

Abstract
We here report the first complete mitochondrial (mt) genome of a skipper, Ctenoptilum vasava Moore, 1865 (Lepidoptera: Hesperiidae: Pyrginae). The mt genome of the skipper is a circular molecule of 15,468 bp, containing 2 ribosomal RNA genes, 24 putative transfer RNA (tRNA), genes including an extra copy of trnS (AGN) and a tRNA-like insertion trnL (UUR), 13 protein-coding genes and an AT-rich region. All protein-coding genes (PCGs) are initiated by ATN codons and terminated by the typical stop codon TAA or TAG, except for COII which ends with a single T. The intergenic spacer sequence between trnS (AGN) and ND1 genes also contains the ATACTAA motif. The AT-rich region of 429 bp is comprised of nonrepetitive sequences, including the motif ATAGA followed by an 19 bp poly-T stretch, a microsatellite-like (AT)3 (TA)9 element next to the ATTTA motif, an 11 bp poly-A adjacent to tRNAs. Phylogenetic analyses (ML and BI methods) showed that Papilionoidea is not a natural group, and Hesperioidea is placed within the Papilionoidea as a sister to ((Pieridae + Lycaenidae) + Nymphalidae) while Papilionoidae is paraphyletic to Hesperioidea. This result is remarkably different from the traditional view where Papilionoidea and Hesperioidea are considered as two distinct superfamilies.
1. Introduction
The taxonomic status and the phylogenetic position of skippers (Hesperiidae) within Lepidoptera remain a controversial issue [1–3]. Due to the distinct differences between the skippers and the typical butterflies/moths in terms of morphological and behavioral characteristics, such as the short stout bodies, hooked antennae, and rapid skipping flight, the skippers were previously proposed to represent a separate group that is distinct from butterflies/moths in lepidopterans. More specifically, the skippers are assigned to the family Hesperiidae in a monotypic superfamily Hesperioidea, a sister lineage to the typical rhopaloceran butterflies, which mostly belong to superfamily Papilionoidea (true butterflies) [2, 4, 5]. In addition, the three superfamilies Hesperioidea, Papilionoidea, and Hedyloidea share numerous morphological characteristics, particularly in their egg, larval, and pupal stages, and thus were considered to be a large natural group [2].
The Lepidoptera is one of the largest groups of insects, accounting for more than 160,000 species. Despite of the huge taxonomic diversity, the current information on the lepidopteran mt genomes is very limited. Only 40 lepidopteran mt genomes were sequenced, including 10 butterfly species such as Coreana raphaelis [6], Artogeia melete [7], Parnassius bremeri [8], Acraea issoria [9], Pieris rapae [10], Eumenis autonoe [11], Calinaga davidis [12], and nearly thirty moth species such as Bombyx mori, Bombyx mandarina [13], Adoxophyes honmai [14], Antheraea pernyi [15], Ochrogaster lunifer [16], Manduca sexta [17], Phthonandria atrilineata [18], Eriogyna pyretorum [19], Antheraea yamamai [20], and Caligula boisduvalii [21]. The sampling is restricted to only six superfamiles (Papilionoidea, Totricoidea, Bombycoidea, Geometroidea, Noctuoidea, and Pyraloidea) to date, a complete mt genome sequence of a skipper from the family Hesperiidae is lacking, despite of a huge diversity of the skippers (>3500 species). The lack of mt genome data from skippers dampens phylogenetic and population genetic studies in skippers and the related species.
The Tawny Angle, Ctenoptilum vasava, is a typical skipper commonly found in southern East Asia, such as China, India, Burma, Thailand, and Vietnam. In this study, we sequenced the complete mitochondrial genome of the skipper, representing the first mt genome sequence from the family Hesperiidae (superfamily Hesperioidea). We next compared this sequence with other lepidopteran mt genomes sequences and examined the phylogenetic relationships within lepidopterans and reevaluated the phylogenetic position of skippers. We show that the skipper shares the general organization and structure of the mt genome with other species from the order Lepidoptera. By examining currently available mt genomes in lepidopterans, we find the Hesperioidea is placed within the Papilionoidea, which may be a paraphyletic group.
2. Materials and Methods
2.1. Sample Collection
Adult individuals of Ctenoptilum vasava (Lepidoptera: Hesperiidae: Pyrginae: Ctenoptilum) were captured from National Natural Conservation Areas of Jiu Lianshan Mountain, Jiangxi Province, China, in August, 2009. After a brief examination for species identification, the fresh tissues were preserved in 100% ethanol immediately for DNA fixation and stored at −20°C until further use for genomic DNA isolation.
2.2. DNA Extraction and PCR Amplification
The whole genomic DNA of C. vasava was isolated from the thoracic muscle of an adult individual using the proteinase K-SiO2 method as described by Hao et al. [32]. Partial sequences of COI, COIII, Cytb, ND4, 16S rRNA, and 12S rRNA genes were amplified using insect universal primers [33]. Polymerase chain reactions (PCRs) were performed under the following condition: 5 minutes of initial denaturation at 95°C, 35 cycles of denaturation at 95°C for 50 seconds, annealing at 45–55°C (depending on primer pairs) for 50 seconds, extension at 72°C for 1 minute, and a final extension at 72°C for 10 minutes. Based on the sequences from the newly acquired gene fragments, the long PCR primers were designed (Table 1) according to the conserved regions by the program Primer premier 5.0 [34], and the entire mt genome of the skipper was in turn amplified in five long fragments (12S-COI, COI-COIII, COIII-ND4, ND4-Cytb, Cytb-12S) by using Takara LA TaqTM (Takara). The long PCR condition is as follow: an initial denaturation at 95°C for 5 min, 15 cycles of denaturation at 95°C for 50 seconds, annealing at 50–55°C (depending on primer pairs) for 50 seconds, extension at 68°C for 150 seconds, additional 15 cycles of denaturation at 95°C for 50 seconds, annealing at 50–55°C for 50 seconds, extension at 68°C for 150 seconds, and a final extension at 68°C for 10 minutes. PCR products were examined by electrophoresis on a 1% agarose gel and purified using a DNA gel extraction kit (Takara). All PCR fragments were directly sequenced in both strands after purification with QIA quick PCR Purification Kit (Qiagen). Long PCR fragments were sequenced using the primer walking strategy (walking primer information will be provided upon request).
Table 1.
Primer sequences for the long PCR amplification used in this study.
| Primers | Upper primer sequence (5′-3′) | Lower primer sequence (5′-3′) |
|---|---|---|
| COI-COIII | GGAAATTGACTTGTGCCT | TTGTATGTTTACCTTGGA |
| COIII-ND4 | AAAGGATTACGATGAGGT | GGTCTTGTTATTGGTGGA |
| ND4-Cytb | CGTCTATGTAAACGCTCA | ATAAGGGTTTTCTACTGGT |
| Cytb-12S | TTTTACATCAAACAGGA | ACTAGGATTAGATACCC |
| 12S-COI | GAAACACTTTCCAGTACCT | CTAAACCAATTCAACATCC |
2.3. Sequence Analysis
The raw sequence files were proof-read and assembled in BioEdit version 7.0 [35]. The concatenated amino acid sequences of the 13 PCGs were obtained and analysed by the ClustalX [36] and the MEGA 3.0 [37] softwares. The structures of the 23 tRNAs and a tRNA-like gene of the skipper were identified by the software tRNAscan-SE version 1.21 [38]. The putative tRNAs, which were not found by tRNAscan-SE, were identified by sequence comparisons between the skipper and other lepidopteran tRNAs. Nucleotide composition was calculated using MEGA 3.0 software [37], and the tandem repeats in the AT-rich region were predicted by the Tandem Repeats Finder available online (http://tandem.bu.edu/trf/trf.html) [39]. The mt genome sequence has been submitted to GenBank database under the accession number JF713818.
2.4. Phylogenetic Analysis
The multiple alignment of the 13 PCG concatenated nucleotide sequences of the 32 lepidopteran mt genome sequences (one is from this study, and 31 were extracted from GenBank, see Table 2) was conducted using Clustal X1.8 [36] and was checked by eye. The phylogenetic trees were reconstructed with the maximum likelihood (ML) and bayesian inference (BI) methods using a hymenopteran species Apis cerana (GenBank accession number NC_014295) as the outgroup. In both phylogenetic analyses, the third position of all the codons was excluded. The ML analyses were conducted in PAUP (version 4.0b8) [40] with searching method of TBR branch swapping (10 random addition sequences), the general time reversible model with invariant sites and among-site variation (GTR+I+Γ) was selected as the best fit model using Modeltest (version 3.06) [41] under the AIC criteria, and the bootstrap values of the ML tree were evaluated via the bootstrap test with 1000 iterations. The Bayesian analysis was performed using MrBayes 3.1.2 [42] with the partitioned strategy, and the best fit substituion model was selected as in the ML analysis. MrBayes 3.1.1 simultaneously initiates 2 Markov Chain Monte Carlo (MCMC) that runs to provide additional confirmation for the convergence of posterior probability distribution. Four simultaneous chains were run, 3 hot and 1 cold. Analyses were run for one million generations until the average standard deviation of split frequencies to be less than 0.01, which means convergence was reached. Chains were sampled every 1,000 generations. Starting trees were random.
Table 2.
Lepidopteran mt genome sequences used in this study.
| Species | Superfamily and family | GenBank acc. no. | References | |
|---|---|---|---|---|
| Hyphantria cunea | Noctuoidea | Arctiidae | NC_014058 | [22] |
| Lymantria dispar | Lymantriidae | NC_012893 | [23] | |
| Helicoverpa armigera | Noctuidae | NC_014668 | [24] | |
| Ochrogaster lunifer | Notodontidae | NC_011128 | [16] | |
| Phalera flavescens | Notodontidae | JF440342 | (Sun et al., unpublished) | |
| Phthonandria atrilineata | Geometroidea | Geometridae | NC_010522 | [18] |
| Spilonota lechriaspis | Tortricoidea | Tortricidae | NC_014294 | [25] |
| Adoxophyes honmai | Tortricidae | NC_008141 | [14] | |
| Grapholita molesta | Tortricidae | NC_014806 | [26] | |
| Antheraea pernyi | Bombycoidea | Saturniidae | NC_004622 | [15] |
| Antheraea yamamai | Saturniidae | NC_012739 | [20] | |
| Eriogyna pyretorum | Saturniidae | NC_012727 | [19] | |
| Caligula boisduvalii | Saturniidae | NC_010613 | [21] | |
| Chinese Bombyx mandarina | Bombycidae | AY301620 | [27] | |
| Bombyx mandarina | Bombycidae | NC_003395 | [13] | |
| Bombyx mori | Bombycidae | NC_002355 | [13] | |
| Manduca sexta | Sphingidae | NC_010266 | [17] | |
| Diatraea saccharalis | Pyraloidea | Crambidae | NC_013274 | [28] |
| Ostrinia furnacalis | Crambidae | NC_003368 | [29] | |
| Ostrinia nubilalis | Crambidae | NC_003367 | [29] | |
| Eumenis autonoe | Papilionoidea | Nymphalidae | NC_014587 | [11] |
| Acraea issoria | Nymphalidae | GQ376195 | [9] | |
| Sasakia charonda kuriyamaensis | Nymphalidae | NC_014223 | (Hakozaki et al., unpublished) | |
| Argyreus hyperbius | Nymphalidae | JF439070 | [30] | |
| Calinaga davidis | Nymphalidae | HQ658143 | [12] | |
| Papilio maraho | Papilionidae | NC_014055 | [31] | |
| Parnassius bremeri | Papilionidae | NC_014053 | [8] | |
| Teinopalpus aureus | Papilionidae | NC_014398 | (Qin et al., unpublished) | |
| Troides aeacus | Papilionidae | EU625344 | (Jiang et al., unpublished) | |
| Coreana raphaelis | Lycaenidae | NC_007976 | [6] | |
| Pieris raphaelis | Pieridae | HM156697 | [10] | |
| Ctenoptilum vasava | Hesperoidea | Hesperiidae | JF713818 | This study |
3. Results and Discussion
3.1. Genome Organization, Gene Arrangement, and Base Composition
The organization of the skipper mt genome was shown in Figure 1. The complete mt genome is 15, 468 bp in length, containing 13 protein-coding genes (ND1-6, ND4L, COI-III, Cytb, ATP6, ATP8), 2 ribosomal RNAs (12S and 16S), 24 putative tRNAs, and an AT-rich region, same characteristics as with those of other butterfly species available (Tables 3 and 4). The size of so far sequenced mt genomes in lepidopteran insects ranges from 15,140 bp in Artogeia melete [7] to 15,928 bp in Bombyx mandarina [13]; our newly sequenced mt genome of the skipper is within the above size range. Mitochondrial genes of the skipper are arranged in the same order and orientation as those of other lepidopterans, except for the presence of an extra copy of trnS (AGN) and a tRNA-like insertion trnL(UUR). Similar to many insect mt genomes, the heavy strand (H-strand) encodes more genes (9 PCGs and 14 tRNAs), whereas the light strand (L-strand) encodes less (4 PCGs, 8 tRNAs, and 2 rRNA genes).
Figure 1.
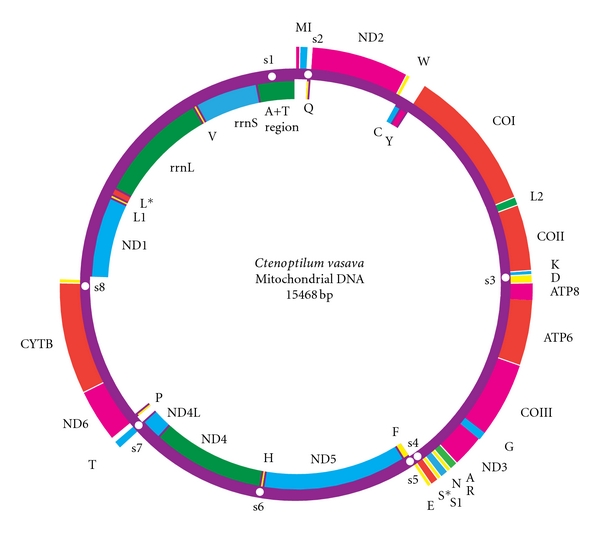
Map of the circular mitochondrial genome of Ctenoptilum vasava. Genes encoded in the H-strand (clockwise orientation) are colored in red or pink. Genes encoded in the L-strand (anticlockwise orientation) are colored in blue or green. The abbreviations for the genes are as follows: COI-III stands for cytochrome oxidase subunits, Cytb for cytochrome b, and ND1-6 for NADH dehydrogenase components. tRNAs are denoted as one-letter symbol according to the IUPAC-IUB single-letter amino acid codes.
Table 3.
Comparison of the mt genome characteristics of all butterfly species available. All genomes share the features of 13 protein-coding genes (ND1-6, ND4L, COI-III, Cytb, ATP6, ATP8), 2 ribosomal RNAs (lsu rRNA (12S) and lsu rRNA (16S)), and an AT-rich region.
| Taxon | Whole genomic DNA | lrRNA | srRNA | A+T-rich region | GenBank accession no. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Size (bp) | A+T % | No. codonsa | PCGb A+T % | Size (bp) | A+T % | Size (bp) | A+T % | Size (bp) | A+T % | ||
| Adoxophyes honmai | 15,680 | 80.4 | 3,748 | 78.5 | 1,387 | 83.6 | 779 | 85.4 | 489 | 94.3 | DQ073916 |
| Antheraea pernyi | 15,575 | 80.2 | 3,732 | 78.5 | 1,369 | 83.9 | 775 | 84.1 | 552 | 90.4 | AY242996 |
| Antheraea yamamai | 15,338 | 80.2 | 3,714 | 79.3 | 1,380 | 83.5 | 776 | 85.9 | 334 | 90.4 | EU726630 |
| Caligula boisduvalii | 15,360 | 80.6 | 3,734 | 79.1 | 1,391 | 84.8 | 774 | 84.1 | 330 | 91.5 | EF622227 |
| Eriogyna pyretorum | 15,327 | 80.8 | 3,711 | 79.3 | 1,338 | 84.6 | 762 | 84.5 | 374 | 91.7 | FJ685653 |
| Bombyx mori | 15,656 | 81.4 | 3,720 | 79.5 | 1,378 | 84.4 | 783 | 85.6 | 494 | 95.5 | AB070264 |
| Manduca sexta | 15,516 | 81.8 | 3,718 | 80.2 | 1,391 | 84.7 | 777 | 86.8 | 324 | 95.1 | EU286785 |
| Phthonandria atrilineata | 15,499 | 81.1 | 3,724 | 79.0 | 1,400 | 85.1 | 803 | 87.5 | 457 | 98.5 | NC010522 |
| Parnassius bremeri | 15,389 | 81.3 | 3,734 | 80.2 | 1,344 | 83.8 | 773 | 85.1 | 504 | 93.6 | FJ871125 |
| Artogeia melete | 15,140 | 79.8 | 3,715 | 78.4 | 1,319 | 83.4 | 777 | 86.9 | 351 | 88.0 | NC010568 |
| Coreana raphaelis | 15,314 | 82.7 | 3,708 | 81.5 | 1,330 | 85.3 | 777 | 85.8 | 375 | 94.1 | DQ102703 |
| Eumenis autonoe | 15,489 | 79.1 | 3,728 | 76.8 | 1,335 | 83.7 | 775 | 85.3 | 678 | 94.5 | GQ868707 |
| Apatura ilia | 15,242 | 80.5 | 3,711 | 78.9 | 1,333 | 86.0 | 776 | 84.9 | 403 | 92.5 | JF437925 |
| Calinaga davidis | 15,267 | 80.4 | 3,737 | 78.8 | 1,337 | 83.8 | 773 | 85.9 | 403 | 92.5 | HQ658143 |
| Argyreus hyperbius | 15,156 | 80.8 | 4,164 | 79.7 | 1,330 | 84.5 | 778 | 85.2 | 349 | 95.4 | JF439070 |
| Acraea issoria | 15,245 | 79.7 | 3,745 | 78.0 | 1,331 | 83.8 | 788 | 83.8 | 430 | 96.0 | GQ376195 |
| Ochrogaster lunifer | 15,593 | 77.9 | 3,746 | 75.7 | 1,351 | 81.4 | 806 | 84.3 | 319 | 92.7 | AM946601 |
| Lymantria dispar | 15,569 | 79.9 | 3,742 | 77.8 | 1,351 | 84.2 | 799 | 85.2 | 435 | 96.1 | FJ617240 |
| Ctenoptilum vasava | 15,468 | 80.5 | 3,698 | 78.9 | 1,343 | 84.1 | 774 | 86.4 | 429 | 88.1 | JF713818 |
Table 4.
Summary of the skipper mt genome. IGNc stands for intergenic nucleotides, and the positive number indicates interval nucleotides (base pairs) between genes, while the negative number indicates the overlapped nucleotides (base pairs) between genes.
| Gene | Direction | Nucleotide no. | Size | IGNc | Start codon | Stop codon |
|---|---|---|---|---|---|---|
| tRNAMet | F | 1–68 | 68 | 0 | ||
| tRNAlle | F | 69–133 | 65 | 0 | ||
| tRNAGln | R | 136–204 | 69 | 2 | ||
| ND2 | F | 266–1270 | 1005 | 61 | ATT | TAG |
| tRNATrp | F | 1269–1335 | 67 | −2 | ||
| tRNACys | R | 1331–1395 | 65 | −5 | ||
| tRNATyr | R | 1407–1471 | 65 | 11 | ||
| COI | F | 1469–2990 | 1522 | −3 | ATT | T-tRNA |
| tRNALeu(UUR) | F | 2991–3057 | 67 | 0 | ||
| COII | F | 3058–3731 | 674 | 0 | ATG | T-tRNA |
| tRNALys | F | 3732–3801 | 70 | 0 | ||
| tRNAAsp | F | 3816–3884 | 69 | 14 | ||
| ATP8 | F | 3885–4052 | 168 | 0 | ATA | TAA |
| ATP6 | F | 4046–4723 | 678 | −7 | ATG | TAA |
| COIII | F | 4723–5505 | 783 | −1 | ATG | TAA |
| tRNAGly | F | 5508–5574 | 67 | 2 | ||
| ND3 | F | 5575–5928 | 354 | 0 | ATT | TAG |
| tRNAAla | F | 5927–5990 | 64 | −2 | ||
| tRNAArg | F | 5996–6061 | 66 | 5 | ||
| tRNAAsn | F | 6061–6125 | 65 | −1 | ||
| tRNASer(AGN) | F | 6133–6194 | 62 | 7 | ||
| tRNASer(AGN) | F | 6208–6267 | 60 | 13 | ||
| tRNAGlu | F | 6279–6344 | 66 | 11 | ||
| tRNAPhe | R | 6343–6409 | 67 | −2 | ||
| ND5 | R | 6409–8145 | 1737 | −1 | ATA | TAA |
| tRNAHis | R | 8161–8228 | 68 | 15 | ||
| ND4 | R | 8228–9568 | 1341 | −1 | ATG | TAA |
| ND4L | R | 9574–9855 | 282 | 5 | ATG | TAA |
| tRNAThr | F | 9869–9933 | 65 | 13 | ||
| tRNAPro | R | 9934–9998 | 65 | 0 | ||
| ND6 | F | 10001–10531 | 531 | 2 | ATA | TAA |
| Cytb | F | 10531–11679 | 1149 | −1 | ATG | TAA |
| tRNASer | F | 11679–11745 | 67 | −1 | ||
| ND1 | R | 11763–12704 | 942 | 17 | ATG | TAA |
| tRNALeu(CUN) | R | 12706–12773 | 68 | 1 | ||
| tRNALeu(UUR) | F | 12774–12852 | 79 | 0 | ||
| 16S | R | 12853–14195 | 1343 | 0 | ||
| tRNAVal | R | 14196–14265 | 70 | 0 | ||
| 12S | R | 14266–15039 | 774 | 0 | ||
| A+T-rich region | 15040–15468 | 429 | 0 |
The composition of A, T, G, C nucleotides in the skipper mt genome L-strand is 39.09%, 41.45%, 7.73%, and 11.72%, respectively, indicating a remarkably high AT content (80.55%) and a low GC content (19.45%). This result is consistent with the AT bias generally observed in other lepidopteran mt genomes, given that the composition of AT is ranging from 77.9% in Ochrogaster lunifer [16] to 82.7% in Coreana raphaelis [6]. The AT and GC skew values in H-strand of the skipper mt genome are −0.0293 and −0.216, respectively, indicating that T and C are favored over A and G, respectively.
3.2. Protein-Coding Genes (PCG)
The 13 PCGs of the skipper mt genome include 7 NADH dehydrogenase subunits, 3 cytochrome c oxidase subunits, 2 ATPase subunits, and one cytochrome b gene. The 13 PCGs are 11,172 bp in length which accounts for 72.23% of the whole mitochondrial genome, encoding 3,724 amino acid residues. All the protein-coding genes except for the COI start with a canonical start codon ATN. More specifically, 7 PCGs (COII, ATP6, COIII, ND4, ND4L, Cytb, and ND1) start with ATG, 2PCGs (ND2, ND3) with ATT, additional 3 PCGs (ATP8, ND5, ND6) with ATA. For the stop codon, 3 PCGs (the ND2, ND3, and ND5) terminate with ATG, 2 PCGs (COI and COII) with a single T, the remaining 8 PCGs with the typical stop codon TAA.
The start codons for COI gene of the lepidopteran insects are not uniform. All lepidopteran species examined to date use Arginine (R), which correspond to the codon CGA, as the initial amino acid for COI [43]. In this study, the CGA is not conserved across all lepidopteran mt genomes, but it has been suggested to serve as the COI start codon [8]. However, in other insect groups, some other canonical codons, such as the TTA [44], TCG [45], TTG [46], ACG [47], were reported as the COI start codons. Additionally, some tetranucleotides, such as the ATAA, ATCA, and ATTA [48], as well as the hexanucleotides, such as the ATTTAA [49–51], TATCTA [52], TTTTAG [13], and TATTAG [29], were also proposed as the start codons for this gene.
The 13 protein-coding genes all possess complete stop codons except for the COI and COII in this study. Most stop codons for COII are single T in most sequenced lepidopteran mt genomes. The single T stop codon is usually located nearby the trnK, the structure of which is recognized by endonucleases processing the polycistronic pre-mRNA transcription, and produces functional stop codons by polyadenylation from its contiguous protein-coding genes [53–55]. Accordingly, partial stop codons observed in most lepidopteran species would be one strategy for the selection of stop codon; in other words, it would minimize the intergenic spacers and gene overlaps from an evolutionary economic perspective.
The amino acids composition and the codon usage of the skipper mt genome are shown in the Tables 5 to 6. The results showed that the codon usage of all the genes has a strong bias, and the RSCU (relative synonymous codon usage) of NNU and NNA codons are greater than 1, indicating that the third positions of the UA have a high frequency of codon usage. The codon usage bias in protein-coding genes and the third position of AT bias (92.1%) are positively correlated. In addition, our analysis also showed that UUU (Phe), UUA (Leu), AUU (Ile), AUA (Met), and AAU (Asn) are the most frequently used codons (45.45%). Together, all observations suggest the strong AT bias of the protein-coding genes in the skipper mt genome.
Table 5.
Amino acid composition of the mt genome of the skipper. Start codons and stop codons were excluded in total codon counts.
| Ala | Cys | Asp | Glu | Phe | Gly | His | Ile | Lys | Leu | Met | Asn | Pro | Gln | Arg | Ser | Thr | Val | Trp | Tyr | Total | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ND2 | 2.10 | 1.20 | 0.30 | 1.50 | 17.72 | 3.30 | 0.60 | 13.81 | 5.11 | 11.11 | 8.41 | 8.41 | 1.50 | 1.20 | 0.90 | 12.01 | 3.00 | 1.20 | 2.40 | 4.20 | 333 |
| COI | 5.72 | 0.39 | 2.76 | 1.58 | 8.88 | 7.89 | 2.96 | 10.65 | 1.78 | 11.64 | 6.34 | 4.93 | 5.13 | 1.97 | 1.38 | 9.47 | 5.52 | 4.14 | 2.56 | 4.34 | 507 |
| COII | 3.59 | 0.9 | 3.59 | 4.48 | 7.17 | 2.69 | 2.24 | 13.9 | 2.24 | 12.11 | 4.48 | 9.87 | 4.48 | 4.04 | 3.14 | 6.73 | 4.48 | 3.59 | 2.69 | 3.59 | 223 |
| ATP8 | 0.00 | 0.00 | 0.00 | 0.00 | 12.96 | 0.00 | 0.00 | 20.37 | 9.26 | 5.56 | 9.26 | 12.96 | 5.56 | 1.85 | 0.00 | 3.70 | 1.85 | 0.00 | 5.56 | 11.11 | 54 |
| ATP6 | 2.23 | 0.00 | 0.45 | 1.79 | 9.82 | 4.46 | 2.23 | 12.95 | 0.89 | 17.41 | 4.91 | 7.14 | 4.91 | 2.23 | 1.34 | 11.16 | 7.14 | 4.02 | 2.23 | 2.68 | 224 |
| COIII | 5.02 | 0.77 | 1.54 | 2.70 | 11.20 | 6.56 | 5.02 | 12.74 | 1.54 | 10.42 | 2.70 | 4.25 | 3.86 | 2.32 | 1.54 | 6.18 | 7.72 | 3.47 | 5.02 | 5.41 | 259 |
| ND1 | 2.88 | 1.28 | 2.24 | 3.21 | 10.26 | 5.13 | 0.00 | 9.62 | 3.53 | 18.59 | 5.13 | 4.49 | 1.92 | 1.28 | 1.60 | 11.86 | 3.21 | 4.49 | 1.92 | 7.37 | 312 |
| Cytb | 4.46 | 0.79 | 1.84 | 1.05 | 9.45 | 5.77 | 2.10 | 14.17 | 2.89 | 14.44 | 3.67 | 7.09 | 5.77 | 2.36 | 1.84 | 5.51 | 5.25 | 3.67 | 3.15 | 4.72 | 381 |
| ND6 | 1.14 | 0.57 | 0.57 | 0.57 | 12.57 | 2.86 | 0.57 | 15.43 | 4.57 | 18.29 | 7.43 | 10.86 | 1.14 | 1.14 | 0.57 | 6.29 | 3.43 | 2.86 | 1.14 | 8.00 | 175 |
| ND4L | 1.09 | 1.09 | 1.09 | 4.35 | 10.87 | 4.35 | 3.26 | 15.22 | 3.26 | 18.48 | 11.96 | 3.26 | 0.00 | 1.09 | 1.09 | 9.78 | 0.00 | 5.43 | 0.00 | 4.43 | 92 |
| ND4 | 3.15 | 2.02 | 0.90 | 2.02 | 9.66 | 6.07 | 1.35 | 9.66 | 0.36 | 17.30 | 10.56 | 4.94 | 2.47 | 1.35 | 1.12 | 8.99 | 1.80 | 3.15 | 2.70 | 7.19 | 445 |
| ND5 | 3.29 | 0.87 | 2.25 | 1.91 | 9.36 | 4.16 | 0.69 | 10.75 | 3.81 | 16.64 | 9.88 | 5.72 | 1.91 | 1.04 | 0.87 | 11.44 | 2.60 | 4.51 | 1.56 | 6.76 | 577 |
| ND3 | 1.72 | 1.72 | 2.59 | 3.45 | 11.21 | 1.72 | 1.72 | 19.83 | 6.03 | 12.96 | 5.17 | 6.03 | 3.45 | 0.86 | 1.72 | 10.34 | 3.45 | 1.72 | 2.59 | 1.72 | 116 |
|
| |||||||||||||||||||||
| Total | 36.39 | 11.60 | 20.12 | 28.61 | 141.13 | 54.96 | 22.74 | 179.10 | 45.27 | 185.0 | 89.90 | 89.95 | 42.10 | 22.73 | 17.11 | 103.12 | 46.00 | 42.25 | 33.52 | 71.52 | 3698 |
Table 6.
The codon usage of the skipper mt genome. n: frequency of codon used; RSCU: relative synonymous codon usage; *stop codon. Start codons and stop codons were excluded in total codon counts.
| Codon (Aa) | n (RSCU) | Codon (Aa) | n (RSCU) | Codon (a) | n (RSCU) | Codon (Aa) | n (RSCU) |
|---|---|---|---|---|---|---|---|
| UUU (F) | 351.0 (1.81) | UCU (S) | 116.0 (2.71) | UAU (Y) | 192.0 (1.90) | UGU (C) | 34.0 (1.94) |
| UUC (F) | 37.0 (0.19) | UCC (S) | 16.0 (0.37) | UAC (Y) | 10.0 (0.10) | UGC (C) | 1.0 (0.06) |
| UUA (L) | 458.0 (5.07) | UCA (S) | 84.0 (1.96) | UAA (*) | 0.0 (0.00) | UGA (W) | 86.0 (1.87) |
| UUG (L) | 13.0 (0.14) | UCG (S) | 7.0 (0.16) | UAG (*) | 0.0 (0.00) | UGG (W) | 6.0 (0.13) |
| CUU (L) | 45.0 (0.50) | CCU (P) | 74.0 (2.45) | CAU (H) | 55.0 (1.72) | CGU (R) | 15.0 (1.20) |
| CUC (L) | 6.0 (0.07) | CCC (P) | 17.0 (0.56) | CAC (H) | 9.0 (0.28) | CGC (R) | 0.0 (0.00) |
| CUA (L) | 19.0 (0.21) | CCA (P) | 29.0 (0.96) | CAA (Q) | 63.0 (1.97) | CGA (R) | 31.0 (2.48) |
| CUG (L) | 1.0 (0.01) | CCG (P) | 1.0 (0.03) | CAG (Q) | 1.0 (0.03) | CGG (R) | 4.0 (0.32) |
| AUU (I) | 431.0 (1.89) | ACU (T) | 94.0 (2.54) | AAU (N) | 209.0 (1.79) | AGU (S) | 36.0 (0.84) |
| AUC (I) | 26.0 (0.11) | ACC (T) | 7.0 (0.19) | AAC (N) | 25.0 (0.21) | AGC (S) | 3.0 (0.07) |
| AUA (M) | 245.0 (1.91) | ACA (T) | 45.0 (1.22) | AAA (K) | 106.0 (1.77) | AGA (S) | 74.0 (1.73) |
| AUG (M) | 12.0 (0.09) | ACG (T) | 2.0 (0.05) | AAG (K) | 14.0 (0.23) | AGG (S) | 6.0 (0.14) |
| GUU (V) | 61.0 (1.86) | GCU (A) | 77.0 (2.44) | GAU (D) | 58.0 (1.81) | GGU (G) | 46.0 (1.00) |
| GUC (V) | 1.0 (0.03) | GCC (A) | 13.0 (0.41) | GAC (D) | 6.0 (0.19) | GGC (G) | 5.0 (0.11) |
| GUA (V) | 61.0 (1.86) | GCA (A) | 33.0 (1.05) | GAA (E) | 71.0 (1.84) | GGA (G) | 109.0 (2.37) |
| GUG (V) | 8.0 (0.24) | GCG (A) | 3.0 (0.10) | GAG (E) | 6.0 (0.16) | GGG (G) | 24.0 (0.52) |
3.3. Ribosomal and Transfer RNA Genes
The srRNA (12S rRNA) and lrRNA (16S rRNA) genes in the skipper mt genome are 774 bp and 1343 bp in size, respectively. Similar to other lepidopteran rRNAs [8, 22], the skipper 12S rRNA is located between trnL (CUN) and trnV, whereas its 16S rRNA resides between trnV and the A+T-rich region. The A+T contents of the two rRNA genes are 86.43% and 84.14%, respectively, which is consistent with those observed in other lepidopterans as well.
The skipper mt genome contains 23 transfer RNAs and a tRNA-like gene (trnL (UUR)), and these genes are interspersed throughout the whole genome and ranged in size from 61 to 79 bp. Twenty-three of them are shown to be folded into the expected clover-leaf secondary structure; however, the trnS (AGN) lacks the dihydrouridine (DHU) loop (Figure 2) as shown in the other insect mt genomes examined to date. Therefore, the incomplete trnS should be considered as one of common features in most, if not all, insect mt genomes. A total of 32 unmatched base pairs were detected in the skipper tRNAs, and 18 of them are GU pairs, which form a weak bond in the tRNAs, the remaining 14 are atypical pairs, including 11 UU pairs, two AC pairs, and one AA pairs (Figure 2).
Figure 2.
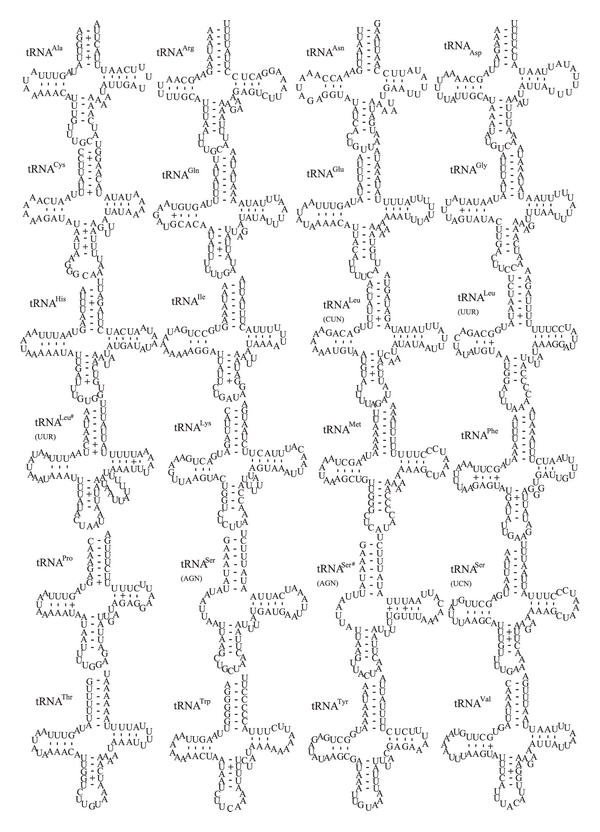
Predicted secondary clover-leaf structure for the tRNA genes of Ctenoptilum vasava. Dashes: Watson-Crick base pairing; centered dots (+): unmatched base pairing.
The extra trnS was detected between trnS (AGN) and trnE in the skipper mt genome (Figure 2), and the tRNA insertions were also observed in other lepidopterans, such as C. raphaelis and A. issoria. The high similarity between the two tandemly repeated copies of the trnS in primary and secondary structures may suggest a recent duplication event [6, 9]. Interestingly, a 79 bp insertion of a tRNA-like gene was detected between the trnL (CUN) and lrRNA genes, and this observation was not shown in any lepidopteran mt genomes to date, despite that tRNA translocation is a frequent event in the evolution of lepidopteran mt genomes [56].
3.4. Intergenic Spacers and Overlapping Sequences
The skipper mt genome contains a total of 607 bp intergenic spacers, which distribute in 11 regions, ranging from 2 to 61 bp in length. Most of the spacers are shorter than 10 bp, only 7 spacers are longer (Table 2). The longest intergenic spacer (61 bp) is located between the trnQ and ND2 genes, with a high AT content (98.4%). This spacer is usually considered as a constitutive synapomorphic feature of lepidopteran mt genomes because of the absence in nonlepidopteran insects [17]. The second longest one (ATACTAAAAATATATTA) is inserted between the trnS2 and ND1 and shared the ATACTAA motif observed in most insects, including lepidopterans [17], and possibly fundamental to site recognition by the transcription termination peptide (mtTERM protein) [57]. The third longest spacer (CAATTTCTTTT) is inserted between the trnC and trnY, which is the same as O. lunifer. The fourth longest spacer (AAATTATTAAATTT) is located between trnK and trnD, which is also found in other lepidopterans. The fifth longest one (TTTTCTTTTCTTT) is resided between the two trnS, which is almost identical to that in C. raphaelis.
The skipper mt genes are overlapped in a total of 51 bp at 12 locations, with the longest overlap of 7 bp, which is located between the ATP8 and ATP6. The overlapped nucleotides are shared in all the lepidopteran mt genomes so far examined and are probably helpful to form the structure of hairpin loop for posttranslation modifications [6, 43]. Other overlap regions include the 5 bp nucleotides between the trnW and trnC, 3 bp nucleotides between the trnY and COI, and the remaining 9 overlapping nucleotides are shorter than 3 bp (Table 4). Of the overlaps, the one between the trnF and ND5 is only 1 bp in size, substantially shorter than those of other lepidopteran species, such as the A. pernyi (15 bp), C. boisduvalii (17 bp), E. pyretorum (17 bp), O. furnacalis (15 bp), O. nubilalis (16 bp), E. autonoe (16 bp), and A. honmai (29 bp).
3.5. The AT-Rich Region
The 429 bp AT-rich region with the AT content of 88.11% is located between the srRNA and trnM. This region includes the ON (origin of minority or light strand replication) which has the motif ATAGA and is followed by 19 bp poly-T that is conserved across all lepidopterans. The AT-rich region also includes a few short microsatellite-like repeat regions such as poly-A, poly-T, and (AT)3 (TA)9 prior to the structural motif ATTTA or ATTA that is common to all the lepidopteran mt genomes. In addition, for the first time, we found two large repeated elements, the 20 bp repeat (TATAATTTAATAAAAAAAATA), in the AT-rich region in a lepidopteran mt genome (Figure 3), similar observation was also reported in satyrid Eumenis autonoe, despite that their functions still remain unknown [11].
Figure 3.

Repeated segments in A+T-rich region in the skipper mt genome. The consensus pattern (20 bp) repeat unit (TATAATTTAATAAAAAAAATA) is highlighted in red and purple. Numbers are shown as the positions of starting nucleotides.
4. Phylogenetic Analysis
A total of 32 lepidopteran species represent seven lepidopteran superfamilies (Tortricoidea, Pyraloidea, Papilionoidea, Hesperioidea, Bombycoidea, Geometroidea, and Noctuoidea). Of them, the superfamilies Papilionoidea and Hesperioidea are usually referred to as the macrolepidopterans together with the superfamilies Bombycoidea, Geometroidea, and Noctuoidea. Their phylogenetic relationships have long been a subject of controversy [58–61]. One of the most compelling hypotheses is that the Hesperioidea and Papilionoidea are closely related to each other [58, 60].
The ML and BI phylogenetic trees in this study based on 13 protein coding genes of the mitochondrial genomes revealed two major clusters: the first includes Bombycoidea, Geometroidea, Noctuoidea, Tortricoidea, and Cramboidea, and the second contains Papilionoidea and Hesperioidea with strong support (Figure 4(a)). Because the first cluster was also revealed by Kim et al. [11] and Mutanen et al. [62], we here focus on the second cluster, in which the superfamily Papilionoidea appears to be paraphyletic (Figure 4(a)) or polyphyletic (Figure 4(b)), and the superfamily Hesperioidea is placed within the Papilionoidea as a sister to ((Pieridae + Lycaenidae) + Nymphalidae). This result is consistent with those reported earlier based on combined data sets of a few nuclear and mitochondrial genes [62–64]. It is, however, remarkably different from the traditional view where Papilionoidea and Hesperioidea are considered as two distinct superfamilies. In conclusion, on the contrary to the common view that Papilionoidea and Hesperioidea are two separate groups in light of morphological and behavioral characteristics, our new mt genome data support that, while the majority of the Papilionoidea lineages is a sister group to Hesperioidea, the Papilionoidae is a separate clade among the two superfamilies. Our results call for more data from Hesperioidea to establish the accurate phylogeny of skippers (Hesperioidea) and true butterflies (Papilionoidea).
Figure 4.
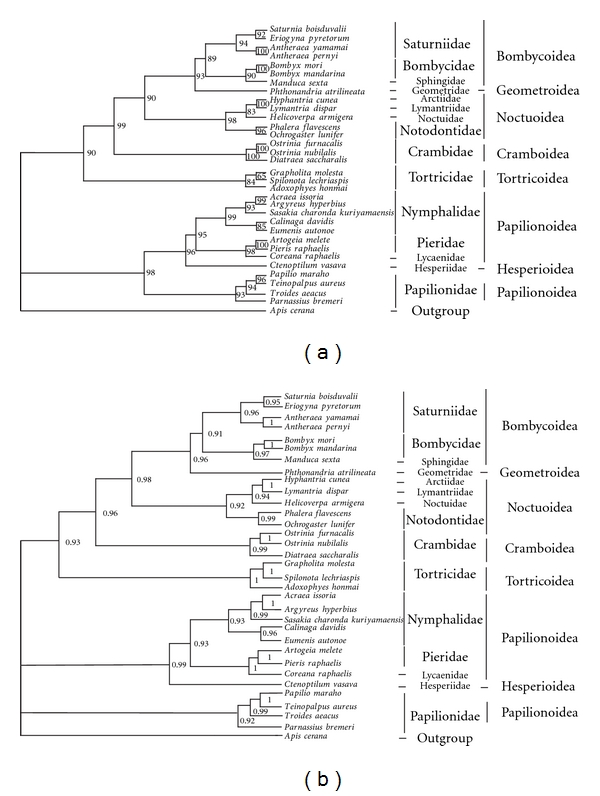
Phylogenetic trees of the lepidopterans based on 13 PCG nucleotide sequences. (a) ML tree. (b) BI tree. Numbers at each node indicate bootstrap percentages of 100 pseudoreplicates for ML analysis and posterior probability for BI analysis.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grants no. 41172004 and 40902004), the CAS/SAFEA International Partnership Program for Creative Research Teams, Chinese Academy of Sciences (Grant no. KZCX22YW2JC104), the Provincial Key Project of the Natural Science Foundation from the Anhui Province, China (Grant no. KJ2010A142), and the Opening Funds from the State Key Laboratory of Palaeobiology and Stratigraphy, Nanjing Institute of Geology and Palaeontology, Chinese Academy of Sciences. The authors also thank Dr. Gang Liu (School of Resources and Environmental Engineering, Anhui University) for his assistance in the laboratory and valuable comments on an earlier version of this paper.
References
- 1.Bridges CA. Catalogue of Hesperiidae (Lepidoptera: Rhopalocera) Urbana, Ill, USA: 1988. [Google Scholar]
- 2.Ackery PR, de Jong R, Vane-Wright RI. The butterflies: hedyloidea, hesperioidea and papilionoidae. In: Kristensen NP, editor. Handbook of Zoology, A Natural History of the Phyla of the Animal Kingdom Vol. 4, Arthropoda: Insecta—Part 35, Lepidoptera, Moths and Butterflies, Evolution, Systematics and Biogeography. Vol. 1. 1999. pp. 263–300. [Google Scholar]
- 3.Gorbunov PY. The butterflies of Russia: classification, genitalia, keys for identification (Lepidoptera: Hesperioidea and Papilionoidea) Ekaterinburg, Russia: Tezis Izdatel Stvo; 2001. Ph.D. thesis. [Google Scholar]
- 4.Chiba H, Eliot JN. A review of the genus Parnara Moore ( Lepidoptera: Hesperiidae ), with special reference to the Asian species. Tyo to Ga. 1991;42(3):179–194. [Google Scholar]
- 5.Chou IO. Classification and Indentification of Chinese Butterflies. Zhengzhou, China: Henan Scientific and Technological Publishing House; 1998. [Google Scholar]
- 6.Kim I, Lee EM, Seol KY, et al. The mitochondrial genome of the Korean hairstreak, Coreana raphaelis (Lepidoptera: Lycaenidae) Insect Molecular Biology. 2006;15(2):217–225. doi: 10.1111/j.1365-2583.2006.00630.x. [DOI] [PubMed] [Google Scholar]
- 7.Hong GY, Jiang ST, Yu M, et al. The complete nucleotide sequence of the mitochondrial genome of the cabbage butterfly, Artogeia melete (Lepidoptera: Pieridae) Acta Biochimica et Biophysica Sinica. 2009;41(6):446–455. doi: 10.1093/abbs/gmp030. [DOI] [PubMed] [Google Scholar]
- 8.Kim MI, Baek JY, Kim MJ, et al. Complete nucleotide sequence and organization of the mitogenome of the red-spotted apollo butterfly, Parnassius bremeri (Lepidoptera: Papilionidae) and comparison with other lepidopteran insects. Molecules and Cells. 2009;28(4):347–363. doi: 10.1007/s10059-009-0129-5. [DOI] [PubMed] [Google Scholar]
- 9.Hu J, Hao JS, Zhang DX, Huang D, Cameron S, Zhu CD. The complete mitochondrial genome of the yellow coaster, Acraea issoria (Lepidoptera: Nymphalidae: Heliconiinae: Acraeini): Sequence, gene organization and a unique tRNA translocation event. Molecular Biology Reports. 2010;37(7):3431–3438. doi: 10.1007/s11033-009-9934-3. [DOI] [PubMed] [Google Scholar]
- 10.Mao ZH, Hao JS, Zhu GP, Hu J, Si MM, Zhu CD. Sequencing and analysis of the complete mitochondrial genome of Pieris rapae Linnaeus (Lepidoptera: Pieridae) Acta Entomologica Sinica. 2010;53(11):1295–1304. [Google Scholar]
- 11.Kim MJ, Wan XL, Kim KG, Hwang JS, Kim I. Complete nucleotide sequence and organization of the mitogenome of endangered Eumenis autonoe (Lepidoptera: Nymphalidae) African Journal of Biotechnology. 2010;9(5):735–754. [Google Scholar]
- 12.Xia J, Hu J, Zhu GP, Zhu CD, Hao JS. Complete mitochondrial DNA sequence of the Calinaga davidis. Acta Entomologica Sinica. 2011;54(5):555–565. [Google Scholar]
- 13.Yukuhiro K, Sezutsu H, Itoh M, Shimizu K, Banno Y. Significant levels of sequence divergence and gene rearrangements have occurred between the mitochondrial genomes of the wild mulberry silkmoth, Bombyx mandarina, and its close relative, the domesticated silkmoth, Bombyx mori . Molecular Biology and Evolution. 2002;19(8):1385–1389. doi: 10.1093/oxfordjournals.molbev.a004200. [DOI] [PubMed] [Google Scholar]
- 14.Lee ES, Shin KS, Kim MS, Park H, Cho S, Kim CB. The mitochondrial genome of the smaller tea tortrix Adoxophyes honmai (Lepidoptera: Tortricidae) Gene. 2006;373(1-2):52–57. doi: 10.1016/j.gene.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 15.Liu Y, Li Y, Pan M, et al. The complete mitochondrial genome of the Chinese oak silkmoth, Antheraea pernyi (Lepidoptera: Saturniidae) Acta Biochimica et Biophysica Sinica. 2008;40(8):693–703. [PubMed] [Google Scholar]
- 16.Salvato P, Simonato M, Battisti A, Negrisolo E. The complete mitochondrial genome of the bag-shelter moth Ochrogaster lunifer (Lepidoptera, Notodontidae) BMC Genomics. 2008;9, article 331 doi: 10.1186/1471-2164-9-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cameron SL, Whiting MF. The complete mitochondrial genome of the tobacco hornworm, Manduca sexta, (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene. 2008;408(1-2):112–123. doi: 10.1016/j.gene.2007.10.023. [DOI] [PubMed] [Google Scholar]
- 18.Yang L, Wei ZJ, Hong GY, Jiang ST, Wen LP. The complete nucleotide sequence of the mitochondrial genome of Phthonandria atrilineata (Lepidoptera: Geometridae) Molecular Biology Reports. 2009;36(6):1441–1449. doi: 10.1007/s11033-008-9334-0. [DOI] [PubMed] [Google Scholar]
- 19.Jiang ST, Hong GY, Yu M, et al. Characterization of the complete mitochondrial genome of the giant silkworm moth, Eriogyna pyretorum (Lepidoptera: Saturniidae) International Journal of Biological Sciences. 2009;5(4):351–365. doi: 10.7150/ijbs.5.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim SR, Kim MI, Hong MY, et al. The complete mitogenome sequence of the Japanese oak silkmoth, Antheraea yamamai (Lepidoptera: Saturniidae) Molecular Biology Reports. 2009;36(7):1871–1880. doi: 10.1007/s11033-008-9393-2. [DOI] [PubMed] [Google Scholar]
- 21.Hong MY, Lee EM, Jo YH, et al. Complete nucleotide sequence and organization of the mitogenome of the silk moth Caligula boisduvalii (Lepidoptera: Saturniidae) and comparison with other lepidopteran insects. Gene. 2008;413(1-2):49–57. doi: 10.1016/j.gene.2008.01.019. [DOI] [PubMed] [Google Scholar]
- 22.Fang L, Wang L, Wu S, et al. The complete mitochondrial genome of the fall webworm, Hyphantria cunea (Lepidoptera: Arctiidae) International Journal of Biological Sciences. 2010;6(2):172–186. doi: 10.7150/ijbs.6.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu YJ, Fang R, Zhou GL, Ye J, Yi JP. The complete sequence determination and analysis of Lymantria dispar mitochondrial genome. National Center for Biotechnology Information. 2009;24(4):6–11. [Google Scholar]
- 24.Yin J, Hong GY, Wang AM, Cao YZ, Wei ZJ. Mitochondrial genome of the cotton bollworm Helicoverpa armigera (Lepidoptera: Noctuidae) and comparison with other Lepidopterans. Mitochondrial DNA. 2010;21(5):160–169. doi: 10.3109/19401736.2010.503242. [DOI] [PubMed] [Google Scholar]
- 25.Zhao JL, Zhang YY, Luo AR, Jiang GF, Cameron SL, Zhu CD. The complete mitochondrial genome of Spilonota lechriaspis Meyrick (Lepidoptera: Tortricidae) Molecular Biology Reports. 2011;38:3757–3764. doi: 10.1007/s11033-010-0491-6. [DOI] [PubMed] [Google Scholar]
- 26.Gong YJ, Shi BC, Kang ZJ, Zhang F, Wei SJ. The complete mitochondrial genome of the oriental fruit moth Grapholita molesta (Busck) (Lepidoptera: Tortricidae) Molecular Biology Reports. 2011;39(3):2893–2900. doi: 10.1007/s11033-011-1049-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pan MH, Yu QY, Xia YL, et al. Characterization of mitochondrial genome of Chinese wild mulberry silkworm, Bomyx mandarina (Lepidoptera: Bombycidae) Science in China C. 2008;51(8):693–701. doi: 10.1007/s11427-008-0097-6. [DOI] [PubMed] [Google Scholar]
- 28.Li WW, Zhang XY, Fan ZX. Structural characteristics and phylogenetic analysis of the mitochondrial genome of the Sugarcane Borer, Diatraea saccharalis (Lepidoptera: Crambidae) DNA and Cell Biology. 2011;30(1):3–8. doi: 10.1089/dna.2010.1058. [DOI] [PubMed] [Google Scholar]
- 29.Coates BS, Sumerford DV, Hellmich RL, Lewis LC. Partial mitochondrial genome sequences of Ostrinia nubilalis and Ostrinia furnicalis. International Journal of Biological Sciences. 2005;1(1):13–18. doi: 10.7150/ijbs.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang XC, Sun XY, Sun QQ, et al. The complete mitochondrial genome of the laced fritillary Argyreus hyperbius (Lepidoptera: Nymphalidae) Zoology Research. 2011;32(5):465–475. doi: 10.3724/SP.J.1141.2011.05465. [DOI] [PubMed] [Google Scholar]
- 31.Feng X, Liu DF, Wang NX, Zhu CD, Jiang GF. The mitochondrial genome of the butterfly Papilio xuthus (Lepidoptera: Papilionidae) and related phylogenetic analyses. Molecular Biology Reports. 2010;37(8):3877–3888. doi: 10.1007/s11033-010-0044-z. [DOI] [PubMed] [Google Scholar]
- 32.Hao JS, Li CX, Sun XY, Yang Q. Phylogeny and divergence time estimation of cheilostome bryozoans based on mitochodrial 16S rRNA sequences. Chinese Science Bulletin. 2005;12:1205–1211. [Google Scholar]
- 33.Simon C, Frati F, Bekenbach A, Crespi B, Liu H, Flook P. Evolution, weighting, and phylogenetic utility of mitochondrial genesequences and a compilation of conserved polymerase chainreaction primers. Annals of the Entomological Society of America. 1994;87(6):651–701. [Google Scholar]
- 34.Singh VK, Mangalam AK, Dwivedi S, Naik S. Primer premier: Program for design of degenerate primers from a protein sequence. BioTechniques. 1998;24(2):318–319. doi: 10.2144/98242pf02. [DOI] [PubMed] [Google Scholar]
- 35.Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symposium Series. 1999;(41):95–98. [Google Scholar]
- 36.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The clustal X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Research. 1997;24:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumar S, Tamura K, Nei M. MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Briefings in Bioinformatics. 2004;5(2):150–163. doi: 10.1093/bib/5.2.150. [DOI] [PubMed] [Google Scholar]
- 38.Lowe TM, Eddy SR. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Research. 1997;25(5):955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Research. 1999;27(2):573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swofford DL. PAUP∗. Phylogenetic Analysis Using Parsimony (∗and Other Methods), Version 4.10. Sunderland, Mass, USA: Sinauer Associates; 2002. [Google Scholar]
- 41.Posada D, Crandall KA. Modeltest: testing the model of DNA substitution. Bioinformatics. 1998;14(9):817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- 42.Huelsenbeck JP, Ronquist F. Mrbayes: bayesian inference of phylogenetic trees. Bioinformatics. 2001;17(8):754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- 43.Fenn JD, Cameron SL, Whiting MF. The complete mitochondrial genome sequence of the Mormon cricket (Anabrus simplex: Tettigoniidae: Orthoptera) and an analysis of control region variability. Insect Molecular Biology. 2007;16(2):239–252. doi: 10.1111/j.1365-2583.2006.00721.x. [DOI] [PubMed] [Google Scholar]
- 44.Yamauchi MM, Miya MU, Nishida M. Complete mitochondrial DNA sequence of the Japanese spiny lobster, Panulirus japonicus (Crustacea: Decapoda) Gene. 2002;295(1):89–96. doi: 10.1016/s0378-1119(02)00824-7. [DOI] [PubMed] [Google Scholar]
- 45.Nardi F, Carapelli A, Dallai R, Frati F. The mitochondrial genome of the olive fly Bactrocera oleae two haplotypes from distant geographical locations. Insect Molecular Biology. 2003;12(6):605–611. doi: 10.1046/j.1365-2583.2003.00445.x. [DOI] [PubMed] [Google Scholar]
- 46.Lutz-Bonengel S, Sanger T, Pollak S. Different methods to determine length heteroplasmy within the mitochondrial control region. International Journal of Legal Medicine. 2004;118(5):274–281. doi: 10.1007/s00414-004-0457-0. [DOI] [PubMed] [Google Scholar]
- 47.Ogoh K, Ohmiya Y. Complete mitochondrial DNA sequence of the sea-firefly, Vargula hilgendorfii (Crustacea, Ostracoda) with duplicate control regions. Gene. 2004;327(1):131–139. doi: 10.1016/j.gene.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 48.Flook PK, Rowell CHF, Gellissen G. The sequence, organization, and evolution of the Locusta migratoria mitochondrial genome. Journal of Molecular Evolution. 1995;41(6):928–941. doi: 10.1007/BF00173173. [DOI] [PubMed] [Google Scholar]
- 49.Beard CD, Hamm SM, Collins FH. The mitochondrial genome of the mosquito Anopheles gambiae: DNA sequence, genome organization, and comparisons with mitochondrial sequences of other insects. Insect Molecular Biology. 1993;2(2):103–124. doi: 10.1111/j.1365-2583.1993.tb00131.x. [DOI] [PubMed] [Google Scholar]
- 50.Mitchell SE, Cockburn AF, Seawright JA. The mitochondrial genome of Anopheles quadrimaculatus species A: complete nucleotide sequence and gene organization. Genome. 1993;36(6):1058–1073. doi: 10.1139/g93-141. [DOI] [PubMed] [Google Scholar]
- 51.Spanos L, Koutroumbas G, Kotsyfakis M, Louis C. The mitochondrial genome of the Mediterranean fruit fly, Ceratitis capitata . Insect Molecular Biology. 2000;9(2):139–144. doi: 10.1046/j.1365-2583.2000.00165.x. [DOI] [PubMed] [Google Scholar]
- 52.Wilson K, Cahill V, Ballment E, Benzie J. The complete sequence of the mitochondrial genome of the crustacean Penaeus monodon: are malacostracan crustaceans more closely related to insects than to branchiopods? Molecular Biology and Evolution. 2000;17(6):863–874. doi: 10.1093/oxfordjournals.molbev.a026366. [DOI] [PubMed] [Google Scholar]
- 53.Ojala D, Merkel C, Gelfand R, Attardi G. The tRNA genes punctuate the reading of genetic information in human mitochondrial DNA. Cell. 1980;22(2):393–403. doi: 10.1016/0092-8674(80)90350-5. [DOI] [PubMed] [Google Scholar]
- 54.Ojala D, Montoya J, Attardi G. tRNA punctuation model of RNA processing in human mitochondria. Nature. 1981;290(5806):470–474. doi: 10.1038/290470a0. [DOI] [PubMed] [Google Scholar]
- 55.Lu C, Liu YQ, Liao SY, et al. Complete sequence determination and analysis of Bombyx mori mitochondrial genome. Journal of Agricultural Biotechnology. 2002;10(2):163–170. [Google Scholar]
- 56.Taylor MFJ, Mckechnie SW, Pierce N, Kreitman M. The lepidopteran mitochondrial control region: structure and evolution. Molecular Biology and Evolution. 1993;10(6):1259–1272. doi: 10.1093/oxfordjournals.molbev.a040075. [DOI] [PubMed] [Google Scholar]
- 57.Taanman JW. The mitochondrial genome: structure, transcription, translation and replication. Biochimica et Biophysica Acta. 1999;1410(2):103–123. doi: 10.1016/s0005-2728(98)00161-3. [DOI] [PubMed] [Google Scholar]
- 58.Minet J. Tentative reconstruction of the ditrysian phylogeny (Lepidoptera: Glossata) Entomologica Scandinavica. 1991;22:69–95. [Google Scholar]
- 59.Minet J. The Bombycoidea: phylogeny and higher classification (Lepidoptera : Glossata) Entomologica Scandinavica. 1994;25(1):63–88. [Google Scholar]
- 60.Nielsen ES. Phylogeny of major lepidopteran groups. In: Fernholm B, Bremer K, Jörnvall H, editors. The Hierarchy of Life. Amsterdam, The Netherlands: Elsevier; 1989. pp. 281–294. [Google Scholar]
- 61.Scott JA. On the monophyly of the macrolepidoptera, including a reassessment of their relationship to cossoidea and castnioidea, and a reassignment of mimallonidae to pyraloidea. The Journal of Research on the Lepidoptera. 1986;25(1):30–38. [Google Scholar]
- 62.Mutanen M, Wahlberg N, Kaila L. Comprehensive gene and taxon coverage elucidates radiation patterns in moths and butterflies. Proceedings of the Royal Society B. 2010;277(1695):2839–2848. doi: 10.1098/rspb.2010.0392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wahlberg N, Braby MF, Brower AVZ, et al. Synergistic effects of combining morphological and molecular data in resolving the phylogeny of butterflies and skippers. Proceedings of the Royal Society B. 2005;272(1572):1577–1586. doi: 10.1098/rspb.2005.3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Regier JC, Cook CP, Mitter C, Hussey A. A phylogenetic study of the ’bombycoid complex’ (Lepidoptera) using five protein-coding nuclear genes, with comments on the problem of macrolepidopteran phylogeny. Systematic Entomology. 2008;33(1):175–189. [Google Scholar]