

Abstract
Broadly neutralizing antibodies (bNAbs), which develop over time in some HIV-1 infected individuals, define critical epitopes for HIV vaccine design. Using a systematic approach, we have examined neutralization breadth in the sera of about 1,800 HIV-1 infected individuals, primarily infected with non-clade B viruses, and selected donors for monoclonal antibody (mAb) generation. We then used a high-throughput neutralization screen of antibody-containing culture supernatants from approximately 30,000 activated memory B cells from a clade A-infected African donor to isolate two potent mAbs that target a broadly neutralizing epitope. The previously undescribed epitope is preferentially expressed on trimeric Envelope protein and spans conserved regions of variable loops of the gp120 subunit. The results provide a framework for the design of new vaccine candidates for the elicitation of bNAb responses.
Despite over two decades of research, a protective vaccine against HIV-1 remains elusive. It is widely accepted that such a vaccine will require the elicitation of both T-cell mediated immunity and a broadly neutralizing antibody response (1-3). All of the known bNAbs provide protection in the best available primate models (4-9) and therefore are considered to be the types of antibodies that should be elicited by a vaccine. Unfortunately, these antibodies tend to display limited breadth and potency against non-clade B viruses, which make up the majority of infections outside North America and Europe, and they recognize epitopes on the virus that have so far failed to elicit broadly neutralizing responses when incorporated into a diverse range of immunogens (10-12). Therefore, in order to develop a successful vaccine, it is of high priority to identify new bNAbs that bind to epitopes that may be more amenable to immunogen design.
We have screened serum from approximately 1,800 HIV-1 infected donors from Thailand, Australia, the United Kingdom, the United States, and several sub-Saharan African countries for neutralization activity and identified donors who exhibit broad and potent neutralizing serum activity (13, 14). Monoclonal antibodies are currently being generated from these donors using different approaches. In this study, we used a high-throughput strategy to screen immunoglobulin (Ig)G-containing culture supernatants from approximately 30,000 activated memory B cells from a clade A infected donor for binding to monomeric recombinant envelope glycoproteins gp120 (HIV-1 primary isolate JR-CSF) and gp41 (HIV-1 strain HxB2) (trimeric gp120 and gp41 complexes, referred to as Env, mediate viral entry) and neutralization activity against HIV-1 primary isolates JR-CSF and SF162 (Table S1) (15). The memory B cells were cultured at near clonal density, which enabled us to reconstitute the authentic antibody heavy and light chain pair from each culture well. Unexpectedly, 97.7% of B cell culture supernatants that neutralized HIV-1JR-CSF and 46.5 % that neutralized HIV-1SF-162 did not bind to monomeric gp120JR-CSF or gp41HxB2, and only 2% of cultures with neutralization activity could neutralize both viruses (fig. S1). Antibody genes were obtained from five B cell cultures that exhibited differing functional profiles; one bound to gp120 and only neutralized HIV-1SF162 (PGC14), two bound to gp120 and weakly neutralized both viruses (PGG14 and PG20), and two potently neutralized HIV-1JR-CSF, failed to neutralize HIV-1SF162, and did not bind to gp120 or gp41 in ELISA (PG9 and PG16) (15). Analysis of the antibody variable genes revealed two pairs of somatic variants, one of which contained long CDRH3 loops (PG9 and PG16) (Table S2). Long CDRH3 loops have been previously associated with polyreactivity (the ability to bind to a variety of structurally dissimilar antigens with moderate affinity) (16), thus we tested PG9 and PG16 for reactivity against a panel of antigens and confirmed that the antibodies were not polyreactive (fig. S2) (15).
All five antibodies were first tested for neutralization activity against a multi-clade 16-pseudovirus panel (table S3) (15). Two of the antibodies that bound to gp120 in the initial screen (PGG14 and PG20) did not show substantial neutralization breadth or potency against any of the viruses tested, and the third antibody that bound to gp120 (PGC14) neutralized 4/16 viruses with varying degrees of potency. In contrast, the two antibodies that failed to bind gp120 or gp41 and did not neutralize HIV-1SF162 in the initial screen (PG9 and PG16) neutralized a large proportion of the viruses at concentrations less than 1 μg/ml. The observation that 93.3% of B cell cultures that neutralized HIV-1JR-CSF did not bind to gp120 or gp41 or neutralize HIV-1SF162 suggests that this donor’s NAb response against HIV-1JR-CSF might be mediated by antibodies of the PG9 and PG16 type. We next evaluated PG9, PG16, and PGC14 on a large multi-clade pseudovirus panel consisting of 162 viruses to further assess the neutralization breadth and potency of these three antibodies (Table 1, tables S4 and S5). The bNAbs b12, 2G12, 2F5, and 4E10, and the donor’s serum, were also included in the panel for comparison. Overall, PG9 and PG16 demonstrated a remarkable combination of neutralization breadth and potency. PG9 neutralized 127 out of 162 and PG16 119 out of 162 viruses with a potency that frequently considerably exceeded that noted for the four control bNAbs. The median IC50 and IC90 values for neutralized viruses across all clades were an order of magnitude lower for PG9 and PG16 than for any of the four existing bNAbs (Table 1A, tables S4 and S5), and both mAbs showed an overall greater breadth of neutralization than b12, 2G12, and 2F5 (Table 1B, tables S4 and S5). At low antibody concentrations, PG9 and PG16 also demonstrated greater neutralization breadth than 4E10 (Table 1B). Furthermore, both mAbs potently neutralized one virus (IAVI-C18) that exhibits resistance to all four existing bNAbs (table S4). Inspection of the mAb neutralization curves revealed that, whereas the PG9 neutralization curves usually exhibited sharp slopes, the neutralization curves for PG16 sometimes exhibited gradual slopes or plateaud at less than 100% neutralization (fig. S3 and table S3). Although neutralization curves with similar profiles have been reported previously (17, 18), the mechanism for this is not well understood. Comparison of the neutralization profile of the serum with the neutralization profile of PG9, PG16 and PGC14 revealed that these three antibodies could recapitulate the breadth of the serum neutralization in most cases (table S4). For example, almost all of the viruses that were neutralized by the serum with an IC50 > 1:500 were neutralized by PG9 and/or PG16 at <0.05 μg/ml. The one case where this did not occur was against HIV-1SF162, but this virus was potently neutralized by PGC14. Although PG9 and PG16 are somatic variants, they exhibited different degrees of potency against a number of the viruses tested. For instance, PG9 neutralized HIV-16535.30 approximately 185 times more potently than PG16, and PG16 neutralized HIV-1MGRM-AG-001 approximately 440 times more potently than PG9. In some cases, the two antibodies also differed in neutralization breadth; PG9 neutralized nine viruses that were not sensitive to PG16, and PG16 neutralized two viruses that were not sensitive to PG9. Based on these results, it appears that this donor’s broad serum neutralization might be mediated by somatic antibody variants that recognize slightly different epitopes and display varying degrees of neutralization breadth and potency against any given virus. Selection for these types of antibodies may reflect the immune system’s response to an ever-evolving viral envelope target.
Table 1.
Neutralization activity of mAbs.
| a) Neutralization potency | ||||||||
|---|---|---|---|---|---|---|---|---|
| Median IC50 (μg/ml) against viruses neutralized with an IC50<50 μg/ml |
||||||||
| Clade* | # viruses | b12 | 2G12 | 2F5 | 4E10 | PG9 | PG16 | PGC14 |
| A | 27 | 6.98 | 17.10 | 5.70 | 6.20 | 0.16 | 0.11 | 41.59 |
| B | 31 | 0.80 | 0.82 | 2.41 | 5.22 | 0.43 | 0.70 | 21.88 |
| C | 27 | 6.46 | 2.93 | 31.51 | 2.97 | 0.22 | 0.25 | 11.97 |
| D | 25 | 1.47 | 7.71 | 3.17 | 4.60 | 0.10 | 0.02 | 38.57 |
| CRF01_AE | 10 | 21.53 | >50 | 0.26 | 0.51 | 0.08 | 0.03 | >50 |
| CRF_AG | 10 | 10.40 | 0.95 | 0.64 | 1.42 | 0.80 | 0.03 | 45.10 |
| G | 15 | 3.07 | 31.03 | 1.24 | 1.44 | 0.29 | 1.21 | >50 |
| F | 15 | >50 | 9.23 | 1.78 | 2.30 | 0.09 | 0.08 | 25.71 |
| Total | 162 | 2.82 | 2.43 | 2.30 | 3.24 | 0.22 | 0.15 | 25.99 |
Boxes are color coded as follows: white, median potency >50 μg/ml; green, median potency between 20 and 50 μg/ml; yellow, median potency between 2 and 20 μg/ml; orange, median potency between 0.2 and 2 μg/ml; red, median potency <0.2 μg/ml.
CRF_07BC and CRF_08BC viruses are not included in the clade analysis, but are counted toward the total number of neutralized viruses, because there was only one virus tested from each of these clades.
| b) Neutralization breadth | ||||||||
|---|---|---|---|---|---|---|---|---|
| % viruses neutralized with an IC50 <50 μg/ml | ||||||||
| Clade* | # viruses | b12 | 2G12 | 2F5 | 4E10 | PG9 | PG16 | PGC14 |
| A | 27 | 30 | 37 | 74 | 96 | 85 | 85 | 11 |
| B | 31 | 58 | 71 | 68 | 97 | 74 | 74 | 29 |
| C | 27 | 33 | 11 | 7 | 96 | 78 | 78 | 19 |
| D | 25 | 48 | 24 | 56 | 96 | 76 | 60 | 8 |
| CRF01_AE | 10 | 30 | 0 | 89 | 100 | 100 | 100 | 0 |
| CRF_AG | 10 | 30 | 50 | 80 | 100 | 80 | 60 | 10 |
| G | 15 | 13 | 20 | 80 | 100 | 87 | 73 | 7 |
| F | 15 | 0 | 21 | 87 | 100 | 67 | 64 | 13 |
| Total | 162 | 35 | 32 | 60 | 98 | 79 | 73 | 15 |
| % viruses neutralized with an IC50 <1.0 μg/ml | ||||||||
| Clade* | # viruses | b12 | 2G12 | 2F5 | 4E10 | PG9 | PG16 | PGC14 |
| A | 27 | 0 | 4 | 4 | 0 | 70 | 63 | 0 |
| B | 31 | 32 | 39 | 23 | 0 | 45 | 42 | 3 |
| C | 27 | 7 | 0 | 0 | 11 | 56 | 48 | 0 |
| D | 25 | 12 | 8 | 12 | 8 | 48 | 44 | 0 |
| CRF01 AE | 10 | 11 | 0 | 88 | 80 | 70 | 70 | 0 |
| CRF AG | 10 | 10 | 30 | 60 | 30 | 40 | 50 | 0 |
| G | 15 | 0 | 0 | 27 | 0 | 60 | 33 | 0 |
| F | 15 | 0 | 14 | 13 | 28 | 80 | 79 | 0 |
| Total | 162 | 11 | 12 | 19 | 12 | 57 | 51 | 1 |
Boxes are color coded as follows: white, no viruses neutralized; green, 1 to 30% of viruses neutralized; yellow, 30 to 60% of viruses neutralized; orange, 60 to 90% of viruses neutralized; red, 90 to 100% of viruses neutralized.
CRF_07BC and CRF_08BC viruses are not included in the clade analysis, but are counted toward the total number of neutralized viruses, because there was only one virus tested from each of these clades.
The inability of PG9 and PG16 to bind monomeric gp120JR-CSF or gp41HxB2 in the initial screen while potently neutralizing HIV-1JR-CSF suggested that the epitope targeted by these antibodies might be preferentially expressed on trimeric HIV Env. This possibility was investigated by comparing the ability of PG9 and PG16 to bind monomeric gp120 from several different strains, artificially trimerized gp140 constructs, and trimeric Env expressed on the surface of transfected cells (15). Although both antibodies bound with high affinity to cell surface Env, PG16 did not bind to any of the soluble gp120 or gp140 constructs and PG9 bound only weakly to monomeric gp120 and trimerized gp140 from certain strains (Fig. 1). We hypothesized that PG9 and PG16 do not exhibit exclusive specificity for cleaved HIV-1 trimers because it has been previously shown that a substantial fraction of cell surface Env is comprised of uncleaved gp160 molecules (19). Indeed, this was confirmed by the fact that both antibodies bound with high affinity to cleavage-defective HIV-1YU2 trimers expressed on the surface of transfected cells (fig. S4) (15). These results suggest that there are significant structural differences between soluble, recombinant gp140 and uncleaved gp160 expressed on the surface of transfected cells.
Fig. 1. Antigen binding properties of PG9 and PG16.
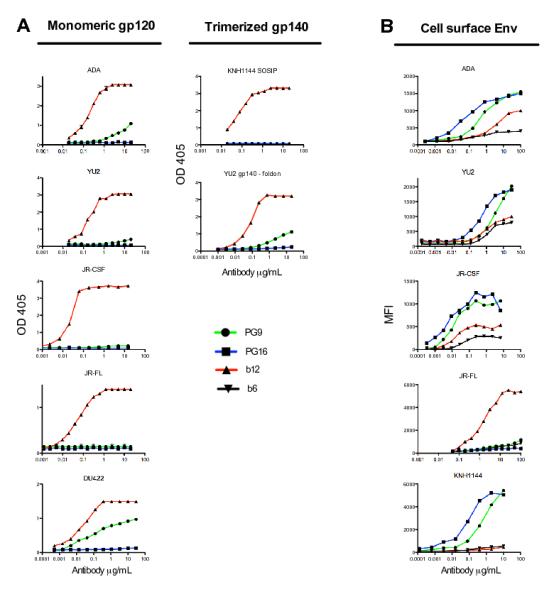
(A) Binding of PG9 and PG16 to monomeric gp120 and artificially trimerized gp140 constructs as determined by ELISA. Antigens were coated directly onto ELISA wells in the experiments shown, but similar results were also obtained when antigens were captured onto wells with antibodies against non-competitive epitopes. (B) Binding of PG9 and PG16 to Env expressed on the surface of 293T cells as determined by flow cytometry. b12 is used as a control for ELISA assays. The bNAb b12, which binds with similar affinity to both cleaved and uncleaved forms of Env, and the non-neutralizing antibody b6, which only binds to uncleaved Env, are included in the cell surface binding assays to show the expected percentages of cleaved and uncleaved Env expressed on the cell surface (19). Experiments were performed in duplicate and data are representative of at least three independent experiments.
We next sought to define the PG9 and PG16 epitopes. We suspected that they would recognize the same or overlapping epitopes because the two antibodies are somatic variants. Indeed, both antibodies competed for binding to HIV-1JR-CSF transfected cells (Fig. 2A) (15). Soluble CD4, a soluble version of the receptor for Env, diminished binding of both PG9 and PG16 to cell surface Env, although neither antibody competed with the CD4-binding site antibody b12 for trimer binding (Fig. 2A-B). This result suggests that conformational changes induced by CD4 binding cause a loss of the epitope targeted by the antibodies. Competition ELISAs revealed that PG9 competed for gp120 binding with antibodies against the V2 and V3 variable loops of gp120, and to a lesser extent, with CD4i antibodies (which recognize an epitope on gp120 dependent on CD4 binding) (Fig. 2C and fig. S5) (15). Neither PG9 nor PG16 bound to HIV-1JR-CSF variants with deleted V1/V2 or V3 variable loops expressed on the surface of transfected cells, which further suggested that the variable loops are critical components of the epitopes (Fig. 2D). To dissect the fine specificity of PG9 and PG16, alanine scanning was performed using a large panel of HIV-1JR-CSF Env alanine mutants that have been described previously (20-23) as well as several new alanine mutants. We generated pseudoviruses incorporating single Env alanine mutations, and PG9 and PG16 were tested for neutralization activity against each mutant pseudovirus (15). Mutations that resulted in viral escape from PG9 and PG16 neutralization were considered important for formation of the PG9 and PG16 epitopes (Table 2 and table S6). On the basis of this data, and consistent with the competition experiments, residues that form the epitopes recognized by PG9 and PG16 are primarily located in conserved regions of the V2 and V3 loops of gp120 (table S6 and fig. S6). Certain co-receptor binding site mutations also had an effect on PG9 and PG16 neutralization, albeit to a lesser extent (table S6). PG9 and PG16 were largely dependent on the same residues, although PG16 was more sensitive to V3 loop substitutions than PG9. Interestingly, although neither antibody bound to wild-type HIV-1JR-FL transfected cells, a E to K mutation at position 168 in the V2 loop of HIV-1JR-FL generated high-affinity PG9 and PG16 recognition (fig. S7). N156 and N160, sites of V2 N-glycosylation, were critical in forming the PG9 and PG16 epitopes because substitutions at these positions resulted in escape from antibody neutralization (Table 2 and fig. S8). HIV-1SF162 contains a rare N to K polymorphism at position 160 (which is non-permissive for N-glycosylation) and mutation of this residue to an N renders this isolate sensitive to PG9 and PG16 (fig. S9). Deglycosylation of monomeric gp120 abolished binding of PG9 (fig. S8), confirming that glycans are important, directly or indirectly, in forming the epitope (15).
Fig. 2. Mapping the PG9 and PG16 epitopes.
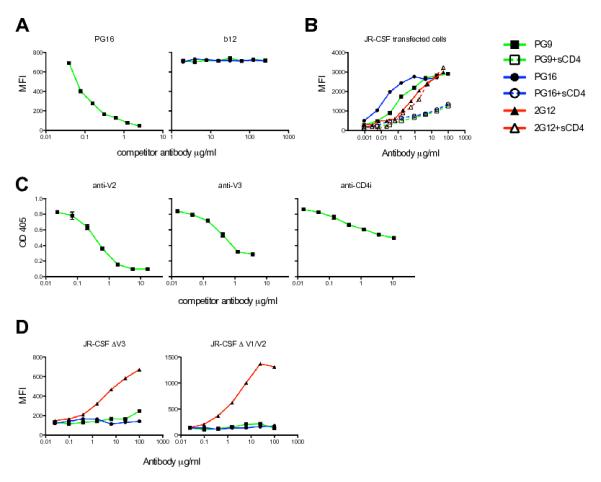
(A) Competition of PG9 and PG16 with each other and with the CD4 binding site (CD4bs) antibody b12 for cell surface Env binding. The competitor antibody is indicated at the top of each graph. (B) Effect of soluble CD4 (sCD4) on the binding of PG9 and PG16 to cell surface Env. 2G12 is included to control for CD4-induced shedding of gp120. (C) Competition of PG9 with antibodies against the V2 loop (10/76b), V3 loop (F425/b4e8), and the CD4i site (X5) for gp120 binding. Antigens were coated directly onto ELISA wells in the experiments shown, but similar results were also obtained when antigens were captured by antibodies against non-competitive epitopes. (D) Binding of PG9 and PG16 to variable loop deleted HIV-1JR-CSF variants expressed on the surface of 293T cells. 2G12 is included to control for cell surface Env expression. All experiments were performed in duplicate and data represents an average of at least two independent experiments.
Table 2.
Alanine mutations that decrease PG9 or PG16 neutralization activity.
| Mutation*† | gp120 domain‡ | Fold IC50 increase relative to wild-type§ | |
|---|---|---|---|
| PG9 | PG16 | ||
| V127A | C1 (V1/V2 stem) | 30 | 57 |
| N134A | V1 | 5 | 23 |
| N156A | C1 (V1/V2 stem) | 280 | 1500 |
| S158A | C1 (V1/V2 stem) | >2000 | >2000 |
| F159A | C1 (V1/V2 stem) | >2000 | >2500 |
| N160K | V2 | >2000 | >2500 |
| T162A | V2 | >2000 | >2500 |
| D167A | V2 | 5 | 30 |
| Y173A | V2 | 1400 | 1000 |
| F176A | V2 | >5000 | >7000 |
| V181A | V2 | 200 | 250 |
| P299A | V3 (base) | 200 | 1400 |
| K305A | V3 (stem) | 50 | 2800 |
| I307A | V3 (tip) | 10 | 3000 |
| I309A | V3 (tip) | 9 | 150 |
| F317A | V3 (tip) | 3 | 1400 |
| Y318A | V3 (tip) | 2 | 1000 |
| N392A | V4 | 7 | 23 |
| I420A | C4 | 9 | 11 |
| I423A | C4 | 40 | 14 |
| I424A | C4 | 10 | 9 |
Amino acid numbering is based on the sequence of HIV-1HxB2.
Boxes are color coded as follows: white, the amino acid is identical among 0 to 49% of all HIV-1 isolates; light blue, the amino acid is identical among 50 to 90% of isolates; dark blue, the amino acid is identical among 90 to 100% of isolates. Amino acid identity was determined based on a sequence alignment of HIV-1 isolates listed in the HIV sequence database at http://hiv-web.lanl.gov/content/hiv-db/mainpage.html.
C refers to constant domains and V refers to variable loops.
Neutralization activity is reported as fold increase in IC50 value relative to WT JR-CSF and was calculated using the equation (IC50 mutant / IC50 WT). Boxes are color coded as follows: green, substitutions which had a negligible effect on neutralization activity; yellow, 4 - 9 fold IC50 increase; orange, 10 - 100 fold IC50 increase; red, >100 fold IC50 increase. Experiments were performed in triplicate and values represent an average of at least three independent experiments.
The preferential binding of PG9 and PG16 to native trimers could either be because their epitopes span more than one gp120 subunit or because the antibodies recognize a single subunit in a conformation that is stabilized on the trimer. To address this question, we studied the binding profiles of PG9 and PG16 to mixed HIV-1YU2 trimers, in which two gp120 subunits contained point mutations to abolish binding of the two antibodies (15). A third substitution that abrogates binding of 2G12, which binds with high affinity to both monomeric gp120 and trimeric Env, was also introduced into the same construct as an internal control. Cell surface binding analysis revealed that all three antibodies bound to the mixed trimers with similar apparent affinity compared to wild-type trimers and all binding was saturated at a similar lower level (fig. S10). This result suggests that the preference of PG9 and PG16 for trimeric Env is due to gp120 subunit presentation in the context of the trimeric spike rather than gp120 cross-linking.
Others have shown that NAbs that bind to epitopes encompassing parts of the V2 or both the V2 and V3 domains can exhibit potency comparable to that of PG9 and PG16, although these antibodies have thus far displayed strong strain-specificity (17, 24). Importantly, the epitopes recognized by these antibodies have been shown to differ from that of the clade B consensus sequence only by single amino acid substitutions, which suggested the existence of a relatively conserved structure within the V2 domain (17, 18). Here, we have confirmed that this region serves as a potent neutralization target and demonstrated that antibodies that recognize conserved parts of V2 and V3 can possess broad reactivity.
Given that this is the first attempt at direct functional screening of such a large number of antibodies, it seems likely that the approach will generate further bNAbs, in particular those that bind poorly to recombinant forms of Env. The neutralization breadth exhibited by PG9 and PG16, particularly against non-clade B isolates, suggests that vaccine-induced antibodies of similar specificity might provide protection against a diverse range of the most prevalent HIV-1 isolates circulating worldwide. Furthermore, the exceptional neutralization potency exhibited by these antibodies in vitro suggests that antibodies of this specificity might provide protection at relatively modest serum concentrations achievable by vaccination. Immunogens designed to focus the immune response on conserved regions of variable loops in the context of the trimeric spike will determine whether these types of antibodies can be readily elicited by a vaccine.
Supplementary Material
Acknowledgments
We thank R. Aguilar-Sino for technical assistance at TSRI and C. Ward, W. Cieplak and M. Branum for technical assistance at Theraclone Sciences. We would also like to thank R. Pejchal for assistance in structural modeling, and S.M. Eagol, C. Corbaci, and C. Williams for assistance with figures. We would like to thank E. Anton from Monogram R&D and the Monogram Clinical reference Laboratory staff, all the study participants and research staff at each of the Protocol G clinical centers, and Protocol G project team members for all of the support that they have provided for this study (individuals listed in Supplementary Notes). We are grateful to A. Pinter for supplying the antibodies 10/76b and c108g, W. Olson and J. Moore for providing the KNH1144 SOSIP trimer, and R. Wyatt for providing the YU2gp140-foldon trimer. This work was supported by the International AIDS Vaccine Initiative (IAVI) through the generous contributions of a number of governmental and private donors who are listed on the website www.iavi.org, as well as IAVI’s Innovation Fund (which is co-funded by the Bill & Melinda Gates Foundation), the United States Agency for International Development (USAID)and NIH AI 33292 (DRB). The contents are the responsibility of the authors and do not necessarily reflect the views of USAID or the United States Government. The authors declare competing financial interests. Protocol G Principal Investigators: G. Miiro, J. Serwanga, A. Pozniak, D. McPhee, O. Manigart, L. Mwananyanda, E. Karita, A. Inwoley, W. Jaoko, J. DeHovitz, L.G. Bekker, P. Pitisuttithum, R. Paris, and S. Allen. Individual affiliations are listed in the Supplementary Notes.
Footnotes
Supporting Online Material www.sciencemag.org Materials and Methods figs. S1 to S10 tables S1 to S6 References
References and Notes
- 1.Johnston MI, Fauci AS. N Engl J Med. 2007;356:2073. doi: 10.1056/NEJMra066267. [DOI] [PubMed] [Google Scholar]
- 2.Barouch DH. Nature. 2008;455:613. doi: 10.1038/nature07352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walker BD, Burton DR. Science. 2008;320:760. doi: 10.1126/science.1152622. [DOI] [PubMed] [Google Scholar]
- 4.Veazey RS, et al. Nat Med. 2003;9:343. doi: 10.1038/nm833. [DOI] [PubMed] [Google Scholar]
- 5.Hessell AJ, et al. PLoS Pathog. 2009;5:e1000433. doi: 10.1371/journal.ppat.1000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parren PW, et al. J Virol. 2001;75:8340. doi: 10.1128/JVI.75.17.8340-8347.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mascola JR. Vaccine. 2002;20:1922. doi: 10.1016/s0264-410x(02)00068-3. [DOI] [PubMed] [Google Scholar]
- 8.Mascola JR, et al. Nat Med. 2000;6:207. doi: 10.1038/72318. [DOI] [PubMed] [Google Scholar]
- 9.Mascola JR, et al. J Virol. 1999;73:4009. doi: 10.1128/jvi.73.5.4009-4018.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Phogat S, Wyatt R. Curr Pharm Des. 2007;13:213. doi: 10.2174/138161207779313632. [DOI] [PubMed] [Google Scholar]
- 11.Montero M, van Houten NE, Wang X, Scott JK. Microbiol Mol Biol Rev. 2008;72:54. doi: 10.1128/MMBR.00020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scanlan CN, Offer J, Zitzmann N, Dwek RA. Nature. 2007;446:1038. doi: 10.1038/nature05818. [DOI] [PubMed] [Google Scholar]
- 13.Simek MD, et al. J Virol. 2009 [Google Scholar]
- 14.Stamatatos L, Morris L, Burton DR, Mascola JR. Nat Med. 2009 doi: 10.1038/nm.1949. [DOI] [PubMed] [Google Scholar]
- 15.Materials and methods are available as supporting material on Science Online.
- 16.Ichiyoshi Y, Casali P. J Exp Med. 1994;180:885. doi: 10.1084/jem.180.3.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Honnen WJ, et al. J Virol. 2007;81:1424. doi: 10.1128/JVI.02054-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pinter A, et al. J Virol. 2005;79:6909. doi: 10.1128/JVI.79.11.6909-6917.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pancera M, Wyatt R. Virology. 2005;332:145. doi: 10.1016/j.virol.2004.10.042. [DOI] [PubMed] [Google Scholar]
- 20.Pantophlet R, et al. J Virol. 2003;77:642. doi: 10.1128/JVI.77.1.642-658.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pantophlet R, Wang M, Aguilar-Sino RO, Burton DR. J Virol. 2009;83:1649. doi: 10.1128/JVI.02046-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Darbha R, et al. Biochemistry. 2004;43:1410. doi: 10.1021/bi035323x. [DOI] [PubMed] [Google Scholar]
- 23.Scanlan CN, et al. J Virol. 2002;76:7306. doi: 10.1128/JVI.76.14.7306-7321.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gorny MK, et al. J Virol. 2005;79:5232. doi: 10.1128/JVI.79.8.5232-5237.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.