

Summary
Toll-like receptors (TLRs) have emerged as one of the most important families of innate immune receptors for initiating inflammation and also for promoting adaptive immune responses. Recent studies have examined the ability of TLRs to promote antibody responses, including T-cell-dependent antibody responses. Initial work suggested that TLR stimulation promotes primarily an extrafollicular antibody response, which rapidly produces moderate affinity antibodies made by short-lived plasma cells. Recent studies, however, have shown that TLRs also can enhance the germinal center response, which produces high affinity class-switched antibody made by long-lived plasma cells. TLR stimulation can increase the magnitude of the latter response and also enhance selection for high affinity IgG. This review summarizes recent advances in understanding the roles of TLRs in B cells and also in other cell types for enhancement of antibody responses, with an emphasis on T-cell-dependent and germinal center antibody responses.
Keywords: Toll-like receptor, B cell, dendritic cell, MyD88
Introduction
The ability of bacterial cell wall components, including lipopolysaccharide (LPS) and fixed Staphylococcus aureus, to serve as very potent stimuli for in vitro B-cell activation and differentiation was appreciated long before the discovery of the Toll-like receptor (TLR) family (1). Early work also established that haptenated derivatives of LPS were potent antigens that can give rise to a substantial immunoglobulin M (IgM) and IgG3 response in vivo in the absence of T cells. After the discovery that TLRs recognize LPS and other bacterial cell wall components and also recognize pathogen-derived nucleic acids, it was found that TLR stimulation enhances T-cell-dependent as well as T-cell-independent antibody responses (2, 3). In this review, we discuss the ways in which TLRs can contribute to specific antibody responses with an emphasis on T-cell-dependent and germinal center (GC) antibody responses.
Antibody responses in secondary lymphoid organs generally exhibit one of two anatomical signatures, which are referred to as extrafollicular responses and GC responses (4). Extrafollicular antibody responses occur during bacterial infections and after injection of polysaccharide immunogens but also typically are a component of the response to injected T-cell-dependent protein antigens (5). Extrafollicular antibody responses are also prominent in some autoimmune models, such as the MRL/lpr mouse (6). This form of antibody response generally occurs rapidly, starting at around 4 days after immunization, and has a moderate degree of class switch to IgG and somatic hypermutation, but less than what occurs in the slower GC response. Thus, the extrafollicular response is viewed as a mechanism that provides rapid production of moderate affinity antibodies over a limited time period (4). The plasma cells generated in this way clonally expand for a short time and are therefore referred to as plasmablasts. Recent evidence indicates that avidity of the plasmablasts strongly affects their degree of clonal expansion and ability to survive (7), so there is a selection for higher affinity antibody clones during an extrafollicular response. The plasmablasts and plasma cells generated in this way remain in the extrafollicular location and mostly have a short half-life, although some of plasma cells generated in this way can compete for survival niches in the spleen and become long-lived (5).
The GC response is slower than the extrafollicular response and involves extensive clonal expansion, somatic hypermutation, and selection for higher affinity clones (4, 8, 9). The more slowly generated but higher quality antibodies produced in this way are mostly class switched isotypes rather than IgM. The antigen-specific B cells selected from a GC response can differentiate into plasma cells that traffic to survival niches in the bone marrow, where they have a very long half-life, probably exceeding one year (10). GC B cells may alternatively become memory B cells that revert to a resting lymphocyte phenotype but can rapidly become activated upon secondary exposure to the antigen (9). However, a significant fraction of memory B cells are generated early in an antibody response, before the initiation of histologically evident GCs and typically before class switch (4, 9, 11, 12). These IgM+ memory B cells can participate in GC responses upon secondary exposure to antigen.
Role of TLRs in antibody responses
Pure TLR ligands serve as excellent adjuvants for antibody responses, as discussed in more detail below, and in such circumstances, the adjuvant activity is dependent on the adapter molecules that mediate TLR signaling, myeloid differentiation factor 88 (MyD88) and/or TIR-domain-containing adapter-inducing interferon-β (Trif) (3). Although TLRs can strongly boost antibody responses, it is clear that TLRs are not necessary for antibody responses induced by standard immunization approaches used in the mouse or those induced by many human vaccines. Nemazee and colleagues (13) took Myd88−/− Trif−/− mice and found that their responses to standard immunizations with haptenated proteins in either alum, incomplete Freund’s adjuvant or complete Freund’s adjuvant were not compromised. We have repeated some of these experiments with Myd88−/− mice and have confirmed these findings (unpublished results). Whether the adjuvant activity of alum depends to some extent on its ability to activate the inflammasome is an area of controversy, although several groups have reported that ASC−/− mice, which are defective in most of the different inflammasome isoforms, do not have decreased antibody responses to alum-based immunization (3). The main function of the inflammasome is to generate active IL-1β and IL-18, the receptors for which signal via MyD88, so normal responses to protein antigen in alum immunizations in Myd88−/− mice would rule out at least these products of the inflammasome as being necessary for such antibody responses. Based on these observations, it seems clear that TLRs are among the innate recognition mechanisms that can promote vigorous antibody responses, but there are other mechanisms as well, consistent with the emerging picture that in addition to TLRs there are several other major families of innate recognition molecules that sense infections or tissue damage and initiate immune responses (14).
Contributions of TLRs in B cells vs. dendritic cells to antibody responses
B cells express TLRs and have strong proliferative and differentiation responses to TLR stimulation in vitro (2). Therefore, it seems likely that B-cell TLRs contribute importantly to antibody responses in vivo, but dendritic cells, macrophages, and other cell types also express TLRs, so the relative contributions of different cell types to antibody responses is not immediately apparent. Investigators have used several different experimental approaches to address this issue. One approach has been to use adoptive transfer of B cells of different genotypes into B-cell deficient mice (for example, μMT or JHT mutant mice) and compare antibody responses. In several reports using this approach, Myd88−/− B cells made decreased antibody responses compared to wild type B cells, indicating an important role for TLR signaling in B cells (15, 16). This approach has the potential disadvantage that the normal architecture of secondary lymphoid organs is dependent on the lymphotoxin-β that is produced by B cells and therefore the recipient mice do not have a normal lymphoid structure of well separated T-cell zones and B-cell follicles (17). Moreover, transfer of B cells into such mice does not correct this anatomical alteration. In one case, however, adoptive transfer of Ig transgenic wildtype or Myd88−/− B cells into wildtype recipients was used, and a requirement for MyD88 for production of the transgene-encoded antibody was seen(18). Taken together, these experiments demonstrate that B-cell TLRs are capable of enhancing antibody responses, and this is true of mice with normal lymphoid architecture in at least some circumstances.
A related approach is the use of mixed bone marrow chimeric mice in which one source of bone marrow is a B-cell-deficient genotype and the other source of bone marrow, which is the origin of all of the B cells, is wildtype or mutant for Myd88 or a particular TLR. In these chimeric mice, the majority of other cell types are normal, but the B cells are all derived from a particular mutant genotype, so a defect in the response is presumably due to the genetic alteration in the B-cell compartment. Experiments using this approach in the context of bacterial infection with Salmonella enterica serovar Typhimurium have demonstrated that a protective T-helper 1 (Th1) response requires MyD88 signaling in B cells (19), as does proper regulation of the innate response to the bacterial infection (20). In both cases, these alterations were ascribed to a role of B cells in producing cytokines, rather than an effect on antibody production.
Studies in human have largely focused on using TLR agonists as adjuvant in vaccine studies (3). In addition, there have been in vitro studies examining the role of human B-cell maturation state in responsiveness to TLR stimulation (2, 21). While immature, transitional B cells and naive recirculating B cells do exhibit some responses to TLR ligands, especially to CpG-containing oligonucleotides (ODNs), TLR responses are considerably stronger for B cells simultaneously stimulated via the BCR and CD40 or for IgM+ memory B cells (2, 21), suggesting that physiologically, human B cell TLRs are most likely to promote antibody responses in the context of other signals promoting B-cell activation and/or in responses of IgM+ memory B cells. In addition, in human as in mouse, TLR stimulation of dendritic cells induces their production of cytokines that can promote antibody responses (21), suggesting an alternative mechanism by which adjuvants based on TLR ligands may contribute to antibody responses.
As a general approach to examine the importance of TLR signaling in particular cell types in mice, we have created a conditional allele of the gene encoding MyD88 by using homologous recombination to place loxP sites on either side of exon 3 of the Myd88 gene (Myd88fl allele). When combined with a CD11c-Cre transgene (22), Myd88 is deleted in approximately 98% of conventional dendritic cells in the spleen and other locations examined, in 80% of plasmacytoid dendritic cells, and in low percentages of other cell types (23), with the exception of several macrophage subpopulations such as alveolar macrophages, which express CD11c and the transgenic Cre at a high level. Similarly, by combination of the Myd88fl allele with a mb1-Cre knockin allele (24), mice are generated that are deficient for MyD88 in at least 98% of B cells. When these mice were immunized intraperitoneally (i.p.) with the protein antigen ovalbumin, either mixed with CpG-containing oligonucleotides (CpG ODNs) or chemically coupled to them, then it was found that the major adjuvant effect of the CpG ODNs was due to TLR signaling in dendritic cells and not in B cells (25). This observation was surprising in the case of the ovalbumin-CpG ODN conjugate, as it would be expected that the B-cell TLR9 would be exposed to its ligand in this case. Nonetheless, a similar result was obtained with a second soluble protein-CpG ODN conjugate, containing the well characterized ragweed pollen allergen Amb a1. A similar result was also obtained after immunization with purified FliC flagellin from Salmonella enterica serovar Typhimurium, which is a ligand for TLR5, and measuring anti-flagellin IgG. In contrast, when MyD88-deficient B cells were adoptively transferred into μMT mice followed by immunization with flagellin, a defect in the anti-flagellin antibody response was seen (15). The reason for the different results in these two systems is not known but may relate to an effect of the altered lymphoid architecture in the latter experimental system. In any case, the results in the cell type-specific MyD88 knockout mice argue that the adjuvant effect of CpG ODNs on the IgG response to a soluble protein antigen is primarily due to enhanced maturation and/or cytokine production by antigen-presenting dendritic cells. We hypothesize that TLR-stimulated dendritic cells induce robust activation of antigen-specific helper T cells, which then promote extrafollicular and GC antibody responses. These experiments did not distinguish between extrafollicular and GC antibody responses, although similar results were seen at day 7 and day 14 of the response and also in secondary responses (25), which is consistent with an effect on the GC response but does not rule out an effect on the extrafollicular response.
In contrast to what was seen with soluble protein-CpG ODN conjugates, multivalent virus-like particles (VLPs) incorporating TLR ligands induced a robust antibody response that was enhanced substantially by MyD88 function in B cells (25). Virus particles typically contain the viral genome inside them, and this nucleic acid can stimulate TLR7 or TLR9, depending on the type of virus. In addition, many virus particles have a highly repetitious structure in which a small number of epitopes are present in a polymeric array on the particle surface and hence have the ability to crosslink many BCRs and induce robust BCR signaling. Virus particles of this type are known to induce a rapid and robust T-cell-independent IgM response together with a T-cell-dependent IgG response (26). Interestingly, the IgG response to the Qβ bacteriophage virus-like particle (VLP) was increased about 30-fold by inclusion of CpG ODNs inside it compared to an empty VLP mixed with CpG ODNs and coinjected i.p. (25). The IgG response to VLPs containing nucleic acid was decreased by 1000 fold or more in Myd88−/− mice compared to wildtype mice. Interestingly, deletion of MyD88 selectively in B cells decreased the IgG anti-VLP response by 30-fold, whereas deletion of Myd88 selectively in dendritic cells had no significant effect (25). The anti-VLP IgG response was nevertheless highly dependent on the presence of T cells and of dendritic cells, as the response was approximately 1000-fold lower in mice lacking TCRα chain or mice in which dendritic cells were transiently depleted by use of a CD11c-driven diphtheria toxin receptor and injection of diphtheria toxin (25). Presumably, the dendritic cells were needed to prime antigen-specific T cells, but they did not need to recognize the nucleic acid with their own TLRs to do so. We hypothesize that in this immunization, the dendritic cells received enough cytokines from other cells to promote their maturation. In support of this possible explanation, genetic deletion of the type 1 interferon receptor in this context greatly attenuated the IgG anti-VLP response, indicating that type 1 interferon, possibly produced by plasmacytoid dendritic cells, was responsible for inducing maturation of conventional dendritic cells (25).
The ability of MyD88 in B cells to enhance the IgG anti-VLP response was accompanied by a 10-fold increase in the number of GC B cells that were specific for the VLPs (25), indicating that MyD88 signaling in antigen-specific B cells enhanced their ability to participate in a GC response. These observations appear to be relevant to antibody responses to enveloped viruses as well: wild type mice or dendritic cells-specific Myd88−/− mice immunized with chemically inactivated influenza virus had equivalent IgG anti-HA responses, whereas the response was decreased fivefold in mice lacking MyD88 selectively in B cells (25).
One possible explanation for the different genetic requirements for the IgG responses to ovalbumin and Qβ VLPs is the different physical nature of these antigens, but there could also be something atypical about the Qβ epitopes. To examine these possibilities, we tried to dissociate the Qβ capsid into soluble monomers but were unable to do so. As an alternative approach, we immunized the various mouse strains lacking MyD88 in particular cell types with a soluble form of the cat dander allergen Fel d1 protein mixed with CpG ODNs or with chemical conjugates of VLPs and Fel d1 (26). Soluble Fel d1 protein mixed with CpG induced a reasonable IgG response that was dependent on MyD88 expression in dendritic cells but was not affected by deletion of MyD88 selectively in B cells (25). Thus, soluble Fel d1 behaved similarly to ovalbumin and the other low valency soluble protein antigens tested. When Fel d1 was conjugated to Qβ VLPs, it was now a much more potent antigen and, in this context, anti-Fel d1 IgG was substantially enhanced by the presence of MyD88 in B cells and did not require MyD88 expression in dendritic cells (25). Thus, it is the VLP physical structure that enables TLR7 or TLR9 in the B cell to greatly enhance the magnitude of the GC response, resulting in much higher titers of IgG.
The results obtained with the Fel d1-Qβ VLP conjugates also provided additional insights into the role of polyvalency of these VLPs for enhancement of the IgG response. Two preparations of Fel d1-VLP conjugates with different amounts of Fel d1 attached to the VLPs were compared, and there was a clear correlation between epitope density and the degree of enhancement of the IgG anti-Fel d1 response by B cell MyD88. The particles with a higher Fel d1 epitope density showed a greater fold enhancement of IgG anti-Fel d1 in the presence of B-cell MyD88. The VLPs with a lower density of Fel d1 still showed an enhancement, but to a lesser extent. The reciprocal effect was seen with the anti-Qβ VLP IgG antibodies, presumably because Fel d1 protein coupled to the VLPs masked the Qβ epitopes, effectively decreasing the valency of the Qβ epitopes accessible to the BCRs on the antigen-specific B cells.
The results summarized above lead to a model whereby CpG ODNs can act as adjuvants for a T-cell-dependent antibody response in either of two ways (Fig. 1): (i) dendritic cells can respond to CpG via their TLR9, enhancing their maturation and ability to activate helper T cells, which then provide help for B cells to make antibody responses, or (ii) antigen-specific B cells take up antigen and internalize the attached CpG to late endosomes, where it comes in contact with B cell TLR9 and induces TLR9 signaling (27-29), which then stimulates the ability of these B cells to participate in a GC response. In the case of a polyvalent virus particle, the ability to crosslink many BCRs and also induce TLR7 or TLR9 signaling evidently promotes a strong GC response of at least some of the antigen-specific B cells.
Fig. 1. TLRs enhance GC IgG responses in two distinct ways.
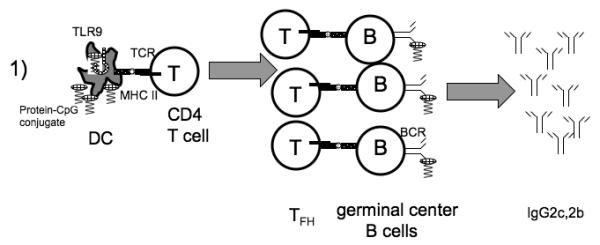
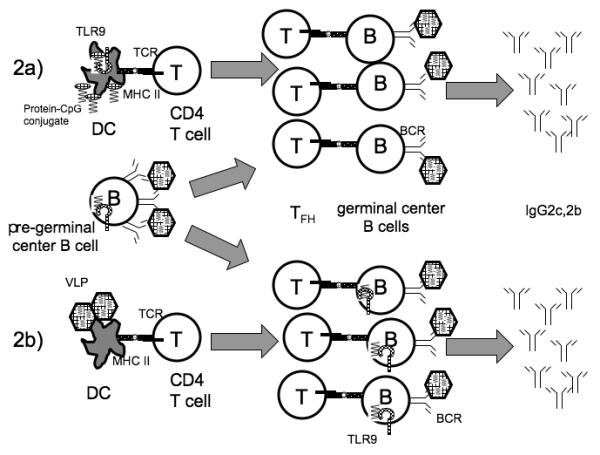
1) TLRs of dendritic cells (DC) can detect TLR ligands such as CpG oligonucleotides either attached to antigens (shown) or simply mixed with them (not shown), and this recognition can enhance DC activation of CD4+ T cells, leading to a vigorous GC response. When the antigen in question is a soluble, low valency protein antigen-CpG conjugate, then recognition by B cell TLR9 does not enhance the IgG response. 2) In some circumstances, TLRs of B cells can promote a T-cell-dependent antibody response. This has been seen most prominently in the case of polyvalent antigens such as virus-like particles (VLPs). The exact mechanism is not established so the figure shows two possible mechanisms. 2a) Combined TLR and BCR signals may cause some antigen-specific B cells to choose a GC fate instead of an extrafollicular fate, which might otherwise be favored in the case of a polyvalent antigen that can induce robust BCR signaling. 2b) B cells at the GC stage may respond in a synergistic fashion to the combination of BCR and TLR stimulation to greatly enhance the magnitude of the GC IgG response. These two possible mechanisms are not mutually exclusive, and TLR signaling in B cells may contribute at both stages to the immune response. It should be noted that in the case of the response to a VLP antigen, the participation of dendritic cells is required but their TLRs do not boost the amount of antibody production. Finally, some types of antigens trigger TLRs in both dendritic cells and in B cells to promote the GC response, as in the case of some oligovalent haptenated protein antigens.
Many questions remain, however. It is unclear whether this synergy between the BCR and intracellular TLRs of the B-cell leading to an enhanced GC response occurs early, when B cells are choosing between extrafollicular and GC fates, or whether it promotes the response of the B cell after it has entered the GC. As described below, it is clear that TLR ligands can also promote extrafollicular antibody responses, so an important question is whether B-cell affinity for antigen influences the choice of fate between extrafollicular response and GC response.
B-cell intrinsic TLR signaling also influences isotype switching of the B cells, promoting class switch to IgG2c and IgG2b and away from IgG1 (25, 30, 31). While the experiments with Salmonella enterica serovar Typhimurium infection suggest that this could be an effect of B cells on T cell polarization (19), there is also evidence to indicate that TLR9 directly induces expression in the responding B cell of the transcriptional activator T-bet, which in turn may promote class switch to IgG2c (32). At this point, it is not clear which of these mechanisms is more important in vivo and in situations where T-cell help is available.
Qualitative effects of a CpG-antigen conjugate on the GC reaction
We have also examined how TLR9 signaling in dendritic cells and B cells affects a GC response that does not require MyD88 signaling. For this purpose, we used biotin and avidin to complex the model antigen (4-hydroxy-3-nitrophenyl) acetyl-conjugated chicken gammaglobulin (NP-CGG) with CpG ODNs or with non-CpG ODNs and immunized mice subcutaneously with the conjugates in PBS. Both conjugates induced strong GC reactions and IgG anti-NP production, although the magnitude of the IgG response was increased several fold by inclusion of a TLR9 ligand ODN (Rookhuizen et al., manuscript in preparation). We hypothesize that the tissue injury caused by tissue injection provides sufficient innate stimulation to serve an adjuvant role in this route of immunization. The increase in IgG anti-NP antibodies resulting from TLR9 stimulation was accompanied by a similar magnitude increase in the number of NP-specific GC-phenotype B cells in the draining lymph node. This increase in IgG titer required the expression of Myd88 in dendritic cells but not in B cells (Rookhuizen et al., manuscript in preparation). Interestingly, the fraction of total anti-NP antibody that was high affinity, as assessed by enzyme-linked immunosorbent assay (ELISA), was ~10-fold higher at day 14 or later times in the mice that received CpG-conjugates, indicating more efficient selection for high affinity clones. The enhanced affinity of anti-NP IgG required MyD88 function in B cells and to a lesser extent in dendritic cells as well. Thus, TLR9/MyD88 signaling in dendritic cells enhanced the magnitude and influenced the quality of the response to the CpG-conjugate, while TLR9/MyD88 signaling in B cells enabled stronger selection for high affinity antibody. Further studies will be needed to define the mechanisms by which TLR signaling in these cell types contributes to the GC response, but enhanced selection in the GCs may relate to improved priming and/or maintenance of follicular helper T cells (Tfh), since immunization with the CpG-conjugate expanded the Tfh population in the draining lymph node by several fold compared to immunization with the non-CpG-conjugate (Rookhuizen et al., manuscript in preparation). In any case, it is clear that TLR/MyD88 signaling in both dendritic cells and B cells can enhance selection for higher affinity IgG antibody, even in a situation where a strong GC response is generated in the absence of TLR/MyD88 signaling.
GC and extrafollicular IgG responses induced by nanoparticles with TLR ligands
Vigorous GC responses were also observed recently by Pulendran and colleagues (16), who used biodegradable virus-size polylactic-co-glycolic acid (PLGA) nanoparticles encapsulating TLR ligands as adjuvant combined with PLGA nanoparticles encapsulating one or another protein antigen. In these experiments, the adjuvant and the antigen were not physically linked but rather were present attached to separate beads that were injected together. For the adjuvant component, they compared monophosphoryl lipid A (MPL), a TLR4 ligand that has been reported to stimulate the Trif pathway well but the MyD88 pathway poorly (33), either alone or together with a synthetic TLR7 ligand (R837). They obtained a considerably more robust response (~5-fold) if the adjuvant nanoparticles contained both MPL and R837, compared to nanoparticles containing only one TLR ligand. This immunization induced a strong GC response that generated high affinity antibodies, excellent B-cell and T-cell memory, and strong protection against subsequent infectious challenge in mice and in primates (16). The authors found that a good response was not obtained if Myd88−/−Trif−/− B cells or TLR4−/−TLR7−/− B cells were adoptively transferred into μMT recipients, indicating a B-cell-intrinsic role for TLR signaling in this response. This is a surprising result, because the antigen and the TLR ligands were present on separate nanoparticles, so the adjuvant beads could not interact with antigen-specific B cells via their BCR. Although the B cells would not take up the nanoparticles by BCR-mediated endocytosis, it is possible that they can take up the beads via another mechanism involving recognition of the nanoparticle material. Given this uncertainty about how the adjuvant would gain access to B cell TLRs (including intracellular TLR7), it is unclear whether TLRs within antigen-specific B cells participate in the response or whether TLRs on bystander B cells may contribute to the response in some way. For example, antigen non-specific B cells might secrete cytokines that promote the response of neighboring antigen-specific B cells. Given the robustness of the antibody response induced by this novel vaccination approach, it will be important to define in better detail how TLRs on B cells contribute to the response.
In a separate study, antigen and a TLR9 agonist were both put onto the same polystyrene nanoparticles, which strongly induced an in vivo extrafollicular antibody response of adoptively transferred MD4 anti-HEL Ig transgenic B cells (34). This extrafollicular antibody required an antigen on the particle with an affinity for the anti-HEL BCR of approximately 1 × 10−7M (KD) or better. With this type of nanoparticle, uptake by the B cells required an antigen-specific BCR, so the TLR9 agonist was specifically delivered to the antigen-specific B cells. Interestingly, when these antigen- and TLR ligand-containing nanoparticles were used to immunize mice with a diverse repertoire of B cells, a strong secondary response was evident (34). This observation suggests that a GC response may have been induced by the antigen- and CpG-containing nanoparticles, but this point was not directly assessed. One interesting possibility is that B cells with an affinity for the epitopes on the bead poorer than 1 × 10−7M can access the GC response in response to these nanoparticles. If so, the IgG response of such B cells may be analogous to the anti-VLP IgG response described above. Future studies comparing the properties of various nanoparticles to VLPs will likely be informative about how synthetic nanoparticles can be formulated to give robust and high quality GC responses.
Role of TLR signaling in B cells for production of autoantibodies in mice that spontaneously develop lupus-like autoimmune disease
Autoantibodies to nuclear antigens, deposition in tissues of their immune complexes with debris from dying cells, and the resulting tissue pathology are hallmarks of the human autoimmune disease systemic lupus erythematosus (SLE). IgG anti-nuclear antibodies, anti-DNA antibodies, and anti-ribonucleoprotein antibodies are spontaneously produced in various mouse strains with genetic susceptibility loci or genetically engineered mutations, including those which alter the regulation of BCR signaling (35, 36). The pioneering in vitro work of Marshak-Rothstein and colleagues (37) first implicated TLR9 signaling in B cells as a likely participant in the production of the anti-nuclear antibodies characteristic of SLE. TLR9 and/or TLR7 have been shown to be critical for the spontaneous production of these autoantibodies in a number of mouse models, presumably reflecting the ability of mammalian DNA to serve as a TLR9 ligand to some extent, and the ability of mammalian RNA in ribonucleoproteins to serve as TLR7 ligands. These studies have been well reviewed recently, and the interested reader is referred to that source (29). In brief, numerous studies of mice with spontaneous or induced lupus-like autoantibody production have examined the effect of genetic deficiency in Tlr7, Tlr9, or Myd88 on autoantibody production and have generally seen a strong diminution of autoantibody production (29). Conversely, increased expression of TLR7 resulting from the duplication of a region of the X chromosome containing TLR7 onto the Y chromosome is seen in a Y-linked autoimmune susceptibility locus called Yaa (Y-linked autoimmune accelerator) (38, 39). Thus, there is strong genetic evidence for the ability of TLR7 and TLR9 to contribute importantly to the production of anti-nuclear antibodies in lupus-like autoimmune mice.
While TLR signaling in B cells has been generally assumed to be largely responsible for the role of TLR7 and TLR9 in the production of SLE-like autoantibodies in these murine studies, it remains possible that TLR signaling in other cell types, such as plasmacytoid dendritic cells, is also important. In one study, TLR9 deficiency decreased anti-chromatin antibodies more than anti-ribonucleoprotein antibodies, and the opposite was seen with TLR7 deficiency (40). This result is most easily explained if TLR signaling in the antigen-specific B cell is important, as predicted by the in vitro data, but the in vivo effects seen were not absolute. This is perhaps not surprising if fragments of apoptotic cells (apoptotic blebs) represent the immunogenic form of nuclear antigens in vivo (41), since apoptotic blebs likely contain both RNA and DNA, but it is also possible that the results reflect a role for TLRs in other cell types in promoting the autoantibody response. For example, plasmacytoid dendritic cells express TLR7 and TLR9 and may respond to immune complexes formed by autoantibodies and the corresponding nuclear antigens by secreting type 1 interferon (29), which in turn may promote autoantibody production by a variety of means, including enhancing antigen presentation by conventional dendritic cells and upregulating TLR7 expression in B cells (29).
An additional question is whether anti-nuclear antibodies are generated primarily from GC responses or from extrafollicular responses. Early work establishing that anti-DNA antibodies contain somatic mutations that enhance their affinity for DNA led to the belief that GC responses were involved. Recent work, however, has shown that the antinuclear antibodies produced in MRL/lpr mice are primarily produced by the extrafollicular response rather than the GC response (6, 42). It is unclear whether this is a conserved feature of the different mouse models of SLE or whether they may differ in this regard.
We have recently addressed the issue of whether MyD88 signaling in B cells or dendritic cells is important for spontaneous autoantibody production in the Lyn−/− model of SLE. Lyn is a Src-family protein tyrosine kinase that participates both positively and negatively in BCR signaling (35). The main effect of Lyn-deficiency in B cells is to increase BCR signaling, because its role in initiating BCR signaling is partially redundant with the other Src family kinases in B cells (Fyn and Blk), whereas its inhibitory role is largely unique (35). Thus, B cells from Lyn-deficient mice exhibit hyperactive BCR signaling, especially once they reach the mature B-cell stage (43). In addition, Lyn is expressed in myeloid cells, and in Lyn−/− mice, these cells are also hyperactive and can promote autoimmunity (44). Previous work established that the autoantibody production and glomerulonephritis in Lyn−/− mice was lost upon deletion of Myd88 (45). We are using the conditional allele of Myd88 to test whether the requirement for MyD88 signaling for autoimmunity in Lyn−/− mice reflects MyD88 signaling in B cells, dendritic cells, or both. Analysis of the phenotype is currently ongoing. Analysis of the Lyn−/− mice lacking Myd88 selectively in B cells (Lyn−/−mb1-Cre Myd88fl/fl) is more nearly complete and demonstrates that IgG anti-nuclear antibodies are greatly reduced by deletion of Myd88 in B cells (Hou et al., manuscript in preparation). This result suggests that the endogenous components that induce anti-nuclear antibody production in Lyn−/− mice, and presumably in other spontaneous mouse models of SLE, can engage both BCR and TLR signaling in a synergistic manner, similarly to what was seen with VLPs and inactivated virus particles and different from what was seen with low valency soluble protein antigens.
Whether the IgG anti-nuclear antibodies produced in Lyn−/− mice are due to GC responses or extrafollicular antibody response is not established, although we believe that a role for GCs is likely. To examine this issue, we generated SAP-deficient (Sh2d1a−) Lyn−/− mice, since SAP is believed to be primarily required for the GC antibody response (46). These mice have substantially decreased IgG anti-nuclear antibody titers compared to their SAP-expressing counterparts (Hou et al., manuscript in preparation), indicating that GC responses are likely to be important for autoantibody production in this model.
Concluding remarks
Engagement of TLRs in B cells by antigens that are physically associated with a TLR ligand, as is often the case for antigens generated during infections, can promote antibody responses by both a T cell-independent route and T cell-dependent responses of both the extrafollicular and GC types. TLR ligands can also promote T-cell-dependent antibody responses in vivo by indirect routes such as stimulation of dendritic cells, probably by promoting stronger activation of helper T cells, including Tfh cells.
Studies of B-cell activation in vitro have generally suggested that TLR stimulation can relieve a requirement for signals coming from Th cells, and this may be valid for the T-cell-independent component of the response. Recent studies indicate a more complex relationship in vivo in which B-cell TLRs may also alter the magnitude and/or quality of a T-cell-dependent antibody response, for example by increasing the GC component of the response or by enhancing the selection for higher affinity antibody production in the context of an ongoing GC response. While much remains to be learned, it is tempting to speculate that the ability of TLRs of B cells to promote a GC response as well as an extrafollicular may represent an evolutionary adaptation to diversify the nature of the antibody response to virus particles and ensure that there is production of high affinity, long-lasting IgG from GC responses as well as the rapidly produced but less protective antibody resulting from extrafollicular responses. While protective in the case of virus infection, when combined with genetic susceptibility alleles and an environmental trigger, these mechanisms may lead to the production of pathogenic anti-nuclear autoantibodies in the human autoimmune disease SLE.
Future studies will help define the rules and mechanisms by which B-cell TLRs can affect T-cell-dependent antibody responses, and this information will likely be useful for development of new strategies for therapeutic intervention in SLE and for design of more efficacious vaccines using TLR ligands as adjuvants.
Acknowledgements
Our work described in this article has been supported by research grants from the US National Institutes of Health to ALD and from the National Natural Science Foundation of China to BH. We thank Matthew Wheeler and Linda Lee for helpful comments on the manuscript. We also thank Martin F. Bachmann, Robert L. Coffman, Philippe Saudan, Gary Ott, Matthew Wheeler, Linda Lee, and Ming Ji for their participation in some of the experiments described here and for informative discussions.
Footnotes
The authors have no conflicts of interest to declare.
References
- 1.Moller G. One non-specific signal triggers B lymphocytes. Transplant Rev. 1975;23:126–37. [PubMed] [Google Scholar]
- 2.Bekeredjian-Ding I, Jego G. Toll-like receptors--sentries in the B-cell response. Immunology. 2009;128:311–323. doi: 10.1111/j.1365-2567.2009.03173.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coffman RL, Sher A, Seder RA. Vaccine adjuvants: putting innate immunity to work. Immunity. 2010;29:492–503. doi: 10.1016/j.immuni.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goodnow CC, Vinuesa CG, Randall KL, Mackay F, Brink R. Control systems and decision making for antibody production. Nat Immunol. 2010;11:681–688. doi: 10.1038/ni.1900. [DOI] [PubMed] [Google Scholar]
- 5.MacLennan ICM, et al. Extrafollicular antibody responses. Immunol Rev. 2003;194:8–18. doi: 10.1034/j.1600-065x.2003.00058.x. [DOI] [PubMed] [Google Scholar]
- 6.Odegard JM, et al. ICOS-dependent extrafollicular helper T cells elicit IgG production via IL-21 in systemic autoimmunity. J Exp Med. 2008;205:2873–2886. doi: 10.1084/jem.20080840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan TD, Gatto D, Wood K, Camidge T, Basten A, Brink R. Antigen affinity controls rapid T-dependent antibody production by driving the expansion rather than the differentiation or extrafollicular migration of early plasmablasts. J Immunol. 2009;183:3139–3149. doi: 10.4049/jimmunol.0901690. [DOI] [PubMed] [Google Scholar]
- 8.Tarlinton DM. Evolution in miniature: selection, survival and distribution of antigen reactive cells in the germinal center. Immunol Cell Biol. 2008;86:133–138. doi: 10.1038/sj.icb.7100148. [DOI] [PubMed] [Google Scholar]
- 9.Good-Jacobson KL, Shlomchik MJ. Plasticity and heterogeneity in the generation of memory B cells and long-lived plasma cells: the influence of germinal center interactions and dynamics. J Immunol. 2010;185:3117–3125. doi: 10.4049/jimmunol.1001155. [DOI] [PubMed] [Google Scholar]
- 10.Hiepe F, Dorner T, Hauser AE, Hoyer BF, Mei H, Radbruch A. Long-lived autoreactive plasma cells drive persistent autoimmune inflammation. Nat Rev Rheumatol. 2011;7:170–178. doi: 10.1038/nrrheum.2011.1. [DOI] [PubMed] [Google Scholar]
- 11.Dogan I, et al. Multiple layers of B cell memory with different effector functions. Nat Immunol. 2009;10:1292–1299. doi: 10.1038/ni.1814. [DOI] [PubMed] [Google Scholar]
- 12.Inamine A, et al. Two waves of memory B-cell generation in the primary immune response. Int Immunol. 2005;17:581–589. doi: 10.1093/intimm/dxh241. [DOI] [PubMed] [Google Scholar]
- 13.Gavin AL, et al. Adjuvant-enhanced antibody responses in the absence of toll-like receptor signaling. Science. 2006;314:1936–1938. doi: 10.1126/science.1135299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;27:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 15.Pasare C, Medzhitov R. Control of B-cell responses by Toll-like receptors. Nature. 2005;438:364–368. doi: 10.1038/nature04267. [DOI] [PubMed] [Google Scholar]
- 16.Kasturi SP, et al. Programming the magnitude and persistence of antibody responses with innate immunity. Nature. 2011;470:543–547. doi: 10.1038/nature09737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fu YX, Chaplin DD. Development and maturation of secondary lymphoid tissues. Annu Rev Immunol. 1999;17:399–433. doi: 10.1146/annurev.immunol.17.1.399. [DOI] [PubMed] [Google Scholar]
- 18.Herlands RA, Christensen SR, Sweet RA, Hershberg U, Shlomchik MJ. T cell-independent and Toll-like receptor-dependent antigen-driven activation of autoreactive B cells. Immunity. 2008;29:249–260. doi: 10.1016/j.immuni.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barr TA, Brown S, Mastroeni P, Gray D. B cell intrinsic MyD88 signals drive IFN-gamma production from T cells and control switching to IgG2c. J Immunol. 2009;183:1005–1012. doi: 10.4049/jimmunol.0803706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nieves P, et al. Signaling via the MyD88 adaptor protein in B cells suppresses protective immunity during Salmonella typhimurium infection. Immunity. 2010;33:1–14. doi: 10.1016/j.immuni.2010.10.016. [DOI] [PubMed] [Google Scholar]
- 21.Lanzavecchia A, Sallusto F. Toll-like receptors and innate immunity in B-cell activation and antibody responses. Curr Opin Immunol. 2007;19:268–274. doi: 10.1016/j.coi.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 22.Caton ML, Smith-Raska MR, Reizis B. Notch-RBP-J signaling controls the homeostasis of CD8-dendritic cells in the spleen. J Exp Med. 2007;204:1653–1664. doi: 10.1084/jem.20062648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hou B, Reizis B, DeFranco AL. Toll-like receptors activate innate and adaptive immunity by using dendritic cell-intrinsic and -extrinsic mechanisms. Immunity. 2008;29:272–282. doi: 10.1016/j.immuni.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hobeika E, et al. Testing gene function early in the B cell lineage in mb1-cre mice. Proc Natl Acad Sci USA. 2006;103:13789–13794. doi: 10.1073/pnas.0605944103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hou B, et al. Selective utilization of Toll-like receptor and MyD88 signaling in B cells for enhancement of the antiviral germinal center response. Immunity. 2011;34:375–384. doi: 10.1016/j.immuni.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jennings GT, Bachmann MF. The coming of age of virus-like particle vaccines. Biol Chem. 2008;389:521–536. doi: 10.1515/bc.2008.064. [DOI] [PubMed] [Google Scholar]
- 27.Chaturvedi A, Dorward D, Pierce SK. The B cell receptor governs the subcellular location of Toll-like receptor 9 leading to hyperresponses to DNA-containing antigens. Immunity. 2008;28:799–809. doi: 10.1016/j.immuni.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Neill SK, et al. Endocytic sequestration of the B cell antigen receptor and toll-like receptor 9 in anergic cells. Proc Natl Acad Sci USA. 2009;106:6262–6267. doi: 10.1073/pnas.0812922106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Green NM, Marshak-Rothstein A. Toll-like receptor driven B cell activation in the induction of systemic autoimmunity. Sem Immunol. 2011;23:106–112. doi: 10.1016/j.smim.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jegerlehner A, Maurer P, Bessa J, Hinton HJ, Kopf M, Bachmann MF. TLR9 signaling in B cells determines class switch recombination to IgG2a. J Immunol. 2007;178:2415–2420. doi: 10.4049/jimmunol.178.4.2415. [DOI] [PubMed] [Google Scholar]
- 31.Ehlers M, Fukuyama H, McGaha TL, Aderem A, Ravetch JV. TLR9/MyD88 signaling is required for class switching to pathogenic IgG2a and 2b autoantibodies in SLE. J Exp Med. 2006;203:553–561. doi: 10.1084/jem.20052438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gerth AJ, Lin L, Peng SL. T-bet regulates T-independent IgG2a class switching. Int Immunol. 2003;15:937–944. doi: 10.1093/intimm/dxg093. [DOI] [PubMed] [Google Scholar]
- 33.Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC. The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science. 2007;316:1628–1632. doi: 10.1126/science.1138963. [DOI] [PubMed] [Google Scholar]
- 34.Ecki-Doma J, Batista FD. BCR-mediated uptake of antigen linked to TLR9 ligand stimulates B-cell proliferation and antigen-specific plasma cell formation. Blood. 2009;113:3969–3977. doi: 10.1182/blood-2008-10-185421. [DOI] [PubMed] [Google Scholar]
- 35.Xu Y, Harder KW, Huntington ND, Hibbs ML, Tarlinton DM. Lyn tyrosine kinase: accentuating the positive and the negative. Immunity. 2005;22:9–18. doi: 10.1016/j.immuni.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 36.Pathak S, Mohan C. Cellular and molecular pathogenesis of system lupus erythematosus: lessons from animal models. Arthritis Res Ther. 2011;241 doi: 10.1186/ar3465. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol. 2006;6:823–835. doi: 10.1038/nri1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006;312:1669–1672. doi: 10.1126/science.1124978. [DOI] [PubMed] [Google Scholar]
- 39.Subramanian S, et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci USA. 2006;103:9970–9975. doi: 10.1073/pnas.0603912103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Christensen SR, Shupe J, Nickerson KM, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 41.Neeli I, Richardson MM, Khan SN, Nicolo D, Monestier M, Radic MZ. Divergent members of a single autoreactive B cell clone retain specificity for apoptotic blebs. Mol Immunol. 2007;44:1914–1921. doi: 10.1016/j.molimm.2006.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.William J, Euler C, Christensen SR, Shlomchik MJ. Evolution of autoantibody responses via somatic hypermutation outside of germinal centers. Science. 2002;297:2066–2070. doi: 10.1126/science.1073924. [DOI] [PubMed] [Google Scholar]
- 43.Gross AJ, Lyandres JR, Panigrahi AK, Prak ET, DeFranco AL. Developmental acquisition of the Lyn-CD22-SHP-1 inhibitory pathway promotes B cell tolerance. J Immunol. 2009;182:5382–5392. doi: 10.4049/jimmunol.0803941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scapini P, et al. Myeloid cells, BAFF, and IFN-gamma establish an inflammatory loop that exacerbates autoimmunity in Lyn-deficient mice. J Exp Med. 2010;207:1757–1773. doi: 10.1084/jem.20100086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Silver KL, Crockford TL, Bouriez-Jones T, Milling S, Lambe T, Cornall RJ. MyD88-dependent autoimmune disease in Lyn-deficient mice. Eur J Immunol. 2007;37:2734–2743. doi: 10.1002/eji.200737293. [DOI] [PubMed] [Google Scholar]
- 46.Cannons JL, Tangye SG, Schwartzberg PL. SLAM family receptors and SAP adaptors in immunity. Annu Rev Immunol. 2011;29:665–705. doi: 10.1146/annurev-immunol-030409-101302. [DOI] [PubMed] [Google Scholar]