

Abstract
Analysis of genetic variation among modern individuals is providing insight into prehistoric events. Comparisons of levels and patterns of genetic diversity with the predictions of models based on archeological evidence suggest that the spread of early farmers from the Levant was probably the main episode in the European population history, but that both older and more recent processes have left recognizable traces in the current gene pool.
Where do the genes of the Europeans come from? A good, but trivial, answer is: From Africa, like everybody else's genes. Paleontologists agree that the long-term human ancestors, a million years ago or so, dwelt in Africa. There is disagreement, however, about what happened after archaic presapiens humans (Homo erectus) spread over much of the Old World. The anatomically archaic populations of Europe, Northeastern Asia, and Southeastern Asia may have gradually evolved into the modern Homo sapiens sapiens populations inhabiting, respectively, Western Eurasia, East Asia, and Australia; this is the multiregional theory of human evolution (1). On the contrary, the Out-of-Africa theory regards all modern populations as descended from an anatomically modern group that dispersed from Africa less than 200,000 years ago and replaced archaic populations (2).
Discussing the relative merits of the two models would be out of place here, but the multiregional model proposes that the Neandertal people are the ancestors of contemporary Europeans. Conversely, we now know that the mitochondrial sequences of Neandertals differed sharply from modern European sequences, and in fact, from all modern human sequences (3, 4). It is more than likely, then, that the Neandertal people left no modern descendants (refs. 5–7; for a different view, see ref. 8).
If there was no Neandertal contribution to the contemporary gene pool, European gene diversity must reflect some combination of the demographic phenomena occurring after Homo sapiens sapiens colonized the continent. However, these phenomena acted upon genetic variation that accumulated both after and before Europe was colonized, because there is no reason to imagine that the first Europeans were all genetically identical. The distinction between histories of populations (which is what this paper is about) and histories of molecules (which are simpler to reconstruct, but are not the same thing) has sometimes been overlooked; we shall come back to it later. As for the European population history, the presence of Homo sapiens sapiens is first documented around 40,000 years ago (9). But which fraction of the modern European gene pool is derived from these first colonizers, and how much, instead, from more recent immigrants?
Genetic Variation as a Clue to Prehistoric Phenomena
The main way to gain insight into past population processes is to analyze and interpret current patterns of genetic variation (10, 11). Data on ancient DNA can also help, but they are scanty now, and will not become abundant in the foreseeable future (12). One difficulty with modern genes lies in the fact that any given pattern of variation may potentially be explained by several different evolutionary phenomena. A cline or gradient, for example, may reflect adaptation to variable environments, or a population expansion at one moment in time, or continuous gene flow between groups that initially differed in allele frequencies. However, it is possible to discard at least some implausible models by jointly analyzing many loci (selection tends to affect single genes, whereas demographic changes determine similar patterns across the genome), and by exploiting nongenetic information, such as archeological and paleobiological data.
Three large-scale phenomena have been inferred from the European archeological record (Fig. 1). In the Upper Paleolithic, around 40,000 years ago, Neandertal people were replaced by anatomically modern humans (9), who moved in from the Levant, and settled in many areas of the continent (13). At the latest glacial maximum, some 18,000 years ago, Northern and Central Europe were largely covered with glaciers. Human presence then seems restricted to the warmest regions, or glacial refugia (14), and only later reappears more to the North, accompanying the retreat of the ice sheet; we shall refer to that postglacial phase as the Mesolithic period. The first evidence of food production (farming and animal breeding—i.e., the so-called Neolithic revolution) dates at around 10,000 years B.P. in the Levant (15, 16). Gradually, Neolithic artifacts spread westwards and northwards, along much the same routes followed by the first Paleolithic colonization. Later demographic shifts affecting Europe as a whole are not documented. Thus, the overall pattern of European genetic diversity probably reflects the effects of the first Paleolithic colonization, or of Mesolithic reexpansions, or of the Neolithic demic diffusion, although the history of each local population must have been much more complicated than that.
Figure 1.

A scheme of the main demographic processes documented in the archeological record of Europe. Numbers are approximate dates, in years before the present. Green arrows, Paleolithic colonization; red arrows, Mesolithic reexpansions (glacial refugia are represented by red circles); blue arrows, Neolithic demic diffusion.
Abundant though it might be, the archeological evidence does not tell us the whole story. For instance, humans may have lived north of the ice limit without leaving archeologically relevant material, and Neolithic artifacts may have spread because early farmers moved, or simply through trading.
More exhaustive information on the demographic impact of prehistoric processes can come only from the study of genes. Many protein markers show broad gradients, spanning from the Levant to Northern and Western Europe (17–20). Allele frequencies of those markers (Fig. 2) correlate with the archeological dates of origin of agriculture, and so Ammerman and Cavalli-Sforza (15) proposed that the European genetic population structure was determined mainly by population dispersal in the Neolithic, a process which they called the Neolithic demic diffusion. Note that population movements do not necessarily produce clines. To generate the observed gradients, four conditions are necessary, namely: (i) that the Neolithic farmers of the Levant differed genetically from the European hunters and gatherers; (ii) that the former were growing in numbers; (iii) that they dispersed westwards and northwards; and (iv) that they did not immediately incorporate in their communities the hunters and gatherers whom they met during the expansion (15). As confirmed by computer simulations (21, 22), only under those conditions could the alleles typical of the Levant end up being distributed in ample gradients.
Figure 2.

A summary of genetic variation in Europe: first principal component. Different shades of gray represent different values of a synthetic variable summarizing allele frequencies at 120 protein loci. (Cavalli-Sforza, L. L., et al., The History and Geography of Human Genes. Copyright © 1994 by PUP. Reprinted by permission of Princeton University Press; ref. 20.)
The model of Neolithic demic diffusion has two important implications. One is that the technologies for food production did not spread by cultural contacts (which would have had no genetic effect), but essentially by population dispersal: farming spread because the farmers did. The second is that a large fraction of the ancestors of current Europeans (at least two-thirds, based on the simulations of refs. 21 and 22) lived in the Levant, not in Europe, 10,000 years ago.
Gene Genealogies and Population Histories
DNA variation is conveniently summarized by gene genealogies. Because of their (complete, or nearly so) absence of recombination, the mitochondrial genome and the Y chromosome are ideal for reconstructing evolutionary trees or networks. Under reasonable assumptions about mutation rates, trees and networks can be put into a time frame, and the age of the molecules at their nodes can be estimated. To the best of our knowledge, a global age of the European mitochondrial genealogy has never been published, and it would be very old anyway, certainly older than the arrival of Homo sapiens sapiens in Europe. However, groups of evolutionarily related alleles have been defined within the genealogy, and their age has been variously estimated between 52,500 (haplogroup U5) and 6,500 years (haplogroup J1a) (23). The fact that the origin of most such haplogroups predates the origin of farming has been taken as evidence that the European mitochondrial pool comes essentially from populations that were already settled in Europe before the Neolithic period (ref. 24, and references therein). The fact that the age of some haplogroups, and hence of the entire genealogy, predates the arrival of Homo sapiens sapiens in Europe has not received much attention.
Although a pre-Neolithic origin of the European gene pool is in contrast with the demic diffusion model, an alternative model has not been explicitly formalized yet. In the first studies of mitochondrial networks (25, 26), the clines observed for non-DNA markers were attributed to repeated founder effects in the course of the initial Paleolithic colonization, a scenario that previous simulations have proven plausible (22). In later papers, however, European patterns of genetic variation were attributed to the effects of large-scale Mesolithic reexpansions from South-Central Europe (24, 27).
When molecular data were analyzed by methods comparing populations, rather than the molecules themselves, broad and significant gradients radiating from the Levant became apparent for both autosomic (28, 29) and Y-linked (30–32) markers. Additional clines were also recognized, on a more limited geographical scale. For instance, biallelic Y-chromosome polymorphisms show a gradient from Northeastern Europe into the South (32), which has also been observed at the protein (17, 18), but not DNA, level, perhaps for lack of suitable samples. For mtDNA, no global cline is evident, but there is a significant gradient of molecular diversity in the Mediterranean region (33).
Evolution by Repeated Founder Effects?
In summary, the clinal distributions of nuclear DNA and protein markers suggest that a directional expansion from the Levant is the main process reflected in the current genetic diversity, and that other phenomena had a lesser impact on modern genetic variation. The direction of the main cline corresponds to the direction of both the initial Paleolithic colonization and the Neolithic demic diffusion, but not to any known Mesolithic process. Conversely, most mtDNA haplogroups coalesce in pre-Neolithic times, which has been interpreted as a consequence of Mesolithic expansions from glacial refugia. Is there any way to reconcile those findings? To understand for good whether the European gene pool derives from Paleolithic or Neolithic ancestors, one should type individuals who lived, respectively, in Europe and in the Near East, say 15,000 years ago. Should these groups prove genetically different, one could infer a Paleolithic origin of the modern gene pool from a closer similarity between modern and ancient Europeans, and a Neolithic origin from a closer similarity between modern Europeans and the ancient inhabitants of the Near East.
That experiment is impossible at present. But similar, albeit more limited, questions can be addressed by analyzing contemporary samples, in the light of theories on the way shared ancestry affects genetic diversity (see ref. 34). When one estimates populations' ages based on molecular trees, the implicit assumption is that population genealogies are well approximated by allele genealogies. In fact, theory shows that that is so only if each population developed from a genetically monomorphic set of founders. Only in that case will all of the existing genetic diversity result from mutations that occurred after the population was established (plus the occasional alleles introduced by gene flow), and therefore will the coalescence time be close to the population's age (35, 36). Thus the question is, are the European populations descended from monomorphic groups of ancestors?
Let us imagine that 10,000 years ago an initially panmictic group split in two groups. If (i) the populations' effective sizes after the split were 5,000 individuals, a conservative value used by other investigators (37, 38), (ii) each generation lasted 20 years, and (iii) 50 sequences are sampled today from each population, the expected number of lineages at the split can be calculated from the simple convolution with itself of the probabilities derived for a single population (39). On the average, about 45 lineages still remain, and 95% of the probability distribution is comprised between 38 and 52. This conclusion is even stronger for larger populations and longer generation times (40), and little changes if we consider expanding populations. Even when the rate of exponential increase is as high as r = 0.02 (which means that the population size 10,000 years ago was a few tens of individuals), 15–20% of lineages coalesce more than 10,000 years ago.
In brief, there is a high chance that populations that separated in Neolithic times and then stayed constant in size or increased contained extensive initial polymorphism. Therefore, any gene genealogy is not expected to portray the recent (i.e., less than 10,000 years ago) population's history, unless founder effects at the origin of each new population eliminated the preexisting polymorphism.
But is it safe to assume that radical founder effects accompanied the origin of farming communities in Europe? The distributions of pairwise mitochondrial sequence differences, or mismatch distributions, are unimodal and smooth in populations that expanded, and multimodal and irregular in populations that were stable in size (41) or shrank (42). On a worldwide scale, mismatch distributions are unimodal in farming populations and multimodal in hunting–gathering communities, suggesting that demographic crises have been common in the latter, not in the former (42). Accordingly, founder effects may have occurred at the origin of specific European farming communities (43), but they really seem an exception, not the rule.
Although interpreting allele trees as population trees is risky (see also ref. 44), mitochondrial data do contain information on the probable timing of population splits. For instance, pairs of populations that separated in Paleolithic times can be expected to share fewer alleles than populations that separated later. We counted the occurrences of identical mitochondrial sequences in pairwise comparisons of populations from a European and Near Eastern dataset (33). The distribution of P0, the frequency of identical sequences in 780 such comparisons, has two distinct peaks (Fig. 3). In the first peak there are 197 pairs of populations with very few sequences in common, if any (P0 ≤ 0.01), and 163 of those comparisons involve Near Easterners, or European populations that have been identified as outliers: Icelanders, Ladins, or Saami (33). In the second peak there are 583 comparisons whose mode is close to 0.03.
Figure 3.

Observed frequency of identical sequences in 780 pairwise comparisons of 40 European mitochondrial population samples. The two boxes refer to the range of expected values, estimated assuming two rates r of population growth (r = 0.005 and r = 0.008) and a Paleolithic (40,000 years ago; box A) or a Neolithic (4,000 years ago; box B) separation of the gene pools.
To estimate the expected value of P0 for populations that diverged T years ago, let us assume a present census size of 107 individuals. Let us also assume that populations have been exponentially increasing to their present size with a rate r = 0.005 or 0.008 (45, 46), and that the mutation rate for the mtDNA first hypervariable control region is 1.65 × 10−7 per site per year (42). Under these conditions, the expected P0 is the product of the probability of 0 differences from the present time back to time T (i.e., the probability of 0 mutations in 2T generations), multiplied by the probability of 0 differences from time T until the coalescence of the two sequences in the ancestral population. This second factor is complex for nonstationary populations, but it can be found by using the theorem of total probability
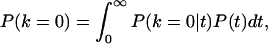 |
where k is the number of substitutions between two sequences. The conditional probability P(k = 0|t) is again the probability of no mutations in 2t generations, whereas an expression for P(t) can be found in ref. 47. Solving numerically the integral above for T = 4,000 years (Neolithic divergence) and T = 40,000 years (Paleolithic divergence), we see that the expected intervals defined by using the two different growth rates overlap with the two peaks of the observed distribution. These calculations are approximate, but they offer a plausible interpretation of the bimodal distribution of Fig. 3. A Neolithic separation of most European groups is expected to result in a range of values corresponding to the second peak (Fig. 3, box B) of the distribution. An older separation of groups that have a non-European origin is expected to result in a lower number of alleles in common with most other samples, which is reflected in the first peak of the distribution (Fig. 3, box A). Groups that evolved in isolation have diverged faster, and they tend to contribute more to the latter than to the former peak.
Future Prospects
Where do the genes of the Europeans come from, and when did they come in? We think the best answer is still: Mostly from the Levant, mostly in Neolithic times, but from other places and in other times as well. Colin Renfrew remarked that the model of Neolithic demic diffusion was initially proposed on the basis of a rather generic resemblance between maps of allele frequencies and radiocarbon dates of early farming sites (48). However, many empirical studies (18, 19, 29–33), and computer simulations (21, 22, 29), have now shown that an origin in the Levant and a Neolithic spread are in excellent agreement with the nuclear data. The only alternative seems to imagine that the European gradients were established during the Paleolithic colonization of the continent, and little of significance happened afterward. That cannot be ruled out at present, but it does not seem a probable scenario either.
Mitochondrial data have been regarded as inconsistent with the Neolithic model. In this paper we claim that calculations based on the coalescent theory may reconcile the mitochondrial evidence with the scenario based on the analysis of nuclear polymorphisms. To clarify this issue further, it is important that models other than the Neolithic demic diffusion be better formalized, so that their predictions may be tested better than one can do now. At present, a model interpreting the current gene pool as the result of Mesolithic expansions from glacial refugia does not seem able to explain the extension, and the number, of clines observed at nuclear loci.
If the spread of early farmers has probably determined the main European pattern of genetic diversity, additional patterns are also apparent, and they point to local phenomena that should be further investigated. Specific mutations have a peculiar geographic distribution, which suggests recent local origin of some alleles (49), or input of genes from sources other than the Levant, notably North Africa (49, 50) and Asia (32). Contacts between specific populations, documented in the historical record, have left a recognizable mark in the distribution of genetic distances (51), whereas geographic and linguistic barriers have led to significant local divergence (52). Both in large-scale and in small-scale studies, it will be important to remember that variation at each locus, and at each variable nucleotide site, represents just one realization of an evolutionary process that contains a strong stochastic component. Different genes are expected to show different modes of variation purely by chance, quite aside from the action of selective pressures upon them. Therefore, we think that the best reconstruction of population history is the one that accounts for the variation observed at the genome, not at the single-locus, level.
Acknowledgments
We thank Henry Harpending and Laurent Excoffier for critical reading of this manuscript.
References
- 1.Wolpoff M H, Wu X, Thorne A G. In: The Origins of Modern Humans: A World Survey of the Fossil Evidence. Smith F H, Spencer F, editors. New York: Liss; 1984. pp. 411–483. [Google Scholar]
- 2.Stringer C B, Andrews P. Science. 1988;239:1263–1268. doi: 10.1126/science.3125610. [DOI] [PubMed] [Google Scholar]
- 3.Krings M, Stone A, Schmitz R W, Krainitzki H, Stoneking M, Pääbo S. Cell. 1997;90:19–30. doi: 10.1016/s0092-8674(00)80310-4. [DOI] [PubMed] [Google Scholar]
- 4.Ovchinnikov I V, Götherström A, Romanova G P, Kharitonov V, Lidén K, Goodwin W. Nature (London) 2000;404:490–493. doi: 10.1038/35006625. [DOI] [PubMed] [Google Scholar]
- 5.Lahr M M. J Hum Evol. 1994;26:23–56. [Google Scholar]
- 6.Foley R. Genome Res. 1998;8:339–347. doi: 10.1101/gr.8.4.339. [DOI] [PubMed] [Google Scholar]
- 7.Jorde L B, Watkins W S, Bamshad M J, Dixon M E, Ricker C E, Seielstad M T, Batzer M A. Am J Hum Genet. 2000;66:979–988. doi: 10.1086/302825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hawks J, Hunley K, Lee S H, Wolpoff M. Mol Biol Evol. 2000;17:2–22. doi: 10.1093/oxfordjournals.molbev.a026233. [DOI] [PubMed] [Google Scholar]
- 9.Straus L G. Nature (London) 1989;342:476–477. [Google Scholar]
- 10.Sokal R R. Annu Rev Anthropol. 1991;20:119–140. [Google Scholar]
- 11.von Haeseler A, Sajantila A, Pääbo S. Nat Genet. 1995;14:135–140. doi: 10.1038/ng1096-135. [DOI] [PubMed] [Google Scholar]
- 12.Cooper A, Poinar H N. Science. 2000;289:1139. doi: 10.1126/science.289.5482.1139b. (lett.). [DOI] [PubMed] [Google Scholar]
- 13.Mellars P A. Phil Trans R Soc London B. 1992;337:225–234. doi: 10.1098/rstb.1992.0100. [DOI] [PubMed] [Google Scholar]
- 14.Otte M. In: The World at 18,000 BP. Soffer O, Gamble C, editors. Vol. 1. London: Unwin; 1990. pp. 54–68. [Google Scholar]
- 15.Ammerman A J, Cavalli-Sforza L L. The Neolithic Transition and the Genetics of Populations in Europe. Princeton, NJ: Princeton Univ. Press; 1984. [Google Scholar]
- 16.Renfrew C. Archaeology and Language: The Puzzle of Indo-European Origins. London: Cape; 1987. [Google Scholar]
- 17.Menozzi P, Piazza A, Cavalli-Sforza L L. Science. 1978;201:786–792. doi: 10.1126/science.356262. [DOI] [PubMed] [Google Scholar]
- 18.Sokal R R, Harding R M, Oden N L. Am J Phys Anthropol. 1989;80:267–294. doi: 10.1002/ajpa.1330800302. [DOI] [PubMed] [Google Scholar]
- 19.Sokal R R, Oden N L, Wilson C. Nature (London) 1991;351:143–145. doi: 10.1038/351143a0. [DOI] [PubMed] [Google Scholar]
- 20.Cavalli-Sforza L L, Menozzi P, Piazza A. The History and Geography of Human Genes. Princeton, NJ: Princeton Univ. Press; 1994. [Google Scholar]
- 21.Rendine S, Piazza A, Cavalli-Sforza L L. Am Nat. 1986;128:681–706. [Google Scholar]
- 22.Barbujani G, Sokal R R, Oden N L. Am J Phys Anthropol. 1995;96:109–132. doi: 10.1002/ajpa.1330960202. [DOI] [PubMed] [Google Scholar]
- 23.Richards M, Macaulay V, Bandelt H J, Sykes B. Ann Hum Genet. 1998;62:241–260. doi: 10.1046/j.1469-1809.1998.6230241.x. [DOI] [PubMed] [Google Scholar]
- 24.Sykes B. Phil Trans R Soc London B. 1999;354:131–139. doi: 10.1098/rstb.1999.0366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richards M, Corte-Real H, Forster P, Macaulay V, Wilkinson-Herbots H, Demaine A, Papiha S, Hedges R, Bandelt H J, Sykes B. Am J Hum Genet. 1996;59:185–203. [PMC free article] [PubMed] [Google Scholar]
- 26.Richards M, Macaulay V, Sykes B, Pettit P, Forster P, Hedges R, Bandelt H J. Am J Hum Genet. 1997;61:251–254. doi: 10.1016/S0002-9297(07)64211-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Torroni A, Bandelt H J, D'Urbano L, Lahermo P, Moral P, Sellitto D, Rengo C, Forster P, Savontaus M L, Bonné-Tamir B, et al. Am J Hum Genet. 1998;62:1137–1152. doi: 10.1086/301822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chikhi L, Destro-Bisol G, Pascali V, Baravelli V, Dobosz M, Barbujani G. Hum Biol. 1998;70:643–657. [PubMed] [Google Scholar]
- 29.Chikhi L, Destro-Bisol G, Bertorelle G, Pascali V, Barbujani G. Proc Natl Acad Sci USA. 1998;95:9053–9058. doi: 10.1073/pnas.95.15.9053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Casalotti R, Simoni L, Belledi M, Barbujani G. Proc R Soc London Ser B. 1999;266:1959–1965. [Google Scholar]
- 31.Quintana-Murci L, Semino O, Minch E, Passarino G, Brega A, Santachiara-Benerecetti A S. Eur J Hum Genet. 2000;7:603–608. doi: 10.1038/sj.ejhg.5200347. [DOI] [PubMed] [Google Scholar]
- 32.Rosser, Z. H., Zerjal, T., Hurles, M. E., Adojaan, M., Alavantic, D., Amorim, A., Amos, W., Armenteros, M., Arroyo, E., Barbujani, G., et al. (2000) Am. J. Hum. Genet., in press. [DOI] [PMC free article] [PubMed]
- 33.Simoni L, Calafell F, Pettener D, Bertranpetit J, Barbujani G. Am J Hum Genet. 2000;66:262–278. doi: 10.1086/302706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Donnelly P. In: Variation in the Human Genome, CIBA Foundation Symposium. Weiss K, editor. Vol. 197. Chichester, UK: Wiley; 1996. pp. 25–50. [Google Scholar]
- 35.Tajima F. Genetics. 1983;105:437–460. doi: 10.1093/genetics/105.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barbujani G, Bertorelle G, Chikhi L. Am J Hum Genet. 1998;62:488–491. doi: 10.1086/301719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takahata N, Satta Y, Klein J. Theor Popul Biol. 1995;48:198–221. doi: 10.1006/tpbi.1995.1026. [DOI] [PubMed] [Google Scholar]
- 38.Aris-Brosou S, Excoffier L. Mol Biol Evol. 1996;13:494–504. doi: 10.1093/oxfordjournals.molbev.a025610. [DOI] [PubMed] [Google Scholar]
- 39.Tavaré S. Theor Popul Biol. 1984;26:119–164. doi: 10.1016/0040-5809(84)90027-3. [DOI] [PubMed] [Google Scholar]
- 40.Tremblay M, Vezina H. Am J Hum Genet. 2000;66:651–658. doi: 10.1086/302770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rogers A R, Harpending H. Mol Biol Evol. 1992;9:552–569. doi: 10.1093/oxfordjournals.molbev.a040727. [DOI] [PubMed] [Google Scholar]
- 42.Excoffier L, Schneider S. Proc Natl Acad Sci USA. 1999;96:10597–10602. doi: 10.1073/pnas.96.19.10597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sajantila A, Salem A H, Savolainen P, Bauer K, Gierig C, Pääbo S. Proc Natl Acad Sci USA. 1996;93:12035–12039. doi: 10.1073/pnas.93.21.12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Templeton A R. Am Anthropol. 1993;95:51–72. [Google Scholar]
- 45.Slatkin M, Rannala B. Am J Hum Genet. 1997;60:447–458. [PMC free article] [PubMed] [Google Scholar]
- 46.Pritchard J K, Seielstad M T, Perez-Lezaun A, Feldman M W. Mol Biol Evol. 1999;16:1791–1798. doi: 10.1093/oxfordjournals.molbev.a026091. [DOI] [PubMed] [Google Scholar]
- 47.Slatkin M, Hudson R R. Genetics. 1991;129:555–562. doi: 10.1093/genetics/129.2.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Renfrew C. Cambridge Archaeol J. 2000;10:7–34. [Google Scholar]
- 49.Malaspina P, Cruciani F, Ciminelli B M, Terrenato L, Santolamazza P, Alonso A, Banyko J, Brdicka R, Garcia O, Gaudiano C, et al. Am J Hum Genet. 1998;63:847–860. doi: 10.1086/301999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arnaiz-Villena A, Iliakis P, Gonzalez-Hevilla M, Longàs J, Gomez-Casado E, Sfyridaki K, Trapaga J, Silvera-Redondo C, Matsouka C, Martinez-Laso J. Tissue Antigens. 1999;53:213–226. doi: 10.1034/j.1399-0039.1999.530301.x. [DOI] [PubMed] [Google Scholar]
- 51.Sokal R R, Oden N L, Walker J, Di Giovanni D, Thomson B A. Hum Biol. 1996;68:873–898. [PubMed] [Google Scholar]
- 52.Barbujani G, Sokal R R. Proc Natl Acad Sci USA. 1990;87:1816–1819. doi: 10.1073/pnas.87.5.1816. [DOI] [PMC free article] [PubMed] [Google Scholar]