

Abstract
Loss of epithelial cell identity and acquisition of mesenchymal features are early events in the neoplastic transformation of mammary cells. We investigated the pattern of expression of a selected panel of genes associated with cell polarity and apical junction complex or involved in TGF-β-mediated epithelial-mesenchymal transition and cell-fate decision in a series of DCIS and corresponding patient-matched normal tissue. Additionally, we compared DCIS gene profile with that of atypical ductal hyperplasia (ADH) from the same patient. Statistical analysis identified a “core” of genes differentially expressed in both precursors with respect to the corresponding normal tissue mainly associated with a terminally differentiated luminal estrogen-dependent phenotype, in agreement with the model according to which ER-positive invasive breast cancer derives from ER-positive progenitor cells, and with an autocrine production of estrogens through androgens conversion. Although preliminary, present findings provide transcriptomic confirmation that, at least for the panel of genes considered in present study, ADH and DCIS are part of a tumorigenic multistep process and strongly arise the necessity for the regulation, maybe using aromatase inhibitors, of the intratumoral and/or circulating concentration of biologically active androgens in DCIS patients to timely hamper abnormal estrogens production and block estrogen-induced cell proliferation.
1. Introduction
Ductal carcinoma in situ (DCIS), also known as intraductal carcinoma, is the most common type of noninvasive breast cancer in women [1]. From the mid-1970s, the incidence of DCIS has sharply increased, primarily because of the adoption of radiographic screening for invasive carcinoma. Currently, it accounts for approximately 25% of newly diagnosed breast cancer cases [1].
DCIS is a nonobligate precursor to invasive breast cancer, and for this reason some members of the 2009 US National Institutes of Health DCIS consensus conference proposed to remove the word “carcinoma” from the term DCIS [2]. Nevertheless, experimental studies have shown the presence of carcinoma precursor cells in DCIS lesions [3–5], and clinical evidence indicates that approximately 50% of cases will progress to invasive breast cancer if untreated [6, 7]. As a result, the malignant nature of DCIS remains debated, primarily because of the limited knowledge of DCIS arising and development. In fact, while several genome-wide studies have compared the gene profiles of DCIS and invasive breast cancer, very few studies have investigated and recognized the molecular alteration that characterize DCIS with respect to normal tissue [8–11].
DCIS is defined as an abnormal proliferation of transformed mammary epithelial cells within the closed environment of a duct, likely in response to microenvironment alterations including hypoxia and nutrient deprivation [4, 12]. Among the processes early affected during mammary cells transformation, those involved in the establishment and maintenance of epithelial cell identity and tissue specificity are of particular relevance. In fact, epithelial mammary cells are characterized by an asymmetric distribution of cytoplasmic and membrane proteins, termed apicobasolateral cell polarity, essential for a correct cell-cell adhesion and the formation of an epithelial sheet. As a result of an epithelial to mesenchymal transition (EMT), during neoplastic transformation, cell polarity and epithelial morphology are early lost polarized and immotile epithelial cells acquire a fibroblast-like morphology and increased cell motility [13].
EMT process can be induced by a variety of signaling pathways among which the main and best characterized is that involving transforming-growth factor-β (TGF-β). TGF-β is a multifunctional cytokine and a powerful tumor suppressor that governs many aspects of mammary epithelial cells physiology and homeostasis [14]. Under abnormal microenvironment conditions; however, some mammary epithelial cells may acquire resistance to TGF-β, circumvent its cytostatic effect and tumor suppressive activity, and activate EMT [15, 16].
Recent studies have demonstrated that EMT may generate cells with stemness-like properties, especially in the transitioning mammary epithelial cell compartment [17, 18]. Therefore, a interrelationship among EMT (TGF-β mediated), disruption of the mechanisms deputed to cell polarity and adhesion control, and acquisition of stemness-like features can be assumed already in DCIS [19, 20].
Taking advantage from the only microarray dataset publicly accessible at the ArrayExpress web site, we investigated the pattern of expression of a selected panel of genes involved in TGF-β-mediated EMT or associated with epithelial cells identity (i.e., cell polarity and apical junction complex) and cell-fate decision in a series of DCIS and corresponding patient-matched histologically normal (HN) epithelium [11]. As the whole-gene expression profile of patient-matched atypical ductal hyperplasia (ADH) was also available, we further compared DCIS and ADH profiles to verify the hypothesis according to which breast cancer progression is a multistep process involving a continuum of changes from normal phenotype through hyperplastic lesions, carcinoma in situ, and invasive carcinoma [21].
2. Materials and Methods
2.1. Materials
As reported in the original paper [11], patient-matched samples (HN, ADH, and DCIS) were isolated via laser capture microdissection from surgical specimens of 12 preoperative untreated patients with an ER-positive (immunohistochemically evaluated) sporadic breast cancer. Gene expression was determined by using the Affymetrix Human Genome HG-U133A GeneChip; corresponding microarray dataset was publicly available at the ArrayExpress web site (http://www.ebi.ac.uk/arrayexpress/) with the Accession number E-GEOD-16873.
2.2. Gene Selection
To select the panel of genes specifically involved in the TGF-β-activated EMT, cell polarity and apical junction complex, and cell-fate decision, we combined Gene Ontology (http://www.geneontology.org) and PubMed (http://www.ncbi.nlm.nih.gov) information. In addition, since cancer cells often increase their autocrine production of TGF-β to activate angiogenesis in response to oxygen and nutrients deprivation [14], we also included some genes coding for angiogenesis-inducing factors. On the whole, a set of 199 genes evolutionarily conserved in Homo sapiens was established (see Supplementary Table 1 available on line at doi 10.1155/2012/984346). However, because 27 genes had no corresponding probe-sets on the HG-U133A GeneChip, the gene set was actually composed of 172 elements, 33 of which involved in EMT activation [13–15, 21–23], 75 involved in cell polarity and apical junction assembly [24–26], 36 involved in cell fate-decisions and in the maintenance of a self-renewal state in tumorigenic adult tissues [27–29], 28 involved in hormone steroid signaling [30–32], angiogenesis activation [33, 34] or used as luminal and basal markers [35–38]. These 172 genes corresponded to 339 Affymetrix probe-sets, as verified by GeneAnnot system v2.0 (http://bioinfo2.weizmann.ac.il/geneannot/), that additionally provided us information about the quality of each probe-set in terms of sensitivity and specificity score [39] (see Supplementary).
2.3. Statistical Analysis
As some genes are recognized by more than a single probe set, each of which characterized by an individual specificity and sensitivity that differently contribute to gene expression value, a gene expression mean value was calculated after weighting each probe-set for its own sensitivity and specificity score. Specifically, each expression value (already log 2 transformed in the original dataset) was multiplied for the semi sum of sensitivity and specificity scores of the corresponding probe set. Given the patient-matched samples study design, all statistical analyses were performed considering a regression model for repeated measures with random effect, and the differential gene expression was evaluated by t-test on regression coefficients. To correct for multiple testing, the false discovery rate (FDR) was used [40].
To evidence latent variables accounting for genes correlations, a factor analysis was applied [41] in the following three comparisons: DCIS and paired ADH, DCIS and paired HN, and ADH and paired HN. The number of retained factors was selected according to the scree test [42]. To facilitate the interpretation of the factors, varimax rotation was applied. Loading values lower than 0.3 were not considered.
All analyses were performed using open source software R 2.11.1 packages HDMD (http://www.R-project.org).
3. Results and Discussion
Genes found differentially expressed (P < 0.05) between DCIS and NH or ADH and NH are reported in Table 1. Specifically, 47 of the 172 selected genes were found differentially expressed between DCIS and NH (11 with an estimated FDR < 0.01) and 28 were found differentially expressed between ADH and NH (only one with an estimated FDR < 0.01). Notably, 24 of the 28 genes found differentially expressed between ADH and NH were found differentially expressed (in a similar manner) also between DCIS and NH. The persistence of this “core” of genes, dysregulated in a similar manner in both invasive breast cancer precursors, seems to support the hypothesis of ADH as the direct precursor of DCIS. In fact, in agreement with the proposed multistep process, DCIS showed an increased number of genes differentially expressed with respect to ADH.
Table 1.
Differentially expressed genes between ADH or DCIS and histologically normal (HN) tissue (ordered according to P value).
| ADH versus HN | DCIS versus HN | ||||
|---|---|---|---|---|---|
| Gene symbol | P value | Variation | Gene symbol | P value | Variation |
| JAM3 | 0.000045 | ↓ | JAM3 | 0.000016 | ↓ |
| JAG2 | 0.000337 | ↓ | EGFR | 0.000103 | ↓ |
| CD24 | 0.000364 | ↑ | SNAI2 | 0.000127 | ↓ |
| SNAI2 | 0.000594 | ↓ | CLDN5 | 0.000136 | ↓ |
| EGFR | 0.001897 | ↓ | JAM2 | 0.000150 | ↓ |
| FOXC1 | 0.002222 | ↓ | FOXC1 | 0.000167 | ↓ |
| EGF | 0.003163 | ↓ | CD24 | 0.000277 | ↑ |
| ID2 | 0.003547 | ↑ | JAG2 | 0.000297 | ↓ |
| JAM2 | 0.005052 | ↓ | KRT5 | 0.000420 | ↓ |
| TJP3 | 0.005070 | ↑ | KRT14 | 0.000425 | ↓ |
| TGFBR3 | 0.005537 | ↓ | GATA3 | 0.000490 | ↑ |
| KRT17 | 0.005624 | ↓ | TGFBR3 | 0.000710 | ↓ |
| GATA3 | 0.005795 | ↑ | TJP3 | 0.001046 | ↑ |
| TP53 | 0.006737 | ↑ | KRT17 | 0.001402 | ↓ |
| CDH4 | 0.006916 | ↓ | SOX4 | 0.001942 | ↑ |
| AKT1 | 0.009044 | ↑ | AKT1 | 0.002867 | ↑ |
| CLDN7 | 0.012026 | ↑ | CLDN8 | 0.003090 | ↓ |
| EPCAM | 0.015207 | ↑ | CDC42 | 0.003643 | ↑ |
| ABCG2 | 0.019210 | ↓ | EGF | 0.003752 | ↓ |
| KRT5 | 0.019854 | ↓ | EPCAM | 0.004605 | ↑ |
| KRT14 | 0.020117 | ↓ | CLDN7 | 0.004749 | ↑ |
| CLDN11 | 0.023005 | ↓ | ESR1 | 0.007188 | ↑ |
| PARD3 | 0.030202 | ↓ | DLG1 | 0.007249 | ↑ |
| SOX4 | 0.036127 | ↑ | KRT19 | 0.007637 | ↑ |
| CDC42 | 0.036404 | ↑ | FOXA1 | 0.007844 | ↑ |
| TIAM1 | 0.038034 | ↑ | KRT18 | 0.010091 | ↑ |
| ESR1 | 0.038527 | ↑ | CDH3 | 0.010638 | ↓ |
| PVR | 0.039104 | ↓ | TIAM1 | 0.010722 | ↑ |
| TGFBR2 | 0.013243 | ↓ | |||
| RHOA | 0.013540 | ↑ | |||
| BRCA1 | 0.014098 | ↑ | |||
| ID4 | 0.017030 | ↓ | |||
| ID2 | 0.018115 | ↑ | |||
| NOTCH4 | 0.018550 | ↓ | |||
| PVRL2 | 0.019914 | ↑ | |||
| AKT3 | 0.020275 | ↓ | |||
| ACTN1 | 0.020619 | ↓ | |||
| PROM1 | 0.022238 | ↓ | |||
| CDH4 | 0.022933 | ↓ | |||
| DLG3 | 0.023798 | ↑ | |||
| F11R | 0.031657 | ↑ | |||
| CTNNA1 | 0.033234 | ↑ | |||
| MTA2 | 0.033999 | ↓ | |||
| ABCG2 | 0.037966 | ↓ | |||
| MPP5 | 0.039023 | ↑ | |||
| HIF1A | 0.039489 | ↑ | |||
| CDKN1A | 0.047769 | ↑ | |||
In bold, genes with an estimated FDR < 0.01.
With respect to normal tissue, both DCIS and ADH showed the overexpression of ESR1, coding for the estrogen receptor; CD24, coding for a mucin-like cell-adhesion molecule positively associated with a terminally differentiated luminal phenotype [43, 44]; GATA3, coding for a transcription factor involved in mammary gland morphogenesis [35, 36]; CLDN7, EPCAM, and TJP3, coding for tight junction components; CDC42 and TIAM1, coding for two small GTPase family members involved in cell polarity and apical junction complex formation. Concomitantly, both breast cancer precursors showed the underexpression of EGFR gene, in which expression is generally negatively associated with ESR1 expression, KRT5, KRT14, and KRT17, all coding for cytokeratins associated with a basal phenotype. On the whole, this pattern of expression clearly indicates that DCIS and ADH are both characterized by a terminally differentiated luminal phenotype. Since all specimens were derived from patients with an ER-positive ductal carcinoma, it is conceivable the hypothesis that the establishment of an estrogen-dependent phenotype, in response to estrogens present in the microenvironment, should be a very early event in the tumorigenic process. Such a finding is in agreement with the model for breast cancer development proposed by Dontu et al. [45], according to which ER-positive cancers should derive from transiently amplifying ERpositive progenitor cells. Escaped from proliferation control as a consequence of genetic and epigenetic alterations in genes involved in cell-fate decision, these ER-positive progenitor cells should generate cells constitutively expressing estrogen receptor. Once established in ADH, this terminally differentiated luminal phenotype seems to consolidate in DCIS as demonstrated by the presence, among the genes exclusively expressed in DCIS, of KRT18, KRT19 (coding for some luminal-associated cytokeratins) and DLG1, DLG3, and MPP5 (coding for some cell polarity complex components).
With respect to both tumor precursors, histologically normal tissue expressed genes coding for some transcription factors involved in EMT (SNAI2 and TGFBR3) and cell-fate decision (FOXC1 and JAG2), and for stemness-associated features (ABCG2). This finding is more evident considering the 47 genes differentially expressed between DCIS and normal tissue among which we found some other genes involved in the TGF-β-mediated EMT (AKT3, ID4 and TGFBR2), and cell-fate decision and self-renewal (NOTCH4 and PROM1).
Such an apparently paradoxical finding, that is, the positive association between histologically normal tissues and EMT- and stemness-related genes (more likely expected in transformed tissues) is not surprising since we observed a similar behavior in an independent cases series composed of primary breast cancers and corresponding patient-matched normal tissue (submitted paper) and in normal pleura with respect to pleural mesothelioma [46]. In agreement with the physiological remodeling of the mammary gland, such a finding should support the persistence of resident stem/progenitor cells in normal tissue [47, 48].
Factor analysis, applied to investigate the latent variables intrinsically associated with the selected 172 genes, corroborated these findings and highlighted some other interesting interrelations. In agreement with the results provided by t-paired test, factor analysis indicated that in both precursors (Figure 1(a) for DCIS and Figure 2(a) for ADH), the first factor (F1) was principally characterized by genes associated with an estrogen-dependent epithelial phenotype. In fact, within the genes with a positive loading value on F1, we found those coding for hormone steroid receptors (AR and ESR1) and transcriptional coactivator for steroid and nuclear hormone receptors (NCOA1), for tight (EPCAM, MAGI1, MAGI2, MPDZ, TJP1, TJP2 and TJP3) and adherens (CTNND1, PFN1 and PVRL2) junction components, and for small GTPase family members involved in epithelial cell polarization processes (CDC42 and RHOA). In addition, we found some genes associated with cell-fate decision: BMI-1, coding for a member of Polycomb group required to maintain the transcriptionally repressive state of many genes throughout embryo development [49] and adult tissues differentiation including mammary gland [50]; FDZ4 and FDZ6, two members of the frizzled gene family involved in β-catenin-signaling transduction and intercellular transmission of polarity information in differentiated tissues [51].
Figure 1.
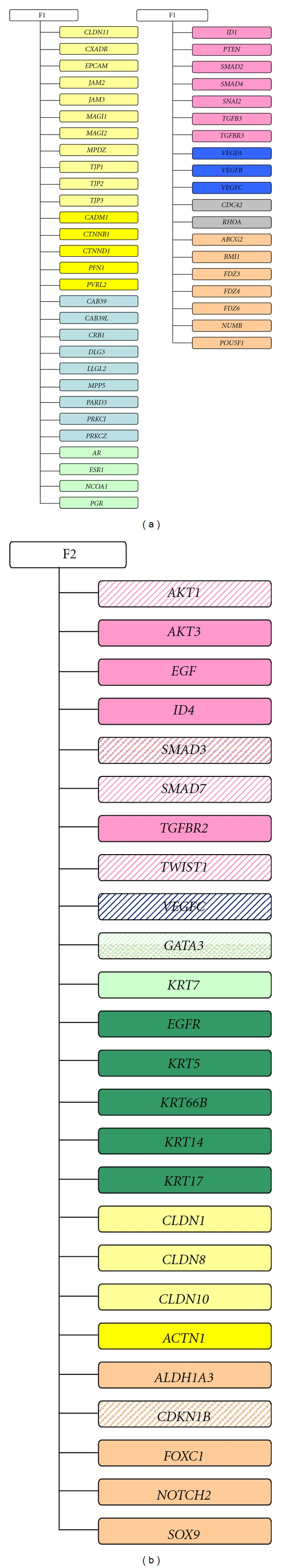
Factor analysis in DCIS subgroup. Schematic representation of genes with a loading value <|0.6| characterizing the first (F1) and the second factor (F2). Solid color indicates a positive loading value whereas dashed color indicates a negative loading value. Color correspondence: light yellow, tight junction components; dark yellow, adherens junction components; light blue, polarity complexes components; dark blue, angiogenesis; orange, cell-fate decision; light green, luminal markers and hormone steroid; dark green, basal markers; pink, epithelial-mesenchymal transition, grey, GTPase family members.
Figure 2.
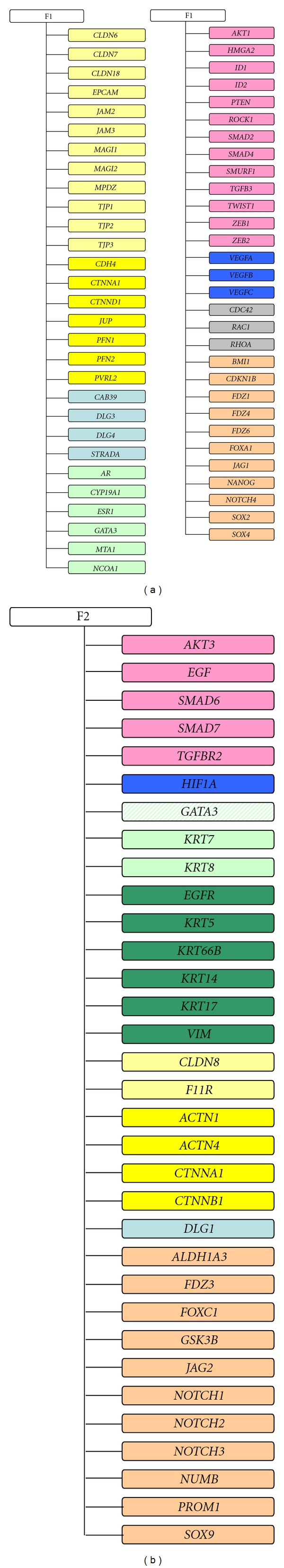
Factor analysis in ADH subgroup. Schematic representation of genes with a loading value <|0.6| characterizing the first (F1) and the second factor (F2). Solid color indicates a positive loading value whereas dashed color indicates a negative loading value. Color correspondence: light yellow, tight junction components; dark yellow, adherens junction components; light blue, polarity complexes components; dark blue, angiogenesis; orange, cell-fate decision; light green, luminal markers and hormone steroid; dark green, basal markers; pink, epithelial-mesenchymal transition, grey, GTPase family members.
Furthermore, in agreement with the notion that ERα, and TGF-β-signaling pathways are major regulators during mammary gland development [52], we found genes coding for TGF-β receptor (TGFBR3) or for proteins involved in canonical (ID1, SMAD2 and SMAD4) and noncanonical (PTEN, RHOA and ROCK1) TGF-β pathways [15, 16].
Finally, probably associated with the adaptation of DCIS and ADH to hypoxic stress caused by the unbalance between cell proliferation and oxygen supply, we also found JAM2, JAM3, VEGFA, VEGFB, and VEGFC, all genes involved in endothelial cell proliferation.
The second factor (F2) identified by factor analysis (Figure 1(b) for DCIS and Figure 2(b) for ADH) was conversely characterized by the positive loading value of several genes coding for mesenchymal markers (EGF, EGFR, KRT5, KRT6B, KRT14 and KRT17), and the negative loading value of GATA3, the gene coding for the transcription factor driving the luminal morphogenesis of the mammary gland [35, 36]. In addition, F2 was characterized by the presence of some genes coding for proteins playing a critical role in cell-fate decision and cell-renewal (ALDH1A3, FOXC1, NOTCH2, PROM1, and SOX9) [53].
Notably, when applied to ADH subgroup, factor analysis identified CYP19A1 as included in the panel of genes characterizing the first factor. This gene encodes for cytochrome P450 (better known as aromatase), the enzyme that catalyzes the conversion from circulating and rostenedione to estrone or testosterone to estradiol, and its presence should support the hypothesis of a very early activation of an autocrine production of estrogen. Furthermore, the concomitant presence of CYP19A1, AR, ESR1, and NCOA1 should provide a transcriptomic confirmation for the clinical evidence that high androgens level may have detrimental effect on breast carcinogenesis and progression due to a persistent local estrogen production that incessantly stimulates epithelial cell proliferation [54–56].
When applied to DCIS subgroup, factor analysis seems to indicate a consolidation of such an estrogen dependence as suggested by the additional presence of PGR, the gene coding for progesterone receptor and which expression is under estrogenic control.
4. Conclusions
Elucidating the initial steps of breast tumorigenesis is of paramount importance to allow an even early diagnosis and consequently an adequate treatment strategy aimed to prevent the malignant transformation of preneoplastic alterations. That is of particular importance for DCIS because of its high incidence [1] and facility in progressing to invasive breast cancer if untreated [6, 7].
Experimental evidence till now accumulated has clearly indicated that at the basis of the neoplastic transformation of mammary epithelial cells there are loss of apicobasal epithelial cell identity and acquisition of a functional mesenchymal morphology. Therefore, we investigated the pattern of expression of a selected panel of genes associated with epithelial cells identity (i.e., cell polarity and apical junction complex) or involved in TGF-β-mediated EMT and cell-fate decision in a series of DCIS and corresponding patient-matched normal tissue. In addition, we compared DCIS profile with that of patient-matched ADH to investigate the hypothesis according to which breast cancer progression is a multistep process involving a continuum of changes from normal phenotype through hyperplastic lesions, carcinoma in situ, and invasive carcinoma [21].
Statistical analysis seems to support this hypothesis because it identified a “core” of genes, mainly associated with a terminally differentiated luminal phenotype, and differentially expressed in both precursors with respect to the corresponding normal tissue. Notably, these alterations in gene expression did not result in a progressive mesenchymal transition but rather in a terminally differentiated luminal phenotype, in agreement with the model according to which ER-positive invasive breast cancer derives from ER-positive progenitor cells [45]. The constitutive expression of ER should make ADH and DCIS forming cells able to exploit the proliferative stimulus induced by estrogens whereas the establishment of an autocrine production of estrogens, through androgens conversion, should provide an additional selective advantage. The detrimental effect of such continuous estrogen stimulation should be corroborated by the observation that all patients included in the present study developed an invasive ER-positive ductal carcinoma.
Experimental evidence supporting the hypothesis that androgens conversion may be involved in DCIS development, and progression has been provided by a recent study in which the intratumoral concentration of estradiol and 5α-dihydrotestosterone (DHT), and the expression of some sex steroid-producing enzymes, including aromatase, has been evaluated in DCIS specimens [57]. The study clearly demonstrated that aromatase expression level was significantly higher in DCIS with respect to nonneoplastic tissue suggesting the self-sustaining process adopted by DCIS.
Taken together all these findings provide transcriptomic confirmation that, at least for the panel of genes considered in present study, ADH and DCIS are part of a tumorigenic multistep process and strongly arise the necessity for the regulation, maybe using aromatase inhibitors, of the intratumoral and/or circulating concentration of biologically active androgens in DCIS patients to timely hamper abnormal estrogens production and block estrogen-induced cell proliferation [58].
There is no doubt that present in silico study suffers for the limitation common to the majority of studies involving gene expression profile, that is, the lack of validation, at protein level, of the modulations observed at mRNA level. In fact, it is well known that mRNA transcript levels do not always reflect protein expression. However, the immunohistochemical data provided in The Human Protein Atlas largely confirms the differential gene expression that we observed between histologically normal and cancerous tissue. For instance, when we considered the 11 genes with an estimated FDR < 0.01 (Table 1, in bold), we found that protein expression of normal breast (glandular cell) and breast cancer tissue mainly paralleled the mRNA differential expression we observed in our dataset, making our preliminary findings more reliable and worthy of further investigation.
Supplementary Material
Table 1: lists the 199 genes selected for the study.
Table 2: lists the 339 Affymetrix probe-sets corresponding to the 172 genes entered the study.
Author's Contribution
D. Coradini and P. Boracchi contributed equally to this paper.
References
- 1.Virnig BA, Tuttle TM, Shamliyan T, Kane RL. Ductal carcinoma in Situ of the breast: a systematic review of incidence, treatment, and outcomes. Journal of the National Cancer Institute. 2010;102(3):170–178. doi: 10.1093/jnci/djp482. [DOI] [PubMed] [Google Scholar]
- 2.Allegra CJ, Aberle DR, Ganschow P, et al. NIH state-of-the-science conference statement: diagnosis and management of ductal carcinoma in situ (DCIS) NIH Consensus and State-of-the-Science Statements. 2009;26(2):1–27. [PubMed] [Google Scholar]
- 3.Castro NP, Osório CABT, Torres C, et al. Evidence that molecular changes in cells occur before morphological alterations during the progression of breast ductal carcinoma. Breast Cancer Research. 2008;10(5, article no. R87) doi: 10.1186/bcr2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Espina V, Mariani BD, Gallagher RI, et al. Malignant precursor cells pre-exist in human breast DCIS and require autophagy for survival. PLoS One. 2010;5(4) doi: 10.1371/journal.pone.0010240. Article ID e10240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sgroi DC. Preinvasive breast cancer. Annual Review of Pathology. 2010;5:193–221. doi: 10.1146/annurev.pathol.4.110807.092306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuerer HM, Albarracin CT, Yang WT, et al. Ductal carcinoma in situ: state of the science and roadmap to advance the field. Journal of Clinical Oncology. 2009;27(2):279–288. doi: 10.1200/JCO.2008.18.3103. [DOI] [PubMed] [Google Scholar]
- 7.Collins LC, Tamimi RM, Baer HJ, Connolly JL, Colditz GA, Schnitt SJ. Outcome of patients with ductal carcinoma in situ untreated after diagnostic biopsy: results from the nurses’ health study. Cancer. 2005;103(9):1778–1784. doi: 10.1002/cncr.20979. [DOI] [PubMed] [Google Scholar]
- 8.Vincent-Salomon A, Lucchesi C, Gruel N, et al. Integrated genomic and transcriptomic analysis of ductal carcinoma in situ of the breast. Clinical Cancer Research. 2008;14(7):1956–1965. doi: 10.1158/1078-0432.CCR-07-1465. [DOI] [PubMed] [Google Scholar]
- 9.Tamimi RM, Baer HJ, Marotti J, et al. Comparison of molecular phenotypes of ductal carcinoma in situ and invasive breast cancer. Breast Cancer Research. 2008;10(4, article no. R67) doi: 10.1186/bcr2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Allred DC, Wu Y, Mao S, et al. Ductal carcinoma in situ and the emergence of diversity during breast cancer evolution. Clinical Cancer Research. 2008;14(2):370–378. doi: 10.1158/1078-0432.CCR-07-1127. [DOI] [PubMed] [Google Scholar]
- 11.Emery LA, Tripathi A, King C, et al. Early dysregulation of cell adhesion and extracellular matrix pathways in breast cancer progression. American Journal of Pathology. 2009;175(3):1292–1302. doi: 10.2353/ajpath.2009.090115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gauthier ML, Berman HK, Miller C, et al. Abrogated response to cellular stress identifies DCIS associated with subsequent tumor events and defines basal-like breast tumors. Cancer Cell. 2007;12(5):479–491. doi: 10.1016/j.ccr.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tomaskovic-Crook E, Thompson EW, Thiery JP. Epithelial to mesenchymal transition and breast cancer. Breast Cancer Research. 2009;11(6, article no. 213) doi: 10.1186/bcr2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Derynck R, Akhurst RJ, Balmain A. TGF-β signaling in tumor suppression and cancer progression. Nature Genetics. 2001;29(2):117–129. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 15.Xu J, Lamouille S, Derynck R. TGF-β-induced epithelial to mesenchymal transition. Cell Research. 2009;19(2):156–172. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heldin CH, Landström M, Moustakas A. Mechanism of TGF-β signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Current Opinion in Cell Biology. 2009;21(2):166–176. doi: 10.1016/j.ceb.2009.01.021. [DOI] [PubMed] [Google Scholar]
- 17.Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morel AP, Lièvre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One. 2008;3(8) doi: 10.1371/journal.pone.0002888. Article ID e2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Creighton CJ, Chang JC, Rosen JM. Epithelial-mesenchymal transition (EMT) in tumor-initiating cells and its clinical implications in breast cancer. Journal of Mammary Gland Biology and Neoplasia. 2010;15(2):253–260. doi: 10.1007/s10911-010-9173-1. [DOI] [PubMed] [Google Scholar]
- 20.Moreno-Bueno G, Portillo F, Cano A. Transcriptional regulation of cell polarity in EMT and cancer. Oncogene. 2008;27(55):6958–6969. doi: 10.1038/onc.2008.346. [DOI] [PubMed] [Google Scholar]
- 21.Simpson PT, Reis-Filho JS, Gale T, Lakhani SR. Molecular evolution of breat cancer. Journal of Pathology. 2005;205(2):248–254. doi: 10.1002/path.1691. [DOI] [PubMed] [Google Scholar]
- 22.Micalizzi DS, Ford HL. Epithelial-mesenchymal transition in development and cancer. Future Oncology. 2009;5(8):1129–1143. doi: 10.2217/fon.09.94. [DOI] [PubMed] [Google Scholar]
- 23.Blick T, Widodo E, Hugo H, et al. Epithelial mesenchymal transition traits in human breast cancer cell lines. Clinical and Experimental Metastasis. 2008;25(6):629–642. doi: 10.1007/s10585-008-9170-6. [DOI] [PubMed] [Google Scholar]
- 24.Wodarz A, Näthke I. Cell polarity in development and cancer. Nature Cell Biology. 2007;9(9):1016–1024. doi: 10.1038/ncb433. [DOI] [PubMed] [Google Scholar]
- 25.Müller HAJ. Genetic control of epithelial cell polarity: lessons from Drosophila. Developmental Dynamics. 2000;218(1):52–67. doi: 10.1002/(SICI)1097-0177(200005)218:1<52::AID-DVDY5>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 26.Martin-Belmonte F, Mostov K. Regulation of cell polarity during epithelial morphogenesis. Current Opinion in Cell Biology. 2008;20(2):227–234. doi: 10.1016/j.ceb.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 27.Liu M, Casimiro MC, Wang C, et al. p21CIP1 attenuates Ras- and c-Myc-dependent breast tumor epithelial mesenchymal transition and cancer stem cell-like gene expression in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(45):19035–19039. doi: 10.1073/pnas.0910009106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lugli A, Iezzi G, Hostettler I, et al. Prognostic impact of the expression of putative cancer stem cell markers CD133, CD166, CD44s, EpCAM, and ALDH1 in colorectal cancer. British Journal of Cancer. 2010;103(3):382–390. doi: 10.1038/sj.bjc.6605762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lawson JC, Blatch GL, Edkins AL. Cancer stem cells in breast cancer and metastasis. Breast Cancer Research and Treatment. 2009;118(2):241–254. doi: 10.1007/s10549-009-0524-9. [DOI] [PubMed] [Google Scholar]
- 30.Manavathi B, Singh K, Kumar R. MTA family of coregulators in nuclear receptor biology and pathology. Nuclear Receptor Signaling. 2007;5, article e010 doi: 10.1621/nrs.05010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fujita N, Kajita M, Taysavang P, Wade PA. Hormonal regulation of metastasis-associated protein 3 transcription in breast cancer cells. Molecular Endocrinology. 2004;18(12):2937–2949. doi: 10.1210/me.2004-0258. [DOI] [PubMed] [Google Scholar]
- 32.Xu J, Wu RC, O’Malley BW. Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nature Reviews Cancer. 2009;9(9):615–630. doi: 10.1038/nrc2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greijer AE, van der Groep P, Kemming D, et al. Up-regualtion of gene expression by hypoxia is mediated predominantly by hypoxia-inducible factor I (HIF-I) Journal of Pathology. 2005;206(3):291–304. doi: 10.1002/path.1778. [DOI] [PubMed] [Google Scholar]
- 34.Schoppmann SF, Tamandl D, Roberts L, et al. HER2/neu expression correlates with vascular endothelial growth factor-C and lymphangiogenesis in lymph node-positive breast cancer. Annals of Oncology. 2009;21(5):955–960. doi: 10.1093/annonc/mdp532. [DOI] [PubMed] [Google Scholar]
- 35.Kouros-Mehr H, Kim JW, Bechis SK, Werb Z. GATA-3 and the regulation of the mammary luminal cell fate. Current Opinion in Cell Biology. 2008;20(2):164–170. doi: 10.1016/j.ceb.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Asselin-Labat ML, Sutherland KD, Barker H, et al. Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nature Cell Biology. 2007;9(2):201–209. doi: 10.1038/ncb1530. [DOI] [PubMed] [Google Scholar]
- 37.Abd El-Rehim DM, Pinder SE, Paish CE, et al. Expression of luminal and basal cytokeratins in human breast carcinoma. Journal of Pathology. 2004;203(2):661–671. doi: 10.1002/path.1559. [DOI] [PubMed] [Google Scholar]
- 38.Hoadley KA, Weigman VJ, Fan C, et al. EGFR associated expression profiles vary with breast tumor subtype. BMC Genomics. 2007;8, article no. 258 doi: 10.1186/1471-2164-8-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferrari F, Bortoluzzi S, Coppe A, et al. Novel definition files for human GeneChips based on GeneAnnot. BMC Bioinformatics. 2007;8, article no. 446 doi: 10.1186/1471-2105-8-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Benjamini Y, Hochberg Y. Controlling the false discovery rate. A practical and powerful approach to multiple testing. Journal of Royal Statistical Society B. 1995;57:284–300. [Google Scholar]
- 41.Husson F, Lê S, Pagès J. Exploratory Multivariate Analysis by Example Using R. Boca Raton, Fla, USA: CRC Press; 2010. (Computer Science and Data Analysis Series). [Google Scholar]
- 42.Jobson JD. Applied Multivariate Data Analysis. Volume 2. Categorical and Multivariate Methods. Berlin, Germany: Springer; 1992. [Google Scholar]
- 43.Sleeman KE, Kendrick H, Ashworth A, Isacke CM, Smalley MJ. CD24 staining of mouse mammary gland cells defines luminal epithelial, myoepithelial/basal and non-epithelial cells. Breast Cancer Research. 2005;8(1, article no. R7) doi: 10.1186/bcr1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ponti D, Costa A, Zaffaroni N, et al. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Research. 2005;65(13):5506–5511. doi: 10.1158/0008-5472.CAN-05-0626. [DOI] [PubMed] [Google Scholar]
- 45.Dontu G, El-Ashry D, Wicha MS. Breast cancer, stem/progenitor cells and the estrogen receptor. Trends in Endocrinology and Metabolism. 2004;15(5):193–197. doi: 10.1016/j.tem.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 46.Casarsa C, Bassani N, Ambrogi F, et al. Epithelial-to-mesenchymal transition, cell polarity and stemness-associated features in malignant pleural mesothelioma. Cancer Letters. 2011;302(2):136–143. doi: 10.1016/j.canlet.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 47.Petersen OW, Polyak K. Stem cells in the human breast. Cold Spring Harbor perspectives in biology. 2010;2(5) doi: 10.1101/cshperspect.a003160. Article ID a003160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tiede B, Kang Y. From milk to malignancy: the role of mammary stem cells in development, pregnancy and breast cancer. Cell Research. 2011;21(2):245–257. doi: 10.1038/cr.2011.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K. Genome-wide mapping of polycomb target genes unravels their roles in cell fate transitions. Genes and Development. 2006;20(9):1123–1136. doi: 10.1101/gad.381706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pietersen AM, Evers B, Prasad AA, et al. Bmi1 regulates stem cells and proliferation and differentiation of committed cells in mammary epithelium. Current Biology. 2008;18(14):1094–1099. doi: 10.1016/j.cub.2008.06.070. [DOI] [PubMed] [Google Scholar]
- 51.Tickenbrock L, Hehn S, Sargin B, et al. Activation of Wnt signalling in acute myeloid leukemia by induction of Frizzled-4. International Journal of Oncology. 2008;33(6):1215–1221. [PubMed] [Google Scholar]
- 52.Band AM, Laiho M. Crosstalk of TGF-β and estrogen receptor signaling in breast cancer. Journal of Mammary Gland Biology and Neoplasia. 2011;16(2):109–115. doi: 10.1007/s10911-011-9203-7. [DOI] [PubMed] [Google Scholar]
- 53.Dontu G, Jackson KW, McNicholas E, Kawamura MJ, Abdallah WM, Wicha MS. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Research. 2004;6(6):R605–615. doi: 10.1186/bcr920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suzuki T, Miki Y, Ohuchi N, Sasano H. Intratumoral estrogen production in breast carcinoma: significance of aromatase. Breast Cancer. 2008;15(4):270–277. doi: 10.1007/s12282-008-0062-z. [DOI] [PubMed] [Google Scholar]
- 55.Hudelist G, Wülfing P, Kersting C, et al. Expression of aromatase and estrogen sulfotransferase in preinvasive and invasive breast cancer. Journal of Cancer Research and Clinical Oncology. 2008;134(1):67–73. doi: 10.1007/s00432-007-0249-2. [DOI] [PubMed] [Google Scholar]
- 56.Díaz-Cruz ES, Sugimoto Y, Gallicano GI, Brueggemeier RW, Furth PA. Comparison of increased aromatase versus ERα in the generation of mammary hyperplasia and cancer. Cancer Research. 2011;71(16):5477–5487. doi: 10.1158/0008-5472.CAN-10-4652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shibuya R, Suzuki T, Miki Y, et al. Intratumoral concentration of sex steroids and expression of sex steroid-producing enzymes in ductal carcinoma in situ of human breast. Endocrine-Related Cancer. 2008;15(1):113–124. doi: 10.1677/ERC-07-0092. [DOI] [PubMed] [Google Scholar]
- 58.Lønning PE. The potency and clinical efficacy of aromatase inhibitors across the breast cancer continuum. Annals of Oncology. 2011;22(3):503–514. doi: 10.1093/annonc/mdq337. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table 1: lists the 199 genes selected for the study.
Table 2: lists the 339 Affymetrix probe-sets corresponding to the 172 genes entered the study.