and in mice. These inhibitors provide a promising scaffold for the development of therapies to treat HPV-mediated pathologies.
INTRODUCTION
The retinoblastoma protein (pRb) was the first protein identified whose mutational inactivation was associated with cancer, a childhood cancer of the eye (Schubert et al., 1994). pRb is now known to have altered activity in many other cancers including osteosarcomas, lung carcinomas and bladder carcinomas (Cordon-Cardo et al., 1997; Hensel et al., 1990; Kitchin and Ellsworth, 1974). pRb is also a target for inactivation by the viral oncoproteins E1a, E7 and T-antigen from adenovirus, human papillomavirus (HPV), and simian virus 40, respectively (Felsani et al., 2006). The normal function of pRb is to regulate the cell cycle, apoptosis and differentiation through its direct binding to and inhibition of the E2F family of transcription factors (Harbour and Dean, 2000; Stevaux and Dyson, 2002). When phosphorylated, pRb releases E2F proteins to transcribe genes necessary for the progression into the S-phase of the cell cycle, as well as for DNA replication (Harbour and Dean, 2000; Harbour et al., 1999; Stevaux and Dyson, 2002). The viral oncoproteins act by binding to hypophosphorylated pRb, disrupting pRb/E2F complexes and thereby leading to dysregulated entry into S-phase of the cell cycle and neoplasia (Ganguly and Parihar, 2009; Munger et al., 2001). HPV-E7 has also been implicated in the degradation of pRb (Boyer et al., 1996; Giarre et al., 2001; Gonzalez et al., 2001).
Each of the viral oncoproteins that inhibit pRb function employ a conserved LxCxE sequence for high affinity pRb binding although they each use other protein regions to contribute to the displacement of pRb/E2F complexes through distinct mechanisms (Felsani et al., 2006; Liu and Marmorstein, 2006). The A and B cyclin fold domains of pRb form the “pocket” region, which forms a groove that makes high affinity contacts to the transactivation domain of E2F (Xiao et al., 2003). The LxCxE motif from viral oncoproteins contribute to disruption of the pRb/E2F complexes by binding to the pRb B domain (Lee et al., 1998). While the A/B pocket of pRb is important for its biological activity, the C-terminal domain is also important for the formation of pRb-E2F complexes and is the target of other regions of the viral oncoproteins. The C-terminal domain of pRb has been shown to make contacts with the marked-box region of E2F, although with a lower affinity (Rubin et al., 2005). This domain of pRb is also subject to cell-cycle dependent posttranslational modifications, such as phosphorylation and acetylation, as well as the recruitment of cyclins/cyclin-dependent kinases (Adams et al., 1999).
Of the viruses that target pRb function, HPV has received considerable attention due to its role in human cancer. In particular, HPV is known to be the causative agent of a number of epithelial cancers, most notably cervical cancer, a leading cause of death for women worldwide (McLaughlin-Drubin and Munger, 2009). HPV infection has also been implicated to have a causative role in about 20% of head and neck cancers as well as several other cancers (Dufour et al., 2011; Sudhoff et al., 2011). There are over 200 HPV genotypes that have been recognized, and they fall under two general forms based on the pathology of the lesions that they cause, low-risk and high-risk, which cause benign tumors and which have the propensity to cause cancer, respectively (Burd, 2003). Two prophylactic vaccines are currently available, Gardasil and Cervarix, which help prevent against infection by the low risk HPV types 6 and 11 and high risk HPV types 16 and 18 (Harper, 2009). While these vaccines target HPV types that cause more than 90% of genital warts and cervical cancer, therapeutic treatments are still needed for those who have already been exposed to the virus.
Towards the development of HPV therapeutics a group of related small molecule compounds have been identified through high throughput screening that can disrupt the HPV E1-E2 interaction and prevent viral replication (White et al., 2011; Yoakim et al., 2003) and optimized to obtain compounds with low nanomolar IC50 values (Goudreau et al., 2007; Wang et al., 2004). Several inhibitors that target the HPV-E6 interaction with E6AP that is required for p53 degradation have also been developed including the Pitx2a protein inhibitor (Wei, 2005), intrabodies(Griffin et al., 2006) and alpha helical peptides (Butz et al., 2000; Liu et al., 2004), however all show modest activity. Ten small molecules inhibitors were also identified by Baleja et al. after pharmacophore development and limited in silico screening (Baleja et al., 2006), however only one compound proved to be active in cells and only at high concentrations.
Although these studies have not progressed to clinical trials, it demonstrates that it is possible to target HPV protein-protein interactions effectively with small molecules. HPV-E7 is a particularly attractive protein target since it is one of the cancer-causing oncoproteins from this virus and it has no human ortholog. To date, there are no known small molecule inhibitors that target HPV-E7.
Here we describe a high-throughput solution screen of about 88,000 compounds resulting in the identification and characterization of a family of small molecule thiadiazolidinedione compounds that we show inhibit the ability of HPV-E7 to disrupt pRb/E2F complexes. We also show that these inhibitors bind directly to pRb with dissociation constants in the mid-high nanomolar range, are competitive for pRb binding to other viral oncoproteins containing an LxCxE motif, and are selectively cytotoxic to cells transformed with high-risk forms of HPV alone and in mice. These inhibitors provide new tools to probe mechanisms involved in HPV transformed cells and may provide a promising chemical scaffold to develop novel therapies to treat HPV-mediated pathologies.
RESULTS
Identification of HPV-E7 inhibitors using a high-throughput solution screen
Approximately 88,000 compounds from several diverse small molecule libraries (Table 1) were screened to search for inhibitors that prevent E7-mediated displacement of E2F from pRb. The protein constructs that were employed include: 6xHis-HPV16-E7CR2-3 (residues 17–98) harboring conserved regions 2 and 3 and the LxCxE motif of HPV-E7 (Figure 1A), GST-pRbABC (residues 376–928) harboring the A/B pocket domain and C-terminal region of pRb and untagged E2FMB-TA (residues 243–437) containing the marked-box and transactivation domains of E2F that make pRb contact. 6xHis-HPV16-E7CR2-3 was modified to improve its solubility and reduce its tendency to aggregate by substituting two nonconserved cysteine residues in its CR3 domain to the corresponding residues found in low–risk HPV1A-E7 (Figure 1A). This mutated form of E7 was confirmed to be properly folded according to its elution profile on gel filtration (data not shown), and it exhibited the ability to bind specifically to pRb and dissociate pRb/E2F complexes, as expected (Figure S1). Furthermore, this E7 mutant was expected to bind pRb with comparable affinity to wild-type E7 since the mutated residues were not located in regions that were shown to mediate pRb binding. The assay used for screening employed an enzyme-linked immunosorbance assay (ELISA) as illustrated in Figure S2A. A GST-pRbABC/ E2FMB-TA complex was bound to a glutathione-coated 384-well microtiter plate and incubated with 6xHis-HPV16-E7CR2-3 in the presence of 1% DMSO (negative control) or 10 µM of compound dissolved in DMSO. Compounds that inhibit HPV-E7-mediated disruption of pRb/E2F complexes maintain E2F bound to the plate through pRb. Therefore, following plate washing, the amount of E2F remaining bound to the plate, as quantified by a bioassay, is correlated to the potency of the compound in inhibiting HPV-E7-mediated disruption of pRb/E2F complexes.
Table 1.
Chemical libraries employed and hits obtained for HPV-E7 inhibitor solution screen.
| Library | # Compounds | # Cherry Picks | Hit Percentage |
ELISA IC50 < 15.6µM |
|---|---|---|---|---|
| Spectrum | 2,000 | 11 | 0.55% | 3 |
| Maybridge HitFinder | 14,400 | 32 | 0.22% | 13 |
| OPS, set 9 | 7,952 | 50 | 0.63% | 16 |
| OPS, sets 3–6, 10, 12–14 | 63,587 | 271 | 0.43% | 88 |
| Total | 87,939 | 364 | 0.41% | 120 |
See also Figure S2.
Figure 1. Characterization of small molecule HPV-E7 inhibitors.

(A) Sequence alignment of E7 from HPV 1A, HPV 16, and HPV 18 used in the experiments. The two residues in red in HPV 16 E7 were mutated to the corresponding residues in HPV 1A E7 for use in the biochemical experiments. (B) IC50 curves for disruption of pRb/E2F complexes by E7 in the presence of a family of thiadiazolidinedione compounds. IC50 curves were generated using the ELISA-based assay described in the Methods. Ten-fold dilutions of inhibitor, starting at 100µM were added to a mixture containing GST-pRbABC/E2FMB-TA and 6xHis-HPV16-E7CR2-3. The amount of E2FMB-TA remaining was determined by adding a primary antibody specific for E2F1. (C) IC50 curves for inhibitor disruption of HPV-E7/pRb complexes. IC50 curves were generated using the ELISA assay with ten-fold dilutions of inhibitor, starting at 100µM were added to a mixture containing GST-pRbABC and 6xHis-HPV16E7CR2-3. The amount of E7 remaining was determined by adding a primary anti-His antibody. (D) Effect of inhibitors on HPV-E7/pRb pulldown. Different concentrations of inhibitor (compound 478166 is shown) were added and the amount of GST-E7FL remaining bound to pRb was probed by using an anti-GST antibody (bottom panel). The top panel shows the loading control of His-pRbABC in each lane. See also Figure S1.
The initial screen resulted in the identification of 364 small molecule HPV-E7 inhibitors. Using liquid stock from the libraries used for screening, we re-tested the activity and potency of all 364 candidate inhibitors in the primary screening assay. These re-test experiments confirmed activity for 120 of the 364 with IC50 values of 15.6 µM or lower. The remaining 244 compounds either did not show reproducible inhibition, or were not sufficiently potent and were discarded from further analyses (Table 1). The 120 confirmed actives were then tested in secondary assays as described below to identify those with selective pharmacological activity in cells. A summary of the process for the identification of confirmed screening hits is shown in Figure S2B. Additional information for interpreting and repeating the screen is provided in Table S1.
A family of thiadiazolidinedione compounds are selective for HPV 16-transformed cells
To reduce the number of compounds from the primary screen for further characterization, the 120 confirmed hits with IC50 values < 16µM from the primary screen were assessed for their ability to be cytotoxic or to inhibit proliferation of cervical cancer cells either transformed with HPV16 (SiHa) or not (C-33A) (Yee et al., 1985). The metabolic viability of cells was measured using an MTS assay (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium). Compounds were tested at a concentration range of 25µM to 100nM. Staurosporine, a non-specific kinase inhibitor, was used as a positive control because it was expected to be toxic in all cells (Ruegg and Burgess, 1989). The 120 confirmed hits were incubated with cells for 48 hours prior to the addition of MTS reagent. The absorbance at 490nm was determined within 3 hours of incubation with MTS reagent. Out of the 120 compounds tested, 25 were either selectively cytotoxic or selectively prevented proliferation of SiHa cells (HPV 16 positive) and not C-33A (HPV negative) cells at concentrations at or below 6µM (data not shown).
Of the 25 compounds that were selectively cytotoxic or prevented proliferation of SiHa cells, seven shared a similar thiadiazolidinedione ring scaffold attached to a phenyl ring with various substitution patterns attached (Table 2) and had IC50 values in the ELISA assay that ranged between 0.34 – 7.6 µM (Figure 1B and Table 2). The remaining compounds were eliminated from further studies due to a variety of reasons including poor reproducibility in activity, significant impurities, and/or structural features suggestive of reactivity, aggregation or other non-specific mechanisms (Table S2). Consequently, our studies focused on characterizing the mechanism of inhibition for the seven thiadiazolidinedione compounds listed in Table 2. The purity and integrity of these seven compounds, ordered as powders, was confirmed by LC/MS studies and 1H NMR of a representative compound (Table S3).
Table 2.
IC50 values for compounds to inhibit HPV-E7-mediated disruption of pRb/E2F complexes and for disrupting pRb/viral oncoprotein complexes
 |
16E7 (500nM) (µM) |
1AE7 (500nM) (µM) |
E1A (100nM) (µM) |
KD (µM) (to pRb) |
Structure | ID |
|---|---|---|---|---|---|---|
| pRb/E2F | 7.6 ± 1.2 | 10.6 ± 1.3 | 2.8 ± 2.2 | 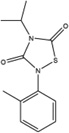 |
LCGC12 478081 | |
| pRb | 11.2 ± 1.3 | 7.9 ± 2.1 | 5.0 ± 1.8 | 0.165 ± 0.052 | ||
| pRb/E2F | 2.2 ± 1.6 | 3.5 ± 1.6 | 0.64 ± 2.3 |  |
LCGC12 478165 | |
| pRb | 0.57 ± 1.2 | 3.0 ± 2.3 | 2.6 ± 1.3 | 0.104 ± 0.025 | ||
| pRb/E2F | 1.9 ± 1.3 | 4.5 ± 1.7 | 0.24 ± 2.0 |  |
LCGC12 478166 | |
| pRb | 0.50 ± 1.5 | 3.4 ± 2.0 | 1.0 ± 2.1 | 0.106 ± 0.034 | ||
| pRb/E2F | 3.2 ± 1.3 | 5.5 ± 1.7 | 1.3 ± 2.2 |  |
LCGC12 478168 | |
| pRb | 4.5 ± 1.5 | 4.7 ± 2.1 | 3.8 ± 1.5 | 0.187 ± 0.022 | ||
| pRb/E2F | 4.6 ± 1.3 | 5.5 ± 1.5 | 1.1 ± 2.5 | 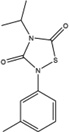 |
LCGC12 478337 | |
| pRb | 3.2 ± 1.3 | 5.5 ± 2.7 | 3.5 ± 1.7 | 0.210 ± 0.051 | ||
| pRb/E2F | 2.3 ± 1.6 | 3.3 ± 1.6 | 1.7 ± 2.7 |  |
LCGC12 478419 | |
| pRb | 0.40 ± 1.4 | 1.3 ± 1.3 | 7.7 ± 1.7 | 0.381 ± 0.031 | ||
| pRb/E2F | 0.34 ± 1.9 | 3.5 ± 1.7 | 3.2 ± 2.7 | 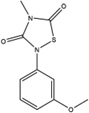 |
LCGC10 478726 | |
| pRb | 0.29 ± 1.7 | 4.0 ± 2.5 | 2.8 ± 2.1 | 0.815 ± 0.070 |
all values indicated are from experiments that were done in triplicate.
See also Tables S1, S3 and S4.
Some structure-activity relationship (SAR) information can be extracted for the seven thiadiazolidinedione compounds. In most of the compounds, there is a phenyl group with various substituents attached at the G2 position (Table 2). Interestingly, compound 478419 has a phenyl group at the G1 position instead of G2. Given that the compounds are pseudo symmetric, it is possible that the phenyl group at the G1 position of compound 478419 may compensate for the phenyl group at the G2 position of the other compounds. This suggests that there may be at least two orientations for the thiadiazolidinedione compounds that allow the phenyl ring to occupy the same binding pocket of its protein target and inhibit HPV-E7. A number of other structural analogs were also tested for inhibition in the ELISA assay (Table S4). These analogs had the sulfur in the heterocycle ring changed to either a carbon, or oxygen, and showed no activity (Table 2). This data suggests that the S heterocycle is necessary for activity, possibly because the larger sulfur atom distorts the ring in such a way to facilitate hydrogen bonding by the oxygens to its protein target or that the S heterocycle has some reactivity that supports activity that the C or O analogs do not.
The HPV-E7 inhibitors function to disrupt HPV-E7 interaction with pRb
Since HPV-E7 interacts with both pRb and E2F for disruption of the pRb/E2F complex, we sought to confirm that the seven active compounds inhibited HPV-E7 activity by directly disrupting HPV-E7 interactions with pRb (Liu et al., 2006; Munger et al., 2001). For these experiments, we modified the ELISA assay to measure the amount of 6xHis-HPV16-E7CR2-3 remaining bound to pRb. We found that an increase in compound concentration led to a displacement of 6xHis-HPV16-E7CR2-3 from GST-pRbABC, suggesting that the compounds prevent the interaction between these two proteins (Figure 1C). To eliminate potential artifacts from this assay format, we tested the ability of the HPV-E7 inhibitors to disrupt HPV-E7/pRb interaction by performing pull-downs on Ni-NTA beads using His-pRbABC and GST-tagged full length 16E7 (GST-16E7FL) (Figure 1D). Consistent with the ELISA assay, the pull-down assay shows that an increase in compound concentration leads to a displacement of GST-E7FL from His-pRbABC. The IC50 values for the amount of respective compound required for 6xHis-HPV16-E7CR2-3 displacement from GST-pRbABC, as determined by the ELISA assay, were within ten-fold of the corresponding IC50 values of E2F displacement from GST-pRbABC in the presence of 6xHis-HPV16-E7CR2-3 (Table 2). These data are consistent with the observation that preventing HPV-E7 binding to pRb inhibits its ability to displace E2F from pRb (Liu et al., 2006; Munger et al., 2001).
The HPV-E7 inhibitors function by binding to pRb through the LxCxE binding motif of viral oncoproteins
Since HPV-E7 mediates high affinity pRb binding through the association of its LxCxE motif in its CR2 domain to the B domain of pRb, we hypothesized that the HPV-E7 inhibitors might bind to either the LxCxE motif of HPV-E7 or the B-domain of pRb. To distinguish between these possibilities, we assayed the ability of the thiadiazolidinedione compounds to inhibit the ability of other LxCxE containing viral oncoproteins from disrupting E2F/pRb complexes: HPV-E7 from a low risk HPV form (type 1A) and Adenovirus E1A proteins. E1A was used as a control because it does not dimerize in solution, unlike E7, and has also been shown to displace E2F via a different mechanism (Felsani et al., 2006). For these studies, we modified our ELISA assay to measure disruption of E2F/pRb complexes by substituting 6xHis-HPV1AE7CR2-3, and 6xHis-Ad5E1ACR1-3. As illustrated in Figure 2A and Table 2, the thiadiazolidinedione compounds show similar levels of inhibition as they did in the presence of 6xHis-HPV16E7CR2-3. The ability of the compounds to prevent an interaction between either 6xHis-HPV1AE7CR2-3 or 6xHis-Ad5E1ACR1-3 with GST-pRbABC was also demonstrated (Figure 2B). The IC50 values from these experiments ranged from 0.2–11.2µM, comparable to the IC50 values for compound inhibition of HPV-16E7 mediated inhibition of E2F/pRb complexes (Table 2). These data suggest that the thiadiazolidinedione inhibitors disrupt the interaction between the pRb B domain and the LxCxE motif of the viral oncoproteins.
Figure 2. In vitro characterization of thiadiazolidinedione inhibitors against viral oncoproteins.

(A) Ability of inhibitors to prevent LxCxE containing viral oncoproteins from disrupting E2F/pRb complexes. Ten-fold dilutions of inhibitor (compound 478166 is shown) were added to GST-pRbABC/6xHis-HPV1AE7CR2-3 or GST-pRbABC/6xHis-Ad5E1ACR2-3. (B) Ability of inhibitors to disrupt complexes between pRb and LxCxE containing viral oncoproteins. Compound 478166 was used for the experiment shown. (C) Binding of inhibitors to pRb as measured by isothermal titration calorimetry. The curve fit for pRb binding to compound 478081 reveals 1:1 binding with a KD of 165nM, and dH of -1237 cal/mol. (D) HPV-E7 binding to pRb in the presence of increasing concentrations of inhibitor. The ELISA-based assay was used to determine the mechanism of binding of the small molecules to pRb. Five-fold dilutions of inhibitor were added to pRb and the amount of E7 that was able to bind to pRb was determined. The calculated apparent KD values for pRB-E7 in the presence of 0.025, 0.25, 0.5 and 5.0 µM of inhibitor 478165 (KD for pRb of 104 nM) were 140 ± 22, 313 ± 21, 304 ± 76 and 764 ± 72 nM, respectively. See also Figure S3.
Because the LxCxE motif from the viral oncoproteins is likely to be extended and flexible when not in complex with partner proteins, we postulated that the small molecule thiadiazolidinedione inhibitors interact with the structured pRb B domain (Lee et al., 1998). To test this hypothesis, we assayed the ability of the HPV-E7 inhibitors to bind directly to a truncated pRb protein construct containing the A and B domains of the pRb pocket (pRbAB) using isothermal titration calorimetry (ITC). The resulting integrated heat-flow spikes confirmed direct binding of inhibitors to pRb with 1:1 stoichiometry and affinities in the sub-micromolar range (Figure 2C and Figure S3A). The dissociation constants obtained range from 100 – 800 nM and are provided in Table 2. The reported KD for the LxCxE E7 peptide binding to pRb is approximately 110nM (Lee et al., 1998) and is comparable to the KD values obtained for pRb binding to the inhibitors. To confirm that inhibitor binding is reversible, one of the pRb/inhibitor complexes (pRb with compound 478166) was dialyzed overnight and ITC was repeated. As before, a binding curve was obtained yielding a similar dissociation constant and stoichiometry, indicating that the inhibitor was still able to interact with pRb in a reversible fashion (Figure S3B). To eliminate the possibility of non-specific binding of the thiadiazolidinediones, ITC was carried out with compound 478166 and 6xHis-HPV16-E7CR2-3 (Figure S3C). The observed heats reveals negligible binding, further demonstrating that the compounds are not binding to pRb non-specifically.
To determine if the inhibitors are competitive with HPV-E7 for pRb binding or work through an allosteric mechanism, we employed the ELISA assay to measure the ability of HPV-E7 to displace the compound from pRb as a function of inhibitor concentration. As shown in Figure 2D, the binding curves of 6xHis-HPV16-E7CR2-3 to GST-pRbABC in the presence of varying concentrations of inhibitor, above and below the dissociation constant of pRb for inhibitor, shows a dependence on the concentration of inhibitor used, where increasing inhibitor concentration is correlated with a rightward shift (higher apparent value) in the Kd values for HPV-E7 binding to pRb. This data suggests that inhibitor and HPV16-E7 bind competitively to pRb. Taking this result together with the observation that these inhibitors are also able to disrupt pRb complexes with HPV1A-E7 and Ad5-E1a (Figure 2A) suggests that these thiadiazolidinedione inhibitors also bind pRb competitively with other LxCxE containing oncoproteins.
HPV-E7 inhibitors selectively cause apoptosis in HPV-transformed cells
Since the seven thiadiazolidinediones listed in Table 2 were cytotoxic or prevented proliferation of SiHa cells due to their role in inactivating pRb, which is mutated in C-33A cells, they were tested in additional cell lines: TC-1, a mouse epithelial line co-transformed with HPV 16 E6/E7 and c-Ha-Ras, HeLa, a human cell line transformed with HPV 18 and HCT116, a human HPV negative colorectal carcinoma cell line containing an intact retinoblastoma gene (DeFilippis et al., 2003; Scheffner et al., 1991; Yee et al., 1985). The levels of cell viability after incubation with compound were determined using the MTS assay as previously described. This time, a concentration range of 100 µM to 3 µM of compound was tested so that the cellular IC50 values could be extracted for these seven compounds, and for the ten inactive analogs. As shown in Figure 3, the thiadiazolidinedione compounds had the greatest effect on SiHa cells, followed by TC-1 cells, and to a smaller extent, HeLa cells. The smallest effect was seen in HCT 116 and C-33A cells. While the inhibitors were cytotoxic in all cell lines at 100 µM, they were selectively cytotoxic in HPV-positive cells at the lower compound concentrations. In general, the IC50 values of the thiadiazolidinedione compounds in SiHa cells varied from between 6.25 µM and 12.5 µM to between 25 µM and 50 µM. The IC50 values in TC-1 and HeLa cells were slightly higher and varied from between 12.5 µM and 25 µM to between 50 µM and 100 µM. The IC50 values in HCT 116 and C-33A cells were all greater than 25 µM. Importantly, the inactive analogs did not demonstrate any effect in any of the cell lines tested (a representative example is shown in Figure 3). Taken together, this data suggests that the seven thiadiazolidinedione HPV-E7inhibitors identified in the primary HTS are either selectively cytotoxic or selectively prevent proliferation of HPV transformed cervical cancer cell lines, with a greater effect in cell lines transformed with HPV 16.
Figure 3. Cellular toxicity of thiadiazolidinedione compounds.

Four different cervical cancer cells lines: SiHa, TC-1, HeLa and C-33A and one non-cervical cancer cell line, HCT116, were employed for these studies. 2-fold compound dilutions starting at 100 µM down to 3.125 µM for the thiadiazolidinediones and starting at 2 µM to 4 nM for staurosporine were incubated for 48 hours with cells before the addition of MTS reagent. After 1–2 hours of incubation with reagent, the absorbance at 490 nm was determined. The percent growth was determined by dividing by the growth in the presence of DMSO control. See also Table S2.
Given that the thiadiazolidinedione inhibitors bind to pRb, a critical regulator of the cell cycle, we asked whether they perturb the cell cycle to prevent proliferation or whether they induce apoptosis in cells transformed with HPV. To carry out these studies, we employed SiHa cells (transformed with HPV 16) since the inhibitors were most effective in this cell line. Cells were treated with either DMSO or 10 µM of two representative thiadiazolidinediones: compounds 478166 and 478168, an inactive analog (compound 44234), or 2 µM of staurosporine, for 48 hours. DNA content was determined by propidium iodine staining and analysis by flow cytometry. In agreement with our biochemical results and the MTS cell viability assay, compounds 478166, 478168, and staurosporine most drastically affected SiHa cells whereas the inactive analog had no effect (Figure 4 and Figure S4). The thiadiazolidinedione inhibitors caused an increase of apoptotic SiHa cells (6.5% and 15.2% of cells were apoptotic when treated with the thiadiazolidinediones 478166 and 478168, respectively, compared to ~1% apoptotic cells that were treated with DMSO or the inactive analog) as did the non-specific kinase inhibitor staurosporine (34.3% of cells were apoptotic) (Figure 4). These results are consistent with the MTS data and in vitro data, together supporting the interpretation that the thiadiazolidinedione inhibitors antagonize the ability of HPV-E7 to maintain the viability of the HPV transformed cells. These results are also consistent with work by others that E7 knockdown can lead to apoptosis in HPV-positive cells (Jiang and Milner, 2002; Sima et al., 2008).
Figure 4. Effects on the cell cycle and apoptosis by the thiadiazolidinediones 478166 and 478168, an inactive analog, and staurosporine.

To determine any effect on cell cycle or apoptosis, SiHa cells were treated with DMSO or 10 µM of compounds 478166, 478168, 44234, or 2 µM staurosporine for 48 hours. After 48 hours, cells were harvested, stained with propidium iodide, and DNA content was analyzed by flow cytometry. The percent of cells in G0/G1, G2/M, S, or apoptotic were determined by the areas under the curves represented by B, C, D, and E, respectively.
A representative compound can reduce tumor size in vivo
Since the thiadiazolidinedione inhibitors exhibited apoptotic activity in cells, we wanted to determine whether or not they would demonstrate anti-tumor activity in vivo. A transplantable tumor model was employed in which TC-1 cells were injected subcutaneously into NOD-SCID mice. After five days, treatment was initiated with compound 478166 (n=6) or vehicle only (DMSO) (n=6) by intraperitoneal injection and repeated daily for a total of 14 days. The tumors were measured once every two days. At the conclusion of treatment, a significant reduction in tumor volumes was observed for mice treated with compound compared to vehicle only (Figure 5). After 14 days, the average tumor volume of mice treated with DMSO was 3950mm3, whereas the average tumor volume of mice treated with drug was 2270mm3 (p < 0.02). Taken together with our results from the cell-based experiments, it appears that the thiadiazolidinedione inhibitor can reduce tumor volume in vivo, with no deleterious effects observed otherwise on animal health.
Figure 5. The anti tumor effect of thiadiazolidinedione compound 478166 in vivo.

A tumor model was constructed by innoculating 2.0 × 105 TC-1 cells into the right flank of 12 NOD SCID female mice. Treatment was started 5 days post-injection; the mice were treated once a day for 14 days, with IP injections of DMSO or compound 478166 at doses of 10mg/kg. Tumor sizes were measured every two days.
DISCUSSION
We have described the identification and characterization of structurally similar thiadiazolidinedione compounds that inhibit the interaction between the LxCxE motif of viral oncoproteins and pRb in a competitive manner with sub-micromolar dissociation constants. The identification of these inhibitors is an important finding given that there are no known inhibitors that specifically block the interaction of pRb with viral oncoproteins. Interestingly, other small molecule compounds that show structural similarity to these thiadiazolidinediones have been implicated in possible treatments against neurodegenerative disorders by targeting glycogen synthase kinase-3 (GSK-3) or the peroxisome proliferator-activated receptor γ (PPARγ) (Luna-Medina et al., 2005; Luna-Medina et al., 2007; Martinex, 2006; Martinez et al., 2005; Martinez et al., 2002; Rosa et al., 2008). The fact that these inhibitors appear to prevent oncoproteins from binding to the LxCxE binding site on pRb suggests that these types of inhibitors provide another route for therapeutics not only against cervical cancer, but also for other diseases caused by viral oncoproteins containing the LxCxE motif.
The B domain of the pRb pocket domain harboring the LxCxE binding site is also the site of interaction with cellular proteins, such as histone deacetylases, cyclin D1, chromatin remodeling enzyme BRG1, and other proteins (Dahiya et al., 2000; Rosa et al., 2008; Singh et al., 2005). However, our studies in cells and in mice show that these inhibitors are not overtly cytotoxic in HPV-negative cells, suggesting that the HPV-E7 inhibitors do not perturb these endogenous interactions, at least to the same extent. The tumors formed by TC-1 cells in mice showed a significant reduction in volume when treated with thiadiazolidinedione 478166, without any noticeable effects on their normal cells, as indicated by a lack of change in animal behavior, implying a potential therapeutic for HPV16-related neoplasms. Furthermore, the cell-based studies reveal that the HPV-E7 inhibitors are more toxic in SiHa cells than HeLa cells. Taken together with our in vitro data showing that the compounds bind competitively with E7 to pRb, this suggests that the compounds are either more effective at disrupting the interaction between pRb and HPV16 E7 than pRb and HPV18 E7 or that the levels of E7 and or pRb are different enough in these cell lines to result in different toxicities. It is also possible that HeLa cell viability is not completely dependent on the HPV-E7 oncoprotein due to additional genetic and epigenetic changes in the tumor genome. Others have shown that the levels of pRb do in fact differ across cervical cancer cell lines, and that there is a difference in the level of pRb phosphorylation, with a greater level of hypophosphorylated pRb in SiHa cells (Scheffner et al., 1991). This observation is consistent with the level of toxicity that we observe in the MTS assay and by cell-cycle analysis, which suggests that the thiadiazolidinedione inhibitors may bind more avidly to hypophosphorylated pRb to prevent the interaction with E7. Furthermore, the increase in apoptosis in SiHa cells upon treatment with the thiadiazolidinedione inhibitors suggests that the inhibitors are antagonizing the ability of E7 to maintain the viability of the HPV-positive cell lines.
While there are reports that E7 has the ability to degrade pRb (Boyer et al., 1996; Giarre et al., 2001; Gonzalez et al., 2001), there are other reports that siRNA or shRNA against E7 results in a de-phosphorylation of pRb, and not an increase in overall pRb levels (Jiang and Milner, 2002; Sima et al., 2008). The de-phosphorylation of pRb by E7 was demonstrated in SiHa and CaSki cells, both of which are transformed with HPV 16. It is possible that the effect on pRb is cell-line dependent, as the former studies (Boyer et al., 1996; Giarre et al., 2001; Gonzalez et al., 2001) were done using different cell lines. We probed for pRb in SiHa, HeLa, TC-1, and HCT 116 cells that were incubated for 48 hours with concentrations of compound 478166 as high as 25 µM and did not see any change in the levels of pRb or in its phosphorylation state (data not shown). pRb was also probed in the mouse tumors that were treated with compound versus those that were not, and again no change in pRb levels or in its phosphorylation state could be observed (data not shown). It is possible that these compounds work differently from the siRNA and shRNA experiments in that they do not cause a detectable change in the levels of pRb or its phosphorylation state in these cell lines.
Our results from ITC confirm that the thiadiazolidinedione inhibitors bind directly to pRb with submicromolar affinity providing a route for structure-based-drug design of more potent and selective HPV inhibitors. The crystal structure of the pocket domain of pRb has been determined and may prove useful for co-crystallization of these small molecules with pRbAB (Balog et al.). Co-crystallization studies of these small molecules with pRbAB may provide further insight to their mode of interaction and guide further optimization. Since HPV mediates cell transformation through the action of two viral oncoproteins, E6 and E7, where E6 targets the p53 tumor suppressor for degradation, it might be particularly advantageous to combine these thiadiazolidinedione inhibitors with inhibitors that prevent E6-mediated p53 degradation to develop a particularly effective therapeutic strategy to treat HPV-mediated pathologies.
SIGNIFICANCE
The retinoblastoma protein, pRb, is an important regulator of cells and can cause neoplastic lesions when inactivated by mutations or by viral oncoproteins. Its inactivation by viral oncoproteins makes it a desirable drug target. In this study, we have identified the first class of small molecule inhibitors that competitively inhibit the interaction of LxCxE motif containing viral oncoproteins with pRb. We show, in vitro, that these thiadiazolidinedione inhibitors bind to pRb and prevent one of the main transforming abilities of these oncoproteins: the premature disruption of the inhibitory pRb/E2F complex. We also employ cell-based and animal studies to demonstrate that these inhibitors exhibit selective cytotoxicity in HPV positive cells. Little or no effect was seen in cancer cells not transformed with HPV. Our in vitro, cell-based-studies, and in vivo results in mice indicate that these thiadiazolidinedione inhibitors may provide a therapeutic strategy for cancers caused by viruses such as HPV.
EXPERIMENTAL PROCEDURES
Expression and purification of proteins
The DNA encoding HPV16-E7CR2-3 (residues 17–98), HPV1A-E7CR2-3 (residues 16–93) and Ad5-E1ACR1-3 (residues 36–189) were cloned into the pRSET vector, containing an N-terminal 6x-histidine tag. HPV-E7 and Ad5-E1A were expressed in E. coli BL21(DE3) cells overnight at 25 °C and 18 °C, respectively. Cells were lysed by sonication in a buffer containing 20mM Tris, 7.5, 500mM NaCl, 35mM imidazole, 10µM Zn(OAc)2, 10mM BME and 1x PMSF. The cell lysate was centrifuged at 18,000 RPM and the resulting supernatant was loaded onto a Ni-NTA column pre-equilibrated with 20mM Tris, 7.5, 500mM NaCl, 35mM imidazole, 10µM Zn(OAc)2, and 10mM BME. The column was washed and the bound protein was eluted using an imidazole gradient from 35mM to 250mM. The proteins were further purified using size exclusion chromatography on a superdex 200 analytical column (GE Healthcare Life Sciences) in a buffer containing 20mM Tris, 7.5, 150mM NaCl, and 10mM BME.
DNA encoding pRbABC (residues 376–928) was cloned into the pFAST-Bac vector, containing an N-terminal GST tag. Protein was expressed in Sf9 cells for 48 hours before harvesting. The protein was purified as described by the manufacturer (Novagen). The plasmid pGex6P-1-E2F1, encoding the marked-box and transactivation domain of E2F1 (residues 243–437) with an N-terminal GST tag, was provided by Dr. Steven Gamblin (MRC, Mill Hill, UK). GST- E2F1MB-TA was expressed in E. coli BL21(DE3) CodonPlus RIL cells (Novagen) for 5–6 hours at 30°C and purified as described elsewhere (Liu et al., 2006). The GST tag was removed using PreScission Protease (GE Healthcare Life Sciences) as described elsewhere to yield an untagged E2F1MB-TA for assay purposes (Liu et al., 2006).
For pull-down studies, GST-tagged full-length HPV-E7 was cloned into the pGEX-4T-1 vector, expressed in E. coli BL21(DE3) cells, and purified as described by the manufacturer (Novagen). 6xHis-pRbABC (residues 376–928) was cloned into the pRSET vector, expressed and purified as described above for the 6xHis-tagged proteins, except that Zn(OAc)2 was excluded from the buffers.
For isothermal titration calorimetry studies, untagged pRbAB (372–787 with the linker from 590–635 removed) was prepared as described elsewhere (Xiao et al., 2003).
Compound libraries
2000 compounds comprising the Spectrum Collection from MicroSource Discovery Systems (Gaylordsville, CT), Inc were tested at a final concentration of 8.3µM. A library of 14,400 chemically diverse compounds from Maybridge HitFinder (Cambridge, UK) were tested at a final concentration of 12.5µM. A third set of compounds, comprising 71,539 small molecules, from the orthogonally pooled screening (OPS) libraries, provided by the Lankenau Chemical Genomics Center (Wynnewood, PA) were tested at a final concentration 6.25µM to 12.5µM. The HitFinder and OPS libraries were orthogonally compressed to contain 5 or 10 compounds per well, respectively.
ELISA assays
IC50 values for inhibition of E2F displacement from pRb by E7 + inhibitors were measured using the same ELISA-based assay as described for the high-throughput screen (supplemental methods), except that the assay was performed manually in 96-well format with all volumes doubled. All compounds were solubilized to 50mM in DMSO and diluted for use in the ELISA-based assay at a final DMSO concentration of less than 5%. The concentrations of the compounds in the IC50 experiment spanned the range of enzyme activity from no inhibition to complete inhibition. To test E1A-pRb and E7-pRb binding, the assay was modified so that GST-pRbABC alone was added to HPV-E7CR2-3 + compound/DMSO, or Ad5-E1ACR1-3 + compound/DMSO. Mouse monoclonal anti-His antibody (Fisher) (1:10,000) and mouse monoclonal Ad5-E1A antibody (Abcam) (1:10,000) were used to detect how much His-E7CR2-3 and E1ACR1-3 remained bound to GST-pRbABC on the plate, respectively. All other steps remained unchanged. To test the mode of inhibition by the inhibitors, each compound was first incubated with pRb for 30–60 min. Different concentrations of HPV- E7CR2-3, ranging from 50µM down to 0.05µM were added to the GST-pRbABC + compound mixture and allowed to incubate for 30–60 min. The reaction mixture was then transferred to a glutathione-coated plate, and shaken for 15–20 min. Mouse monoclonal anti-His antibody (Fisher) (1:10,000) was used to detect how much HPV-E7CR2-3 remained bound to GST-pRbABC on the plate. Three independent IC50 measurements were performed for each compound and the average and standard deviation values are reported. All data was imported into the GraphPad Software (Prism) for IC50 or KD determination. To calculate the IC50 or KD values, the dose-response curves were fit to one-site (Hill slope = 1) sigmoidal-dose-response curves. The error bars were obtained from the standard errors generated by the GraphPad software.
Pull-Down assays
10µg His-tagged protein pRbABC was incubated with 10µl Ni-NTA beads (Fisher) in a buffer containing 20mM Tris, 7.5, 150mM NaCl, 35mM Imidazole and 0.05% Tween20 for 15 min. Then, inhibitor and an equimolar amount of GST-HPV16-E7FL to pRb were added and allowed to incubate at 4° C for 1 hr with gentle agitation. The beads were then washed three times with 1mL buffer (20mM Tris, 7.5, 150mM NaCl, 35mM Imidazole and 0.05% Tween20) and subjected to SDS-page analysis. The samples were transferred to PVDF membrane to be visualized by western blotting. Anti-GST mouse monoclonal antibodies (1:2000) (Calbiochem) and anti-His mouse monoclonal antibodies (1:5000) (Fisher) were used. Bands were visualized by chemiluminescence (Pierce) and exposure to film (Kodak).
Isothermal titration calorimetry
ITC was done using a MicroCal VP-ITC isothermal titration calorimeter (MicroCal, Inc). Proteins were dialyzed against a buffer containing 20mM Hepes, 7.5, 150mM sodium chloride and 0.1mM Tris carboxy ethyl phosphene prior to analysis. 8–12µL injections of 750–1500µM compound (final DMSO concentration of 1.5 %) were titrated into 50–150µM pRbAB (containing the same percentage of DMSO) pre-equilibrated to 22 °C. After subtraction of dilution heats, calorimetric data were analyzed with the MicroCal ORIGIN V5.0 (MicroCal Software, Northampton, MA). Error values obtained from the MicroCal ORIGIN V5.0 software were averaged and reported.
Cell culture
C-33A and SiHa cell lines were purchased from ATCC and grown in 1x minimal eagle’s media (MEM, Cellgro) supplemented with 10% fetal bovine serum (Hyclone), 10ug/ml penicillin-streptomycin (Cellgro), 2mM L-glutamine (Cellgro), 1mM sodium pyruvate (Cellgro), and 100uM non-essential amino acids (Gibco). HeLa, and HCT116 cell lines were generous gifts from the laboratories of Susan Janicki, and Meenhard Herlyn, respectively, and maintained in the same way.
MTS cell proliferation assay
Cultured cell lines were seeded in 384-well, clear, tissue culture plates (NUNC) at 10,000, 1,000, 1,000, 1000, and 2,000 cells/well for C-33A, SiHa, HeLa, TC-1, or HCT116 cells, respectively. The next day, compound dissolved in a final DMSO concentration of 0.5% was added to each well and incubated for 48 hr. Cell viability was then monitored by addition of 8µL of MTS reagent (Promega) and measurement at A490 using an Envision multilabel plate reader within 3 hr of MTS addition.
Flow cytometry
Cultured cell lines were seeded in 60mm tissue culture dishes (Falcon) at 1×105 cells/well. The next day, 10µM compound or DMSO were added and incubated for 48 hr. Cells were then trypsinized, washed with 1.0 mL phosphate-buffered saline (PBS), and fixed in 80% ethanol. Fixed cells were spun at 500g for 5 min, and washed with PBS. Cells were stained with 250µL propidium iodide (PI), which was prepared by adding 100µL 2mg/ml PI (Sigma) and 10 µg RNase A (Sigma) into 10mL PBS. Cells were then analyzed at the Wistar Institute Flow Cytometry Core Facility.
Mouse Studies
A tumor model was constructed by innoculating 2.0 × 105 TC-1 cells into the right flank of 12 NOD SCID female mice (Jackson Laboratory, Bar Harbor, ME). Treatment was started 5 days post-injection, as tumors became palpable. The mice were treated once a day for 14 days, with intraperitoneal injections of DMSO (0.1%) or compound 478166 at doses of 10mg/kg. Tumor sizes were measured every two days with calipers and tumor volume, V, (in mm3) was calculated using “V = l×w2 × π/6.” At the end of the experiment, all mice were sacrificed and the weights of the detached tumors were measured. The experiments were performed twice with similar results. Statistical analysis was done using the paired Student's t test. Errors were obtained by calculating the standard deviations from all the mice in each set. All animal experiments were approved by the Wistar Institutional Animal Care and Use Committee and performed in accordance with relevant institutional and national guidelines.
HIGHLIGHTS.
Thiadiazolidinedione inhibitors prevent E7-mediated E2F displacement from pRb
The inhibitors prevent the interaction between pRb and viral oncoproteins
The inhibitors bind to the LxCxE binding site of pRb with mid-high nanomolar dissociation constants
The inhibitors are selectively cytotoxic to HPV transformed cells alone and in mice
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by NIH grant CA094165 and a Hiliary Koprowski, M.D. Professorship awarded to R.M. and NIH grant R43EB009626 awarded to M.R. D.F was supported by NIH training grant GM071339. We acknowledge support of the Protein Expression and Libraries and Flow Cytometry core facilities at the Wistar Institute. The Protein Expression and Libraries core facility at the Wistar Institute was supported by NIH grant CA010815. We thank Jason Bodily and Laimonis Laimins (Northwestern University) for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adams PD, Li XT, Sellers WR, Baker KB, Leng XH, Harper JW, Taya Y, Kaelin WG. Retinoblastoma protein contains a C-terminal motif that targets it for phosphorylation by cyclin-cdk complexes. Mol Cell Biol. 1999;19:1068–1080. doi: 10.1128/mcb.19.2.1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baleja J, Cherry J, Liu Z, Gao H, Nicklaus M, Voigt J, Chen J, Androphy E. Indentification of inhibitors to papillomavirus type 16 E6 protein based on three-dimensional structures of interacting proteins. Antiviral Res. 2006;72:49–59. doi: 10.1016/j.antiviral.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balog ER, Burke JR, Hura GL, Rubin SM. Crystal structure of the unliganded retinoblastoma protein pocket domain. Proteins. 79:2010–2014. doi: 10.1002/prot.23007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer SN, Wazer DE, Band V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996;56:4620–4624. [PubMed] [Google Scholar]
- Burd EM. Human papillomavirus and cervical cancer. Clin Microbiol Rev. 2003;16:1–17. doi: 10.1128/CMR.16.1.1-17.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordon-Cardo C, Sheinfeld J, Dalbagni G. Genetic studies and molecular markers of bladder cancer. Semin Surg Oncol. 1997;13:319–327. doi: 10.1002/(sici)1098-2388(199709/10)13:5<319::aid-ssu5>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Dahiya A, Gavin MR, Luo RX, Dean DC. Role of the LXCXE binding site in Rb function. Mol Cell Biol. 2000;20:6799–6805. doi: 10.1128/mcb.20.18.6799-6805.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFilippis RA, Goodwin EC, Wu L, DiMaio D. Endogenous human papillomavirus E6 and E7 proteins differentially regulate proliferation, senescence, and apoptosis in HeLa cervical carcinoma cells. J Virol. 2003;77:1551–1563. doi: 10.1128/JVI.77.2.1551-1563.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour X, Beby-Defaux A, Agius G, Lacau St Guily J. HPV and head and neck cancer. Eur Ann Otorhinolaryngol Head Neck Dis. 2011 doi: 10.1016/j.anorl.2011.05.004. [DOI] [PubMed] [Google Scholar]
- Felsani A, Mileo AM, Paggi MG. Retinoblastoma family proteins as key targets of the small DNA virus oncoproteins. Oncogene. 2006;25:5277–5285. doi: 10.1038/sj.onc.1209621. [DOI] [PubMed] [Google Scholar]
- Ganguly N, Parihar SP. Human papillomavirus E6 and E7 oncoproteins as risk factors for tumorigenesis. J Biosciences. 2009;34:113–123. doi: 10.1007/s12038-009-0013-7. [DOI] [PubMed] [Google Scholar]
- Giarre M, Caldeira S, Malanchi I, Ciccolini F, Leao MJ, Tommasino M. Induction of pRb degradation by the human papillomavirus type 16 E7 protein is essential to efficiently overcome p16INK4a-imposed G1 cell cycle Arrest. J Virol. 2001;75:4705–4712. doi: 10.1128/JVI.75.10.4705-4712.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez SL, Stremlau M, He X, Basile JR, Munger K. Degradation of the retinoblastoma tumor suppressor by the human papillomavirus type 16 E7 oncoprotein is important for functional inactivation and is separable from proteasomal degradation of E7. J Virol. 2001;75:7583–7591. doi: 10.1128/JVI.75.16.7583-7591.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudreau N, Cameron DR, Deziel R, Hache B, Jakalian A, Malenfant E, Naud J, Ogilvie WW, O'Meara J, White PW, et al. Optimization and determination of the absolute configuration of a series of potent inhibitors of human papillomavirus type-11 E1-E2 protein-protein interaction: a combined medicinal chemistry, NMR and computational chemistry approach. Bioorg Med Chem. 2007;15:2690–2700. doi: 10.1016/j.bmc.2007.01.036. [DOI] [PubMed] [Google Scholar]
- Griffin H, Elston R, Jackson D, Ansell K, Coleman M, Winter G, Doorbar J. Inhibition of Papillomavirus Protein Function in Cervical Cancer Cells by Intrabody Targeting. J Mol Biol. 2006;355:360–378. doi: 10.1016/j.jmb.2005.10.077. [DOI] [PubMed] [Google Scholar]
- Harbour JW, Dean DC. The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Develop. 2000;14:2393–2409. doi: 10.1101/gad.813200. [DOI] [PubMed] [Google Scholar]
- Harbour JW, Luo RX, Santi AD, Postigo AA, Dean DC. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell. 1999;98:859–869. doi: 10.1016/s0092-8674(00)81519-6. [DOI] [PubMed] [Google Scholar]
- Harper DM. Currently approved prophylactic HPV vaccines. Expert Rev Vaccines. 2009;8:1663–1679. doi: 10.1586/erv.09.123. [DOI] [PubMed] [Google Scholar]
- Hensel CH, Hsieh CL, Gazdar AF, Johnson BE, Sakaguchi AY, Naylor SL, Lee WH, Lee EY. Altered structure and expression of the human retinoblastoma susceptibility gene in small cell lung cancer. Cancer Res. 1990;50:3067–3072. [PubMed] [Google Scholar]
- Jiang M, Milner J. Selective silencing of viral gene expression in HPV-positive human cervical carcinoma cells treated with siRNA, a primer of RNA interference. Oncogene. 2002;21:6041–6048. doi: 10.1038/sj.onc.1205878. [DOI] [PubMed] [Google Scholar]
- Kitchin FD, Ellsworth RM. Pleiotropic effects of the gene for retinoblastoma. J Med Genet. 1974;11:244–246. doi: 10.1136/jmg.11.3.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JO, Russo AA, Pavletich NP. Structure of the retinoblastoma tumour-suppressor pocket domain bound to a peptide from HPV E7. Nature. 1998;391:859–865. doi: 10.1038/36038. [DOI] [PubMed] [Google Scholar]
- Liu X, Clements A, Zhao KH, Marmorstein R. Structure of the human Papillomavirus E7 oncoprotein and its mechanism for inactivation of the retinoblastoma tumor suppressor. J Biol Chem. 2006;281:578–586. doi: 10.1074/jbc.M508455200. [DOI] [PubMed] [Google Scholar]
- Liu X, Marmorstein R. When viral oncoprotein meets tumor suppressor: a structural view. Genes Develop. 2006;20:2332–2337. doi: 10.1101/gad.1471706. [DOI] [PubMed] [Google Scholar]
- Luna-Medina R, Cortes-Canteli M, Alonso M, Santos A, Martinez A, Perez-Castillo A. Regulation of inflammatory response in neural cells in vitro by thiadiazolidinones derivatives through peroxisome proliferator-activated receptor gamma activation. J Biol Chem. 2005;280:21453–21462. doi: 10.1074/jbc.M414390200. [DOI] [PubMed] [Google Scholar]
- Luna-Medina R, Cortes-Canteli M, Sanchez-Galiano S, Morales-Garcia JA, Martinez A, Santos A, Perez-Castillo A. NP031112, a thiadiazolidinone compound, prevents inflammation and neurodegeneration under excitotoxic conditions: Potential therapeutic role in brain disorders. J Neuroscience. 2007;27:5766–5776. doi: 10.1523/JNEUROSCI.1004-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinex A. TDZD: Selective GSK-3 inhibitors with great potential for Alzheimer disease. Neurobiol Aging. 2006;27:S13–S13. [Google Scholar]
- Martinez A, Alonso M, Castro A, Dorronsoro I, Gelpi JL, Luque FJ, Perez C, Moreno FJ. SAR and 3D-QSAR studies on thiadiazolidinone derivatives: Exploration of structural requirements for glycogen synthase kinase 3 inhibitors. J Med Chem. 2005;48:7103–7112. doi: 10.1021/jm040895g. [DOI] [PubMed] [Google Scholar]
- Martinez A, Alonso M, Castro A, Perez C, Moreno FJ. First non-ATP competitive glycogen synthase kinase 3 beta (GSK-3 beta) inhibitors: Thiadiazolidinones (TDZD) as potential drugs for the treatment of Alzheimer's disease. J Med Chem. 2002;45:1292–1299. doi: 10.1021/jm011020u. [DOI] [PubMed] [Google Scholar]
- McLaughlin-Drubin ME, Munger K. Oncogenic activities of human papillomaviruses. Virus Res. 2009;143:195–208. doi: 10.1016/j.virusres.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger K, Basile JR, Duensing S, Eichten A, Gonzalez SL, Grace M, Zacny VL. Biological activities and molecular targets of the human papillomavirus E7 oncoprotein. Oncogene. 2001;20:7888–7898. doi: 10.1038/sj.onc.1204860. [DOI] [PubMed] [Google Scholar]
- Rosa AO, Kaster MP, Binfare RW, Morales S, Martin-Aparicio E, Navarro-Rico ML, Martinez A, Medina M, Garcia AG, Lopez MG, et al. Antidepressant-like effect of the novel thiadiazolidinone NP031115 in mice. Prog Neuro-Psychopharmacology Biol Psychiatry. 2008;32:1549–1556. doi: 10.1016/j.pnpbp.2008.05.020. [DOI] [PubMed] [Google Scholar]
- Rubin SM, Gall AL, Zheng N, Pavletich NP. Structure of the RbC-terminal domain bound to E2F1-DP1: A mechanism for phosphorylation-induced E2F release. Cell. 2005;123:1093–1106. doi: 10.1016/j.cell.2005.09.044. [DOI] [PubMed] [Google Scholar]
- Ruegg UT, Burgess GM. Staurosporine, K-252 and UCN-01: potent but nonspecific inhibitors of protein kinases. Trends Pharmacol Sci. 1989;10:218–220. doi: 10.1016/0165-6147(89)90263-0. [DOI] [PubMed] [Google Scholar]
- Scheffner M, Munger K, Byrne JC, Howley PM. The state of the p53 and retinoblastoma genes in human cervical carcinoma cell lines. Proc Natl Acad Sci U S A. 1991;88:5523–5527. doi: 10.1073/pnas.88.13.5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert EL, Hansen MF, Strong LC. The retinoblastoma gene and its significance. Ann Med. 1994;26:177–184. doi: 10.3109/07853899409147887. [DOI] [PubMed] [Google Scholar]
- Sima N, Wang W, Kong D, Deng D, Xu Q, Zhou J, Xu G, Meng L, Lu Y, Wang S, et al. RNA interference against HPV16 E7 oncogene leads to viral E6 and E7 suppression in cervical cancer cells and apoptosis via upregulation of Rb and p53. Apoptosis. 2008;13:273–281. doi: 10.1007/s10495-007-0163-8. [DOI] [PubMed] [Google Scholar]
- Singh M, Krajewski M, Mikolajka A, Holak TA. Molecular determinants for the complex formation between the retinoblastoma protein and LXCXE sequences. J Biol Chem. 2005;280:37868–37876. doi: 10.1074/jbc.M504877200. [DOI] [PubMed] [Google Scholar]
- Stevaux O, Dyson NJ. A revised picture of the E2F transcriptional network and RB function. Curr Opin Cell Biol. 2002;14:684–691. doi: 10.1016/s0955-0674(02)00388-5. [DOI] [PubMed] [Google Scholar]
- Sudhoff HH, Schwarze HP, Winder D, Steinstraesser L, Gorner M, Stanley M, Goon PK. Evidence for a causal association for HPV in head and neck cancers. Eur Arch Otorhinolaryngol. 2011;268:1541–1547. doi: 10.1007/s00405-011-1714-8. [DOI] [PubMed] [Google Scholar]
- Wang Y, Coulombe R, Cameron DR, Thauvette L, Massariol MJ, Amon LM, Fink D, Titolo S, Welchner E, Yoakim C, et al. Crystal structure of the E2 transactivation domain of human papillomavirus type 11 bound to a protein interaction inhibitor. J Biol Chem. 2004;279:6976–6985. doi: 10.1074/jbc.M311376200. [DOI] [PubMed] [Google Scholar]
- Wei Q. Pitx2a binds to human papillomavirus type 18 E6 protein and inhibits E6-mediated P53 degradation in HeLa cells. J Biol Chem. 2005;280:37790–37797. doi: 10.1074/jbc.M502974200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White PW, Faucher AM, Goudreau N. Small molecule inhibitors of the human papillomavirus E1-E2 interaction. Curr Top Microbiol Immunol. 2011;348:61–88. doi: 10.1007/82_2010_92. [DOI] [PubMed] [Google Scholar]
- Xiao B, Spencer J, Clements A, Ali-Khan N, Mittnacht S, Broceno C, Burghammer M, Perrakis A, Marmorstein R, Gamblin SJ. Crystal structure of the retinoblastoma tumor suppressor protein bound to E2F and the molecular basis of its regulation. Proc Natl Acad Sci U S A. 2003;100:2363–2368. doi: 10.1073/pnas.0436813100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee C, Krishnan-Hewlett I, Baker CC, Schlegel R, Howley PM. Presence and expression of human papillomavirus sequences in human cervical carcinoma cell lines. Am J Pathol. 1985;119:361–366. [PMC free article] [PubMed] [Google Scholar]
- Yoakim C, Ogilvie WW, Goudreau N, Naud J, Hache B, O'Meara JA, Cordingley MG, Archambault J, White PW. Discovery of the first series of inhibitors of human papillomavirus type 11: inhibition of the assembly of the E1-E2-Origin DNA complex. Bioorg Med Chem Lett. 2003;13:2539–2541. doi: 10.1016/s0960-894x(03)00510-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.