

Abstract
A new small molecule inhibitor of bacterial cell division has been discovered using a high-throughput screen in E. coli. Although the lead screening hit (534F6) exhibited modest inhibition of the GTPase activity of FtsZ (20±5% at 100 μM primary target for bacterial cell division inhibitors, several analogs caused potent bacterial growth inhibition with negligible antagonism of FtsZ GTPase activity. A library of analogs has been prepared and several alkyne-tagged photoaffinity probes have been synthesized for use in experiments to elucidate the primary target of this compound.
The emergence of antibiotic-resistant strains of bacteria has prompted a worldwide effort to seek new avenues for fighting infectious disease.1 Most antibiotics discovered to date target a narrow range of biochemical processes in bacteria.2 FtsZ, the prokaryotic analog of tubulin,3 has been examined as a potential new target for antimicrobial chemotherapy. Although FtsZ has been the primary target for small molecules that inhibit bacterial cell division,4 it is likely that other proteins essential for bacterial cytokinesis can also be targeted.5 A high-throughput screen has recently been developed to identify compounds that cause lethal cell filamentation in E. coli.6,7 This screen revealed new inhibitors of FtsZ and at the same time yielded several compounds that caused cell filamentation without inducing the SOS response or without significantly inhibiting the GTPase activity of FtsZ. Herein we describe our preliminary SAR studies of 534F6, an N-benzyl-3-sulfonamidopyrrrolidine (Figure 1) and our initial preparation of photoaffinity reagents for the identification of this compound’s protein target(s).
Figure 1.
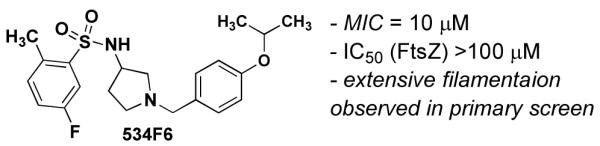
Lead compound (534F6) from phenotypic HTS for compounds that induce lethal filamentation in E. coli.
534F6 displayed weak inhibition of FtsZ GTPase (20±5% at 100 μM), did not affect steady-state FtsZ polymerization as assayed by high-speed sedimentation, and induced SOS-independent E. coli cell filamentation (data not shown). Despite a certain degree of similarity to sulfonamide antibiotics, such as sulfamethoxazole, compound 534F6 exhibited markedly different effects on E. coli. Sulfamethoxazole showed modest lethality (MIC > 80μM) and did not cause E. coli (AcrAB efflux pump knockout strain DRC 394d) to filament. Based on these observations, we set out to develop a library synthesis of 534F6 in an effort to optimize potency and eventually determine the protein target of this compound.
We began by investigating the SAR of 534F6. Since the configuration of this compound was unknown, we prepared each of the two enantiomers as a test for the influence stereochemistry on activity. Although no synthesis of 534F6 had previously been reported, we were able to convert commercially available (S)-1 and (R)-1 in to (S)- and (R)-534F6, respectively, in three
Steps (Scheme 1). We were delighted to find that the (R)-enantiomer caused lethal E. coli filamentation with an MIC of 10 μM, whereas the (S)-enantiomer neither induced cell filamentation nor killed E. coli up to 80 μM. This result suggests that 534F6 is reasonably selective in its interaction with its target or targets.
Scheme 1.
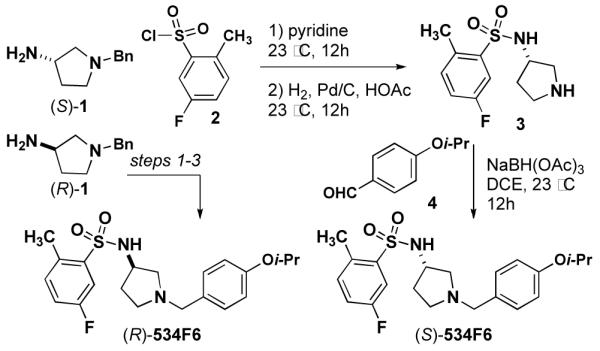
Synthesis of the enantiomers of 534F6
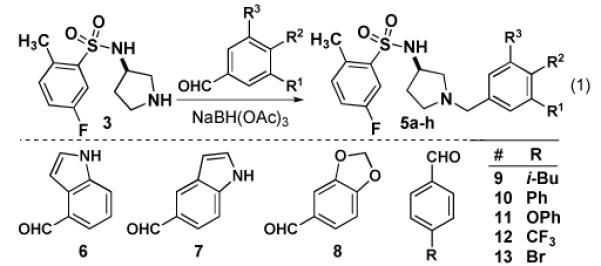
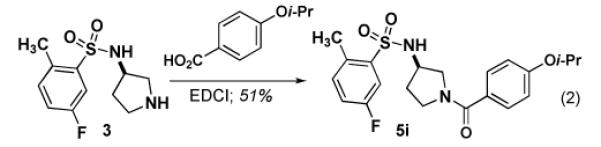
Using our synthetic route to 534F6, we were able to prepare an initial series of analogs to establish the influence of the N-benzyl substituent. Intermediate 3 was condensed with a series of aromatic aldehydes to produce a series of analogs featuring different ortho-, meta-, and para substituents (eq 1). As Table 1 shows, introduction of fused rings or polar para substituents was deleterious to the activity of these compounds. Replacement of the isopropxy group with either the isosteric isobutyl group, a phenyl ring, or a phenoxy group retained activity. Replacement of the benzyl amine with a benzoyl amide greatly diminished the antimicrobial activity of this compound (eq 2).
Table 1.
GTPase Inhibition and antimicrobial activities of 5a-i.
Based on these preliminary results, we developed a solid phase synthesis of 210 analogs of 534F6, mindful of the importance of the N-benzyl substituent. Since the
 |
 |
substituent at the para position had proven to be crucial for activity, we elected to keep this structural feature invariant. We replaced the isopropoxy group of 534F6 with a hydroxyethyl group as a point of attachment to solid phase synthesis resin.8 Approximately 35 mg of resin were employed in Iroritm kans and the synthesis was tracked using 2D-barcoding.9 The first step of the synthesis was a reductive amination with a protected amine that would later be functionalized. We initially explored phthaloyl (phth) and tetrachlorophthaloyl (tcph) protecting groups for the primary amine, but found the former to be preferable once the conditions were adjusted to account for the precipitated phthalyl hydrazide. In order to explore the structural elements that might contribute to activity, we prepared protected amine cores A1-A7. These were condensed onto the aldehyde starting material in six different reaction batches, then pooled for deprotection, and split for attachment of the sulfonyl groups. A series of sulfonyl chlorides were employed in the last step to yield a total of 210 compounds after cleavage. The compounds were tested for growth inhibition and cell filamentation in E. coli. None showed improved activity and the majority appeared to be less potent than 534F6. Although we expected diminished activity from the hydroxyethoxy substituent, we had hoped that an optimized core and sulfonyl substituent would compensate.
We next focussed on a narrower selection of core structures with a single benzyl substituent. Amine core structures A2, A4, and A6 were selected and each was N-benzylated using p-isobutylbenzyaldehyde. These three amines were converted in parallel to the corresponding sulfonamides using excess quantities of sulfonyl chlorides B1-B15 and a scavenger resin for removal of the excess reagent.10 These 45 compounds were tested and compound 14 was found to be the most potent, with an MIC of 10 μM. Examination of the E. coli culture treated with 5 μM 14 showed extensive filamentation (fig. 2A). Compound 15, featuring the same sulfonamide and benzyl groups on a different core, exhibited an MIC of 20 μM, but little filamentation was observed at 10 μM (fig. 2B).
Figure 2.
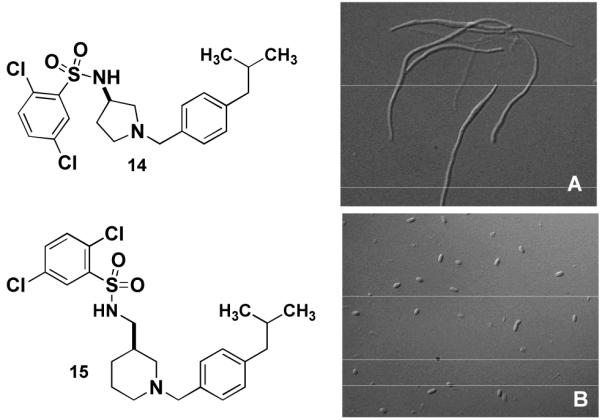
E. coli DRC39 treated for 15h with A) 5 μM 14 15.
In a parallel effort, we have prepared several derivatives of 534F6 for use in target identification. We designed several compounds that would serve as photoaffinity reagents to modify their protein targets.11 In addition, we incorporated terminal alkyne substituents as chemical tags that would allow us to separate the modified target from the cellular lysate (scheme 3).12
Scheme 3.
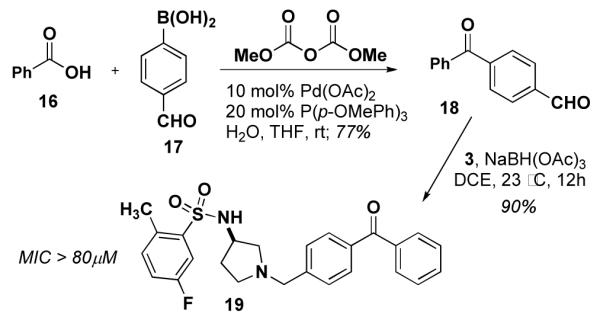
Synthesis of 19, a benzophenone derivative of 534F6.
We initially explored the possibility of incorporating a benzophenone group in the para position of the N-benzyl substituent. The requisite 4-formyl benzophenone (18) was prepared using the palladium coupling reported by Winkel.13 Reductive amination of 18 yielded 19. This compound’s weak activity (MIC >80 μM) prompted us to installing a photoreactive group.
Our next compound was designed to use an aryl azide as the photoreactive group. 21a was prepared by reductive amination of 20. This compound was converted to sulfonamide 22a, which was carried on to alkyne 23a. Sulfonamidopyrrolidine 23a exhibited an MIC of <12 μM, alkyne did not affect the activity. Encouraged by this result, we proceeded with the synthesis of 23b by a parallel synthetic route. This synthesis was enabled by the ligand- and copper-free Sonagashira reaction reported Examination by Verkade,14 which avoids of reduction the and 5 cycloaddition μM of the aryl azide. Compund-23b exhibited an MIC of >40 μM, of a lipophilic group at this site for activity. The activity little of 23a established the viability of an alkyne on sulfonamide portion of the molecule.
The activities of 23a and 23b encouraged us to explore the possibility of a hybrid of these two compounds with 534F6. Sulfonamide 25 was prepared in two steps from N-Boc-(S)-3-aminopyrrolidine (20). The Boc group was removed and the 3-nitro-4-isopropoxy benzyl group was installed by reductive amination.15 The nitro group was reduced to the corresponding aniline, which was then diazotized and displaced with azide. After deprotection with TBAF, compound 29 was examined for antimicrobial activity and found to have an MIC of >64 μM. Although sulfonamide only slightly lowered the activity of 23a relative to 5a, it is apparent that the combined effect of the ortho azide and the alkyne greatly diminishes the activity of 29. We are currently preparing an affinity matrix with 23a using “clickable” agarose in hopes of pulling down the protein target from a cell lysate of E. coli.
In summary, we have discovered a new compound (534F6) that appears to inhibit bacterial cell division without inhibiting FtsZ as the primary target. Initial attempts to prepare alkyne tagged photoaffinity reagents have revealed regions of the molecule that are not suitable for structural variation. We are currently examining a variety of alternative approaches for identifying the target of this compound.
Scheme 2.

Solid- and solution-phase synthesis of analogs of 534F6.
Scheme 4.

Synthesis of 23a and 23b, alkyne-tagged derivatives of 5a.
Scheme 5.
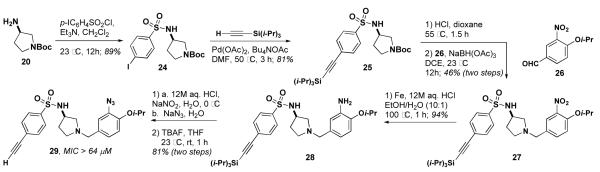
Synthesis of 29, an alkyne-tagged and azide-appended derivative of 534F6.
Acknowledgements
J.T.S. thanks the National Institute for Allergy and Infectious Disease (NIAID, RO3 AI062905) and the Broad Institute Scientific Planning and Allocation of Resources Committee (SPARC) for funding of this research. D.R.C. acknowledges DARPA, the Charles A. Dana Foundation, and the National Institute of General Medical Sciences (NIGMS; RO1 GM068025) for research support. The Hudson-Alpha Institute for Biotechnology is acknowledged for support of S.M. as a visiting scientist. The Harvard College Research Program (HCRP) is acknowledged for a fellowship to T.M. We thank Profs. Marc Kirschner and Tim Mitchison (Harvard Medical School) for insightful discussions. A portion of this work was conducted in Prof. Kirschner’s Laboratory. This project has been funded in part with Federal funds from the NCI’s Initiative for Chemical Genetics, NIH, under Contract No. N01-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the U.S. Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Walsh C. Nature Reviews Microbiology. 2003;1:65–70. doi: 10.1038/nrmicro727. [DOI] [PubMed] [Google Scholar]
- 2.Walsh C. Antibiotics: Actions, Origins, Resistance. ASM Press; Washington DC: 2003. [Google Scholar]
- 3a).Romberg L, Levin PA. Ann. Rev. Microbiol. 2003;57:125–154. doi: 10.1146/annurev.micro.57.012903.074300. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Margolin W. Nat. Rev. Mol. Cell Biol. 2005;6:862–871. doi: 10.1038/nrm1745. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Dajkovic A, Lutkenhaus J. J. Mol. Microbiol. Biotechnol. 2006;11:140–51. doi: 10.1159/000094050. [DOI] [PubMed] [Google Scholar]
- 4a).White EL, Suling WJ, Ross LJ, Seitz LE, Reynolds RC. J. Antimicrob. Chemother. 2002;50:111–114. doi: 10.1093/jac/dkf075. [DOI] [PubMed] [Google Scholar]; b) Reynolds RC, Srivastava S, Ross LJ, Suling WJ, White EL. Bioorg. Med. Chem. Lett. 2004;14:3161–3164. doi: 10.1016/j.bmcl.2004.04.012. [DOI] [PubMed] [Google Scholar]; c) Wang J, Galgoci A, Kodali S, Herath KB, Jayasuriya H, Dorso K, Vicente F, Gonzalez A, Cully D, Bramhill D, Singh S. J. Biol. Chem. 2003;278:44424–44428. doi: 10.1074/jbc.M307625200. [DOI] [PubMed] [Google Scholar]; d) Margalit DN, Romberg L, Mets RB, Hebert AM, Mitchison TJ, Kirschner MW, RayChaudhuri D. Proc. Natl. Acad. Sci. U. S. A. 2004;101:11821–11826. doi: 10.1073/pnas.0404439101. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Urgaonkar S, Pierre H. S. La, Meir I, Lund H, Chaudhuri D. Ray, Shaw JT. Org. Lett. 2005;7:5609–5612. doi: 10.1021/ol052269z. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Stokes NR, et al. J. Biol. Chem. 2005;280:39709–39715. doi: 10.1074/jbc.M506741200. [DOI] [PubMed] [Google Scholar]; g) Jaiswal R, Beuria TK, Mohan R, Mahajan SK, Panda D. Biochemistry. 2007;46:4211–4220. doi: 10.1021/bi602573e. [DOI] [PubMed] [Google Scholar]
- 5.Vollmer W. Appl. Microbiol. Biotechnol. 2006;73:37–47. doi: 10.1007/s00253-006-0586-0. [DOI] [PubMed] [Google Scholar]
- 6.RayChaudhuri D, Kirschner MW. 7,011,946 B2 US Patent. 2006 Mar 14; An efflux pump knockout strain was employed. See also: Hsieh P-C, Siegel SA, Rogers B, Davis D, Lewis K. Proc. Natl. Acad. Sci. U. S. A. 1998;95(12):6602–6606. doi: 10.1073/pnas.95.12.6602.
- 7.Hebert AM, RayChaudhuri D, et al. This screen was conducted at the Institute of Chemistry and Cell Biology. (ICCB, Harvard Medical School), which merged with several other institutions to form the Broad Institute of Harvard and MIT; 2004. manuscript in preparation. [Google Scholar]
- 8.Tallarico JA, Depew KM, Pelish HE, Westwood NJ, Lindsley CW, Shair MD, Schreiber SL, Foley MA. J. Comb. Chem. 2001;3:312–318. doi: 10.1021/cc000107i. [DOI] [PubMed] [Google Scholar]
- 9a).We have previously used the silicon linker-based macrobeads in kans for the preparation of hundreds of compounds: Mitchell JM, Shaw JT. Angew. Chem., Int. Ed. 2006;45:1722–1726. doi: 10.1002/anie.200503341. Ng PY, Tang Y, Knosp WM, Stadler HS, Shaw JT. Angew. Chem., Int. Ed. 2007 doi: 10.1002/anie.200700762. in press.
- 10.PS-N(i-Pr)2 = PL-DIPAM MP, 150-300 μM, 1.6 N/g (Varian/Polymer Laboratories). PS(NHCH2CH2)2-NH2 = PL-DETA MP, 150-300 μm, 4.5 mmol N/g (Varian/Polymer Laboratories).
- 11.Hatanaka Y, Sadakane Y. Curr. Top. Med. Chem. 2002;2:271–288. doi: 10.2174/1568026023394182. [DOI] [PubMed] [Google Scholar]
- 12.Evans MJ, Saghatelian A, Sorensen EJ, Cravatt BF. Nature Biotechnology. 2005;23:1303–1307. doi: 10.1038/nbt1149. [DOI] [PubMed] [Google Scholar]
- 13.Goossen LJ, Paetzold J, Winkel L. Synlett. 2002:1721–1723. [Google Scholar]
- 14.Urgaonkar S, Verkade JG. J. Org. Chem. 2004;69:5752–5755. doi: 10.1021/jo049325e. [DOI] [PubMed] [Google Scholar]
- 15.The requisite aldehyde is prepared in one step (91% yield) from 4-hydroxy-3-nitrobenzaldehyde, i-PrBr, and K2CO3 in CH3CN by heating to 150 °C with microwave irradiation.