

Abstract
Aging is an intricate process that increases susceptibility to sarcopenia and cardiovascular diseases. The accumulation of mitochondrial DNA (mtDNA) mutations is believed to contribute to mitochondrial dysfunction, potentially shortening lifespan. The mtDNA mutator mouse, a mouse model with a proofreading-deficient mtDNA polymerase γ, was shown to develop a premature aging phenotype, including sarcopenia, cardiomyopathy and decreased lifespan. This phenotype was associated with an accumulation of mtDNA mutations and mitochondrial dysfunction. We found that increased expression of peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α), a crucial regulator of mitochondrial biogenesis and function, in the muscle of mutator mice increased mitochondrial biogenesis and function and also improved the skeletal muscle and heart phenotypes of the mice. Deep sequencing analysis of their mtDNA showed that the increased mitochondrial biogenesis did not reduce the accumulation of mtDNA mutations but rather caused a small increase. These results indicate that increased muscle PGC-1α expression is able to improve some premature aging phenotypes in the mutator mice without reverting the accumulation of mtDNA mutations.
INTRODUCTION
Aging is the progressive decline in cellular, tissue and organ function (1–3). This complex process often manifests as loss of muscular strength and cardiovascular function (2). The mitochondrial theory of aging suggests that the accumulation of mitochondrial DNA (mtDNA) mutations leads to mitochondrial dysfunction, loss of organ function and consequently a decrease in lifespan (4,5). This theory is appealing as there is a correlation between age-dependent alterations in mtDNA and an increased risk for developing cardiovascular diseases, neurodegenerative disorders and myopathy (1,6–8). Accordingly, there is an association between the accumulation of mtDNA point mutations and deficiencies in mitochondrial oxidative phosphorylation system (OXPHOS) in aging muscle fibers and hippocampal neurons (9).
The mtDNA mutator mouse, a mouse model with a proofreading-deficient mtDNA polymerase γ (POLG), is a valuable model system to study the contribution of mitochondrial dysfunction to aging (10–12). Animals homozygous for this mutant POLG (mtDNA mutator mice, designated Mut mice) age prematurely, have reduced lifespan and show increased accumulation of mtDNA point mutations (11,12). Reminiscent of normal human aging, Mut mice develop cardiomyopathy and sarcopenia (loss of skeletal muscle mass), which is associated with mitochondrial dysfunction in the heart and skeletal muscle, respectively (11–13).
Peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) is a master regulator of mitochondrial biogenesis. Overexpression of PGC-1α in the mouse skeletal muscle and heart has been shown to increase mitochondrial biogenesis and function (14,15). In addition, PGC-1α induces fiber-type switch from glycolytic to oxidative fibers (16), angiogenesis (17) and retards protein degradation and atrophy in the skeletal muscle (18,19). Furthermore, PGC-1α has been shown to positively regulate cardiac function (20). Since Mut mice have mitochondrial dysfunction, sarcopenia and cardiomyopathy and PGC-1α has been shown to increase mitochondrial biogenesis and function, we tested whether increased PGC-1α expression could compensate for the mitochondrial dysfunction in Mut mice and improve their aging phenotype. Here, we show that Mut mice with increased expression of PGC-1α under the muscle creatine kinase (MCK) promoter have increased mitochondrial biogenesis and function and improved skeletal muscle and heart function.
RESULTS
MCKPGC-1αMut mice have increased PGC-1α in the skeletal muscle
We created Mut mice transgenically expressing PGC-1α under the MCK promoter (heretofore referred to as MCKPGC-1αMut mice). In MCKPGC-1αMut mice, PGC-1α overexpression in the skeletal and cardiac muscle begins at birth.
We allowed MCKPGC-1αMut mice to age until they were 10-month-old, the age when most of the phenotypes reported in the Mut mice become evident (11,12). To ensure that 10-month-old MCKPGC-1αMut mice had increased PGC-1α levels in the skeletal muscle, RNA and total homogenates were prepared from the quadricep of male MCKPGC-1αMut, MCKPGC-1αWT (wild-type mice transgenically expressing PGC-1α under the MCK promoter), Mut and WT mice and used for quantitative reverse transcriptase–Polymerase chain reaction (PCR) and western blot analysis, respectively. We found that 10-month-old MCKPGC-1αMut mice had a 4-fold increase in PGC-1α mRNA levels compared with Mut and our positive controls, MCKPGC-1αWT mice had a 20-fold increase inPGC-1α mRNA compared with WT mice (Fig. 1A). MCKPGC-1αMut mice also had a 2-fold increase in PGC-1α protein in the quadricep compared with Mut, and MCKPGC-1αWT mice had a 3-fold increase compared with WT mice (Fig. 1B and C). These results indicate that 10-month-old MCKPGC-1αMut mice have increased PGC-1α in the skeletal muscle; however, the levels are lower than that of MCKPGC-1αWT mice. Unless otherwise stated, we performed all our experiments with 10-month-old male MCKPGC-1αMut mice and age-matched controls.
Figure 1.

MCKPGC-1αMut mice have increased PGC-1α levels in the skeletal muscle. (A) Gene expression of PGC-1α relative to WT in the quadriceps of 10-month-old male mice. mRNA levels are normalized to GAPDH. (B) Western blot showing PGC-1α protein levels in the quadriceps of 10-month-old male mice with loading control actin. (C) Quantification of western blot in (B) showing PGC-1α protein levels normalized to actin. n = 4/group; Student's t-test: *P < 0.05 and ***P < 0.001. Error bars represent the SEM.
Increased PGC-1α expression increases mitochondrial biogenesis and improves mitochondrial function in the skeletal muscle of Mut mice
To assess changes in mitochondrial biogenesis, we measured mitochondrial protein levels and mtDNA levels in the quadricep of MCKPGC-1αMut mice and controls. We found that MCKPGC-1αMut mice had increased levels of several subunits of the mitochondrial OXPHOS (Fig. 2A and B) and increased mtDNA levels in the quadriceps compared with Mut mice (Fig. 2G). MCKPGC-1αMut mice also had increased levels of these mitochondrial proteins in the gastrocnemius compared with Mut mice (Supplementary Material, Fig. S1A and B).
Figure 2.

Increased PGC-1α expression increases mitochondrial biogenesis and function in the skeletal muscle of Mut mice. (A) Western blot showing the levels of mitochondrial proteins in the quadriceps of mice and loading control actin. ATP synthase subunit 5α (ATP5A; subunit of complex V), ubiquinol-cytochrome c reductase core protein 2 (UQCRC2; subunit of complex III), mitochondrial cytochrome c oxidase subunit 1 (MTCO1; subunit of complex IV), succinate dehydrogenase subunit A and B, or SDHA and SDHB (subunits of complex II) and NADH dehydrogenase (ubiquinone) 1β subcomplex subunit 8 (NDUFB8; subunit of complex I; n = 4/group). (B) Quantification of western blot in (A) showing protein levels normalized to actin. (C) Histology of the quadriceps showing COX (complex IV) and SDH (complex II) activity staining (n = 3/group). (D) Complex I and III activity, (E) complex IV activity and (F) CS activity in total quadricep homogenate (n = 4/group). (G) MtDNA levels relative to WT in the quadriceps based on the ND1 copy number (subunit of complex I) normalized to GAPDH (n = 4/group). Analyzed 10-month-old male mice. Student's t-test: *P< 0.05, **P< 0.01 and ***P< 0.001. Error bars represent the SEM.
To determine if this increase in mitochondrial biogenesis increased mitochondrial function in the quadricep of MCKPGC-1αMut mice, we measured the activity of mitochondrial electron transport chain (ETC) and citric acid cycle-associated enzymes. Frozen transverse sections were prepared from the quadricep of the mice and activity stainings were performed to detect the activity of cytochrome c oxidase (COX), complex IV and succinate dehydrogenase (SDH), complex II of the mitochondrial ETC. We found that MCKPGC-1αMut mice had more fibers positively stained for COX activity (dark brown stain) and SDH activity (blue stain) compared with Mut mice (Fig. 2C). These findings indicate that PGC-1α overexpression increased mitochondrial function in the quadriceps of Mut mice. Total homogenate from quadriceps also showed an increased activity of complexes I and III (Fig. 2D), complex IV (Fig. 2E) and citrate synthase (CS; a marker of mitochondrial mass, Fig. 2F) measured spectrophotometrically. These results indicate that, as expected, MCKPGC-1αMut mice have increased mitochondrial biogenesis and function in the skeletal muscle.
Increased PGC-1α expression induces fiber-type switch and improves skeletal muscle function of Mut mice
It was previously shown that Mut mice develop decreased quadricep and gastrocnemius weight (12). Therefore, we compared the quadricep and gastrocnemius weight of MCKPGC-1αMut to Mut mice to test if increased PGC-1α expression could protect the mice from loss of skeletal muscle mass. Surprisingly, we found that increased PGC-1α expression had no effect on the skeletal muscle weight of the mice (Fig. 3A). However, Mut mice fell more often when running on a treadmill than MCKPGC-1αMut mice (Fig. 3B). This was observed not only at 3 and 5 months, but also at 9–10 months of age when Mut mice develop sarcopenia. This suggests that the increased expression of PGC-1α in the skeletal muscle of the Mut mice is able to improve mitochondrial function, boost endurance and skeletal muscle function.
Figure 3.

Increased PGC-1α expression has no effect on skeletal muscle weight but improves skeletal muscle function of Mut mice. (A) Weight of quadricep and gastrocnemius of 10-month-old male mice (n = 4–5/group). Student's t-test: *P< 0.05 and **P< 0.01. Error bars represent the SEM. (B) The number of falls of mice when put to run on a treadmill for 3 min at 9 m/min (n = 5–10/group). *P< 0.05 represents the difference between Mut and MCKPGC-1αMut; ##P< 0.01 and ###P< 0.001 represents the difference between WT and Mut; ++P< 0.01 and +++P< 0.001 represents the difference between MCKPGC-1αWT and Mut; φP< 0.05 represents the difference between WT and MCKPGC-1αMut. Difference between MCKPGC-1αWT and MCKPGC-1αMut is significant at the 9-month time point only and the difference between WT and MCKPGC-1αWT is not significant at any time point. Statistics represent two-way ANOVA followed by the Bonferroni post-tests. Error bars represent the SEM.
PGC-1α has been shown to regulate the conversion of muscle fibers from glycolytic (type IIB) to oxidative (type I and IIA) fiber types (16). Oxidative muscle fibers are characterized by high mitochondrial density and resistance to fatigue (21). Therefore, we sought to determine if the improved treadmill performance of MCKPGC-1αMut mice was associated with the formation of more oxidative fiber types. We performed immunohistochemistry for myosin heavy chain (MHC) I and MHC IIA (markers of type I and IIA oxidative fibers, respectively) in frozen sections from the quadriceps of 10-month-old male mice. We found that MCKPGC-1αMut mice had more type I and IIA positive skeletal muscle fibers than Mut mice (Supplementary Material, Fig. S2), which explains their improved treadmill performance despite no increase in their skeletal muscle weight.
Increased PGC-1α expression improves the heart function of Mut mice
We found that similar to WT controls, MCKPGC-1αMut mice showed a trend toward increased PGC-1α mRNA levels in the heart compared with Mut mice (Fig. 4A). In addition, they showed a trend toward increased heart mtDNA levels (Fig. 4D). To determine the effect of this mild increase in PGC-1α expression and mtDNA levels on mitochondrial protein levels in the heart, we performed western blot to detect the levels of subunits of mitochondrial OXPHOS enzymes. We found that both MCKPGC-1αMut and MCKPGC-1αWT mice had no detectable increase in the levels of mitochondrial markers in the heart, which suggests that there is no increase in heart mitochondrial biogenesis (Supplementary Material, Fig. S3A and B). Next, we measured the activity of COX and CS to determine if the function of heart mitochondria was improved. Our results showed that MCKPGC-1αMut mice had increased COX activity in total heart homogenate (Fig. 4C) and frozen transverse sections from the heart (Supplementary Material, Fig. S3C) when compared with Mut mice. However, they had no increase in heart CS activity (Fig. 4B). These results indicate that mild PGC-1α overexpression in the myocardium of MCKPGC-1αMut mice along with systemic effects of increased PGC-1α expression in the skeletal muscle improved heart mitochondrial function without causing a major increase in mitochondrial biogenesis.
Figure 4.
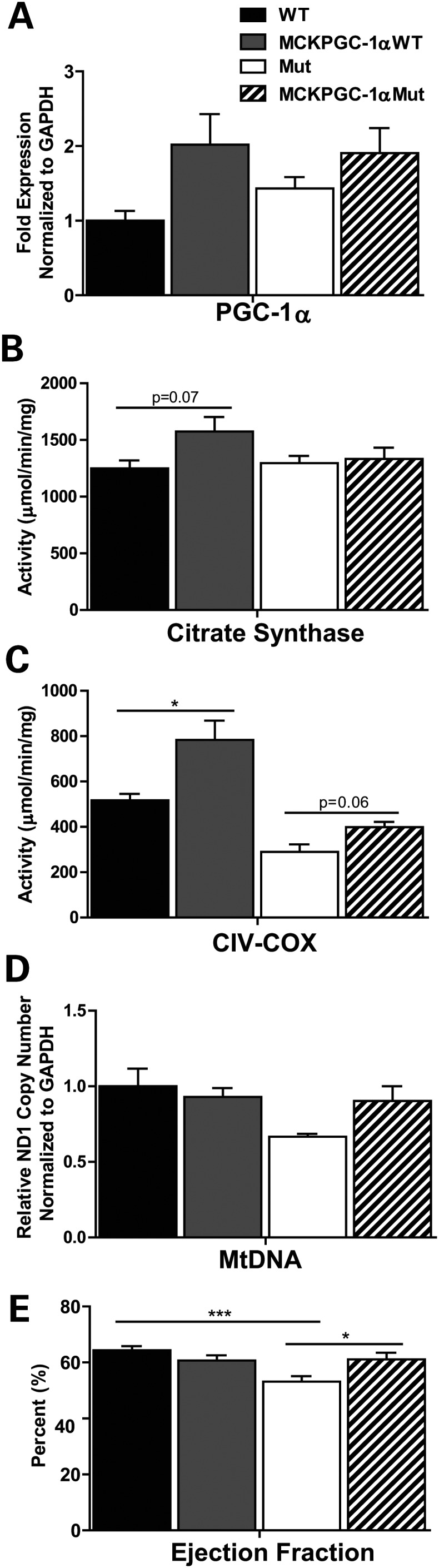
Increased PGC-1α expression stabilizes mtDNA levels and improves mitochondrial function and ejection fraction in the heart of Mut mice. (A) Gene expression of PGC-1α in the heart relative to WT (n = 4/group). mRNA levels are normalized to GAPDH. (B) CS activity and (C) COX (complex IV) activity in total heart homogenate (n = 4/group). (D) MtDNA levels in the heart relative to WT based on the ND1 copy number (subunit of complex I) normalized to GAPDH (n = 4/group). We analyzed 10-month-old male mice. (E) Percent heart ejection fraction based on echocardiogram of the mouse heart (n = 11–12, 10-month-old male and female mice per group). Student's t-test: *P< 0.05 and ***P< 0.001. Error bars represent the SEM.
We next examined if improved mitochondrial function in MCKPGC-1αMut mice had any effect on overall cardiac function. To measure heart function, we performed echocardiograms on 10-month-old male and female mice and determined ejection fraction as a quantitative measure of overall cardiac function. We found that compared with WT mice, Mut mice had a decrease in ejection fraction (Fig. 4E); however, this was restored in MCKPGC-1αMut mice to levels comparable with WT animals (Fig. 4E). Interestingly, MCKPGC-1αMut mice had no change in heart size and ventricle lumen diameter compared with Mut mice (not shown).
Many studies have linked cardiac fibrosis, increase in heart collagen content, to heart aging (22). Therefore, we asked if the improved heart function in MCKPGC-1αMut mice was associated with a reduction in heart collagen levels. We performed immunohistochemistry to detect collagen I in frozen sections from the heart of 10-month-old male mice. Surprisingly, we found that collagen I was reduced along muscle fibers and around blood vessels in the heart of Mut mice and was restored in MCKPGC-1αMut mice (Supplementary Material, Fig. S4). Since collagen I is needed for proper ventricular diastolic function in the heart (23), its increase likely contributes to improved heart ejection fraction in MCKPGC-1αMut mice. It is possible that increased mitochondrial function or systemic effects resulting from increased PGC-1α expression in the skeletal muscle affects heart collagen I levels.
Increased PGC-1α expression increases the abundance of mtDNA point mutations in the skeletal muscle of Mut mice
The premature aging phenotype of Mut mice is believed to be caused by the accumulation of mtDNA mutations resulting from their error-prone POLG (11,24). Therefore, we determined the effect of increased PGC-1α expression on the abundance of mtDNA point mutations in the skeletal muscle of Mut mice. Total DNA was prepared from the quadricep of the mice and we amplified two amplicons spanning the entire mtDNA. We then sequenced this mtDNA using next generation sequencing (NGS) to determine sequence variations in MCKPGC-1αMut mice compared with Mut, MCKPGC-1αWT and WT mice.
As previously shown (25), we found that mice with a Mut background had more mtDNA sequence variations than WT mice (Fig. 5A). NGS demonstrated that the increased levels of point mutations were evenly distributed throughout the whole mitochondrial genome of the Mut mouse. An interesting exception was the displacement loop (D-loop), which showed a cold spot for point mutations (position ∼16 000 in Figure 5A) (26). Mut and MCKPGC-1αMut mice had similar overall abundance of mtDNA point mutations (Fig. 5B). Further analysis showed that all the mice tested (WT, MCKPGC-1αWT, Mut and MCKPGC-1αMut mice) shared similar abundance of their most frequent mtDNA point mutations. Highly abundant point mutations (ranked as #1 to #100 in abundance; 1–40% heteroplasmy) are likely inherited mutations (Fig. 5C), as these mice were littermates born to parents heterozygous for the D257A mutation in POLG, which have increased levels of mtDNA mutations in their germ line (27). Interestingly, WT and MCKPGC-1αWT had much lower levels of the less abundant mtDNA point mutations (ranked as #100–#10 000 in abundance) compared with Mut and MCKPGC-1αMut mice (Fig. 5C). These rare mutations likely represent somatic mutations accumulated during life. In Mut mice, they are caused by the error-prone POLG function in muscle mtDNA. Although individually these mutations are less abundant, there are many more of them, explaining the increased total load of mutations in the Mut mice (Fig. 5A and B). When we took a closer look at low-frequency mutations (ranked from #100 to #10 000), we found that MCKPGC-1αMut mice had a higher average abundance of these mutations compared with mutator mice (Fig. 5D). These results indicate that increased PGC-1α expression increases the abundance of somatically generated mtDNA point mutations in the skeletal muscle of Mut mice. This increase is likely a result of increases in mtDNA replication and mitochondrial biogenesis orchestrated in the skeletal muscle by PGC-1α.
Figure 5.
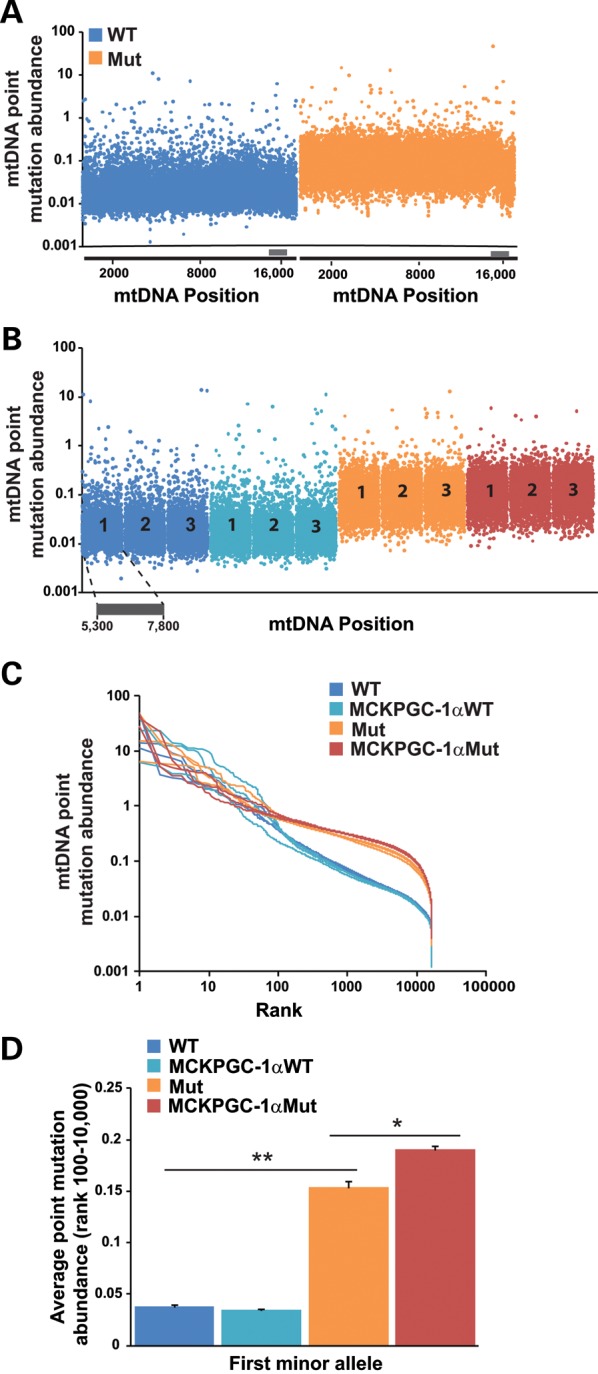
Increased PGC-1α expression increases the average abundance of somatic mtDNA point mutations in the skeletal muscle of Mut mice. (A) Representative plot showing the first minor allele frequency of variants throughout the entire mtDNA from the quadricep of WT and Mut mice. (B) Data as in (A) for variants between positions 5300 and 7800 of the mtDNA [which spans MTCO1, tRNAs1 (Trns1), tRNAd (Trnd), MTCO2 and tRNAk (Trnk) genes] for three different mice (numbered 1–3) for each group. (C) Graph showing abundance versus rank abundance for first minor allele variants in the quadriceps. (D) Average abundance of rare (rank #100–#10 000) variants in the quadriceps. n = 3/group 10-month-old male mice. Student's t-test: *P< 0.05 and **P< 0.01. Error bars represent the SEM.
Increased PGC-1α expression does not increase control region multimers in the heart of Mut mice
The mtDNA control region, which includes the D-loop, regulates mtDNA replication and transcription (28). We recently showed that Mut mice have a signature genetic abnormality, namely, high levels of control region multimers (CRMs), in heart and brain mtDNA (25). CRMs contain novel recombination sites that allow them to be detected and quantified by PCR. We were able to amplify CRMs in the heart of Mut and MCKPGC-1αMut mice but not in WT mice (Supplementary Material, Fig. S5A). Quantitative PCR showed that increased PGC-1α expression in the heart had no effect on the relative CRM levels in MCKPGC-1αMut mice compared with Mut mice (Supplementary Material, Fig. S5B). These results indicate that increased PGC-1α levels in the heart of Mut mice did not affect mtDNA structure in this tissue. They also suggest that CRMs do not contribute to the heart phenotype of the Mut mice.
Increased PGC-1α expression has mild systemic effects on Mut mice
We also assessed the effect of muscle PGC-1α overexpression on non-muscle phenotypes in the Mut mouse. We found that MCKPGC-1αMut mice and Mut mice had very similar appearance (not shown) and weights at each age (Supplementary Material, Fig. S6A). In addition, we found no change in bone mineral density and content (Supplementary Material, Fig. S6D and E), body area, lean mass, total fat and body fat percentage of 10-month-old male MCKPGC-1αMut and Mut mice (Supplementary Material, Fig. S6F). This indicates that overexpression of PGC-1α in the skeletal muscle of Mut mice did not have a major influence on these parameters.
We also compared the blood cell count and blood chemistry of the mice at 10 months to search for systemic effects. Our results showed that there was no change in liver or kidney function markers (not shown). Although there was no change in blood cell count in MCKPGC-1αMut compared with Mut mice (Supplementary Material, Fig. S6B), MCKPGC-1αMut mice had decreased mean corpuscular volume (MCV) and mean corpuscular hemoglobin (MCH) compared with Mut mice (Supplementary Material, Fig. S6C). Because MCV and MCH are increased in Mut mice due to severe anemia, these findings suggest that increased muscle PGC-1α expression ameliorates these blood parameters in Mut mice.
We also compared the survival of MCKPGC-1αMut mice with Mut mice. We found that while MCKPGC-1αMut mice did not live longer than Mut mice, they started dying 6 weeks after Mut mice (Supplementary Material, Fig. S6G). This indicates that increased PGC-1α levels in the skeletal muscle and heart may delay onset of death in the MCKPGC-1αMut group of mice.
DISCUSSION
Phenotypic changes triggered by increased muscle PGC-1α in the Mut mouse
Recent studies have highlighted an association between decreased mitochondrial biogenesis and function, decreased PGC-1α expression and aging (29–31). Consequently, methods of improving mitochondrial biogenesis and function have become topics of interest. We previously showed that in a mouse model with mitochondrial muscle myopathy, increased expression of PGC-1α leads to an increase in mitochondrial mass, respiration and ATP production (14). We also showed that increased expression of PGC-1α in the skeletal muscle of aging WT mice protects from mitochondrial dysfunction and sarcopenia associated with normal aging (32). In this study, we tested whether increased PGC-1α expression in the Mut mice could compensate for mitochondrial dysfunction and confer protection against premature aging phenotypes.
We found that although MCKPGC-1αMut mice had increased PGC-1α levels in the skeletal muscle, this increase was blunted when compared with their WT counterparts. We do not have an explanation for this difference, but it may be related to feedback mechanisms controlling PGC-1α stability in different bioenergetic states. In any case, the increase in mitochondrial biogenesis and associated increase in oxidative skeletal muscle fibers in the Mut muscle was sufficient to promote protection against some of the aging phenotypes.
Similar increases in mtDNA levels, mitochondrial protein levels and COX activity were observed in the skeletal muscle of endurance trained Mut mice (33). This is not surprising as it is known that exercise induces PGC-1α expression in skeletal muscle and PGC-1α has been shown to regulate the adaptation of skeletal muscle to exercise (34,35). However, while endurance exercise rescued the decreased quadriceps and gastrocnemius weight in the Mut mice (33), we found that increased PGC-1α expression had no effect on skeletal muscle weight. The increase in skeletal muscle weight observed in endurance exercised Mut mice may result from effects of exercise that are parallel to the PGC-1α pathway.
Furthermore, we found that MCKPGC-1αMut mice trended to have increased COX activity and mtDNA levels in the heart despite having only a small increase in PGC-1α expression in this tissue. This small increase may be optimal since high levels of PGC-1α in the heart can lead to cardiomyopathy (36). We also observed that MCKPGC-1αMut mice had increased percent heart ejection fraction, suggesting that increased PGC-1α expression and mitochondrial function in the heart improved heart function. This finding supports the role of PGC-1α in regulating heart energy metabolism and function (20). Besides its role in increasing mitochondrial biogenesis, PGC-1α can also increase the levels of specific factors that improve mitochondrial activity, anti-oxidant function and microvascularity (37). Moreover, increased PGC-1α expression in the muscle can lead to the release of myokines that can impact heart function (38).
Despite having improvements in the skeletal muscle and heart, MCKPGC-1αMut mice did not display changes in physical appearance, body weight, bone mineral density or fat/lean mass that was observed in endurance exercised Mut mice (33). However, similar to endurance exercised Mut mice, MCKPGC-1αMut mice had decreased MCV and MCH, suggesting that increased PGC-1α expression improved some blood parameters in the Mut mice (33). Although we did not observe an extension in the lifespan of MCKPGC-1αMut mice as was observed in exercised Mut mice (33), the increased longevity in that study was determined by analyzing very few animals, and confirmation in larger groups would be required. In any case, the overall health of MCKPGC-1αMut mice appeared improved, as the early deaths in this group were observed 6 weeks after the early deaths in the Mut mice.
Therefore, increased expression of PGC-1α in muscle conferred protection mostly to the muscle and heart, likely by increasing the mitochondrial pool, which minimizes the partial defects associated with increased levels of mtDNA mutations. There were also improvements in some blood parameters, which may be associated with the release of myokines (38).
Genotypic changes triggered by increased muscle PGC-1α in the Mut mouse
When we analyzed mtDNA point mutations in the skeletal muscle, we found that increased PGC-1α expression in Mut mice did not decrease the abundance of somatic point mutations, but instead led to a small but significant increase. This finding suggests that increased PGC-1α expression induces mtDNA replication in Mut mice, leading not only to increased mtDNA levels but further accumulation of mtDNA point mutations with each round of replication, since these mice have a proofreading-deficient POLG. In contrast, endurance exercised Mut mice were reported to have decreased frequency of mtDNA point mutations in the skeletal muscle (33). We do not have an explanation for this difference, but they may be due to either different methodology or to systemic effects of exercise that possibly mediate an increase in mtDNA repair or removal of dysfunctional mitochondria in the skeletal muscle of the Mut mice. It has been shown that skeletal muscle of the Mut mice have a lower abundance of mtDNA point mutations in the D-loop compared with the rest of the mtDNA (26). This pattern was preserved in MCKPGC-1αMut mice. The reason for the existence of this ‘cold spot’ remains unexplained.
We previously showed that CRMs are present in the heart of Mut and not WT mice and can be used as markers of mtDNA damage (25). Here, we observed that MCKPGC-1αMut mice had similar levels of CRMs in the heart as Mut mice. This finding suggests that increased PGC-1α expression did not increase mtDNA damage in the heart, although we observed an increase in mtDNA levels.
Our findings highlight the potential of PGC-1α in circumventing mitochondrial dysfunction in aging and age-related conditions. We showed that increased PGC-1α expression under the MCK promoter increases mitochondrial biogenesis and function and improves skeletal muscle and heart function of Mut mice despite not improving their mtDNA mutation load.
MATERIALS AND METHODS
Generation of MCKPGC-1αMut mice
Transgenic mice expressing PGC-1α driven by the muscle/heart-specific MCK promoter (MCKPGC-1α) were described previously (16). MtDNA mutator mice (PolgD257A/D257A) were characterized previously (11,12). We crossed MCKPGC-1α+/− with PolgD257A/+ mice to obtain MCKPGC-1α+/− PolgD257A/+ mice. The MCKPGC-1α+/− PolgD257A/+ mice were then crossed with PolgD257A/+ to obtain MCKPGC-1αMut. The resulting littermates of WT, MCKPGC-1α+/− in a WT background (MCKPGC-1αWT) and PolgD257A/D257A (Mut) were used as controls.
Animal husbandry
Mice were housed in a virus-antigen-free facility of the University Of Miami Division Of Veterinary Resources in a 12-h light/dark cycle at 22°C and fed ad libitum with the irradiated standard mouse diet.
Quantitative PCR
Total RNA was isolated from the snap-frozen quadriceps and heart using the RNeasy Fibrous Tissue Mini kit (Qiagen). cDNA was synthesized from 1 μg of RNA using the Superscript III reverse transcriptase kit (Invitrogen). Quantitative real-time PCR, with specific primers for PGC-1α (5′-CTGCGGGATGATGGAGACA and 5′-AGCAGCGAAAGCGTCACA) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 5′-GCAGTGGCAAAGTGGAGATT and 5′-GAATTTGCCGTGAGTGGAGT), was performed using Maxima SYBR Green/ROX PCR Master Mix (Fermentas).
Total DNA was extracted from the frozen quadriceps and heart with phenol:chloroform and mtDNA copy number was quantified using NADH dehydrogenase subunit 1 (ND1; 5′-CAGCCTGACCCATAGCCATA and 5′-ATTCTCCTTCTGTCAGGTCGAA) and GAPDH primers. MtDNA D-loop (control region) levels were quantified using control region primers (CRMF, CCCCTTCCCCATTTGGTCTATT; CRMB, TTGATGGCCCTGAAGTAAGAACC) and COX1 primers (CO1F, AGGCTTCACCCTAGATGACACA; CO1B, GTAGCGTCGTGGTATTCCTGAA). The ΔΔCt method was used to analyze the abundance of PGC-1α, mtDNA and D-loop.
Polymerase chain reaction
For the detection of CRMs, total DNA was isolated from the frozen heart by phenol:chloroform extraction. CRMs were amplified with Expand Long Template PCR kit (Roche) using primers 15720F (CACCAATGCCCCTCTTCTCG) and 16022B (TTGGGTTTTGCGGACTAATGAT). Reaction was performed with Buffer 2 using an extension time of 2 min.
Western blot
Western blot analysis was performed as described previously (39). Primary antibodies used were PGC-1α (Santa Cruz H-300), succinate dehydrogenase subunit A (SDHA; Mitosciences), Total OXPHOS Rodent Cocktail (Mitosciences) and Actin (Sigma). All primary antibodies were used at 1:1000 dilution and incubated overnight at 4°C. Secondary antibodies used were infrared-conjugated anti-rabbit 700 (1:3000) and anti-mouse 800 (1:5000) (Rockland). Blots were visualized with Odyssey Infrared Imaging System (LI-COR Biosciences) and band intensity/optical density was quantified with default software supplied by LI-COR.
Histochemistry and immunohistochemistry
Deeply anesthetized mice were perfused with cold phosphate-buffered saline (PBS), and the quadriceps and heart were removed and immediately frozen in isopentane cooled in liquid nitrogen. For histochemistry, triplicate 10-µm thick cross-sections of these frozen tissues were stained for SDH and COX activities as described previously (40). The stained sections were analyzed using a light microscope. For immunohistochemistry, frozen sections were blocked in PBS 5% bovine serum albumin for 30 min. For heart sections, primary antibody for collagen I (1:200; Abcam) was incubated overnight at 4°C and wheat germ agglutinin conjugated to Texas Red-X (1:100; Molecular Probes) was added for 10 min. Secondary antibody conjugated to Alexa 488 (1:400; Molecular Probes) was added for 1 h in the dark. Sections were mounted with VECTASHIELD mounting medium with 4′,6-diamidino-2-phenylindole (Vector Laboratories) and then analyzed with an LSM710 confocal microscope (Zeiss). Alexa 488 intensity was quantified using the default confocal microscope software (Zeiss). For fiber typing in the quadriceps, primary antibodies recognizing MHC IIA (SC-71-c) and MHC I (A4.951-c) (DSHB) were incubated overnight at 4°C. Secondary antibody conjugated to Alexa 488 (1:400; Molecular Probes) was added and incubated for 2 h in the dark. Sections were analyzed with a fluorescent microscope.
Spectrophotometric assays
The activity of complex IV and CS was determined spectrophotometrically in total homogenate from the quadriceps and heart of mice as described previously (41). The activity of complexes I and III was determined spectrophotometrically in total homogenate from the quadriceps of mice as described previously (42). However, complex I reaction was started with coenzyme Q1 and inhibited with rotenone, and complex III reaction was started with coenzyme Q2 and inhibited with stigmatellin. Assay results were normalized to protein concentration obtained by the Bradford method.
Treadmill test
Mice were acclimated to the treadmill (Columbus Instruments, Columbus, OH, USA) prior to testing; they were put to run for ∼1–2 min until they knew how to perform the test. For the test, mice were put to run on the treadmill at 9 m/min for 3 min. Mice performance was measured by the number of times a mouse falls off the running belt onto the grid during the test.
Echocardiogram
Transthoracic echocardiography was performed on 10-month-old mice anesthetized with 1% isoflurane as described previously (43). The VisualSonics 770 system (Toronto, Canada) was used for this procedure and percent ejection fraction was calculated from the obtained images.
Next generation sequencing
Sequencing libraries were prepared from two amplicons covering the entire mtDNA using the Kapa high fidelity PCR system. Fragments were amplified using 5′-blocked (C6-TFA) primers 12728F (CTGTACCCACGCATTCTTCA), 4200B (GGATAGGCCTATTAATGTTATGT), 4075F (AGCAGCAACAAAATACTTCGTCACAC) and 12886B (GTGAGGGCGAGGCTTCCGATTAC). Products were cleaned using the Sigma Genelute PCR cleanup kit, quantified with an Invitrogen Qubit fluorometric quantitation system and mixed in equimolar amounts. Sequencing libraries were prepared using Illumina TruSeq sample prep and two times 100 bp paired-end sequencing was done on an Illumina HiSeq 2000 system. Read files were created using CASAVA 1.8 without duplicate removal. CLCBio 4.7.2 was used for assembly and variant detection. Reads were imported and quality trimmed at default settings with a length cutoff of 60 bp to produce working files which were assembled with a cutoff of 0.9 similarities and 0.95 lengths with paired-end distance optimized for each sample. Variants were detected using the CLCBio single-nucleotide polymorphism (SNP) detection algorithm with default quality scoring and no significance scoring. Thus, values reported are sum variance (biological and technical) at each position and differences in mutation frequency between WT and other mice are assumed to reflect differences in actual mutation relative to WT variance. SNP tables were analyzed and graphics produced in MS Excel. Only the first minor allele frequencies were considered.
Blood analysis
Mice were deprived of food overnight and the following day anesthetized and blood was collected by cardiac puncture. Blood was sent to the University of Miami Comparative Pathology Laboratory for complete blood cell count and liver/kidney panel analysis.
DEXA scan
The body composition of mice was determined as described previously (44). In brief, mice were anesthetized and weighed and bone mineral density and content along with fat and lean body mass was measured using a Lunar PIXImus II Densitometer (GE Medical Systems, Waukesha, WI, USA).
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the National Institutes of Health Grant EY010804, AG036871 the Muscular Dystrophy Association and the James & Esther King Biomedical Research Program (C.T.M.).
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Ms Sofia Garcia for assistance with various aspects of the mouse experiments, Dr Teresa Zimmers for fiber typing antibodies, Dr Wayne Balkan for access to the DEXA scanner, Dr Bruce M. Spiegelman (Dana-Farber Cancer Institute) for the MCKPGC-1α mouse and the University of Miami Comparative Pathology Laboratory for blood work analysis.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Figueiredo P.A., Mota M.P., Appell H.J., Duarte J.A. The role of mitochondria in aging of skeletal muscle. Biogerontology. 2008;9:67–84. doi: 10.1007/s10522-007-9121-7. doi:10.1007/s10522-007-9121-7. [DOI] [PubMed] [Google Scholar]
- 2.Meissner C., Bruse P., Oehmichen M. Tissue-specific deletion patterns of the mitochondrial genome with advancing age. Exp. Gerontol. 2006;41:518–524. doi: 10.1016/j.exger.2006.03.010. doi:10.1016/j.exger.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 3.Meissner C. Mutations of mitochondrial DNA—cause or consequence of the ageing process? Z. Gerontol. Geriatr. 2007;40:325–333. doi: 10.1007/s00391-007-0481-z. doi:10.1007/s00391-007-0481-z. [DOI] [PubMed] [Google Scholar]
- 4.Taylor R.W., Turnbull D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005;6:389–402. doi: 10.1038/nrg1606. doi:10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loeb L.A., Wallace D.C., Martin G.M. The mitochondrial theory of aging and its relationship to reactive oxygen species damage and somatic mtDNA mutations. Proc. Natl Acad. Sci. USA. 2005;102:18769–18770. doi: 10.1073/pnas.0509776102. doi:10.1073/pnas.0509776102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fukui H., Moraes C.T. The mitochondrial impairment, oxidative stress and neurodegeneration connection: reality or just an attractive hypothesis? Trends Neurosci. 2008;31:251–256. doi: 10.1016/j.tins.2008.02.008. doi:10.1016/j.tins.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Polisecki E.Y., Schreier L.E., Ravioli J., Corach D. Common mitochondrial DNA deletion associated with sudden natural death in adults. J. Forensic. Sci. 2004;49:1335–1338. [PubMed] [Google Scholar]
- 8.Mohamed S.A., Hanke T., Erasmi A.W., Bechtel M.J., Scharfschwerdt M., Meissner C., Sievers H.H., Gosslau A. Mitochondrial DNA deletions and the aging heart. Exp. Gerontol. 2006;41:508–517. doi: 10.1016/j.exger.2006.03.014. doi:10.1016/j.exger.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 9.Trifunovic A. Mitochondrial DNA and ageing. Biochim. Biophys. Acta. 2006;1757:611–617. doi: 10.1016/j.bbabio.2006.03.003. doi:10.1016/j.bbabio.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 10.Wiesner R.J., Zsurka G., Kunz W.S. Mitochondrial DNA damage and the aging process: facts and imaginations. Free Radic. Res. 2006;40:1284–1294. doi: 10.1080/10715760600913168. doi:10.1080/10715760600913168. [DOI] [PubMed] [Google Scholar]
- 11.Trifunovic A., Wredenberg A., Falkenberg M., Spelbrink J.N., Rovio A.T., Bruder C.E., Bohlooly Y.M., Gidlof S., Oldfors A., Wibom R., et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. doi:10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 12.Kujoth G.C., Hiona A., Pugh T.D., Someya S., Panzer K., Wohlgemuth S.E., Hofer T., Seo A.Y., Sullivan R., Jobling W.A., et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. doi:10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 13.Hiona A., Sanz A., Kujoth G.C., Pamplona R., Seo A.Y., Hofer T., Someya S., Miyakawa T., Nakayama C., Samhan-Arias A.K., et al. Mitochondrial DNA mutations induce mitochondrial dysfunction, apoptosis and sarcopenia in skeletal muscle of mitochondrial DNA mutator mice. PLoS One. 2010;5:e11468. doi: 10.1371/journal.pone.0011468. doi:10.1371/journal.pone.0011468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wenz T., Diaz F., Spiegelman B.M., Moraes C.T. Activation of the PPAR/PGC-1alpha pathway prevents a bioenergetic deficit and effectively improves a mitochondrial myopathy phenotype. Cell Metab. 2008;8:249–256. doi: 10.1016/j.cmet.2008.07.006. doi:10.1016/j.cmet.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Lehman J.J., Barger P.M., Kovacs A., Saffitz J.E., Medeiros D.M., Kelly D.P. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Invest. 2000;106:847–856. doi: 10.1172/JCI10268. doi:10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin J., Wu H., Tarr P.T., Zhang C.Y., Wu Z., Boss O., Michael L.F., Puigserver P., Isotani E., Olson E.N., et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. doi:10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- 17.Arany Z., Foo S.Y., Ma Y., Ruas J.L., Bommi-Reddy A., Girnun G., Cooper M., Laznik D., Chinsomboon J., Rangwala S.M., et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature. 2008;451:1008–1012. doi: 10.1038/nature06613. doi:10.1038/nature06613. [DOI] [PubMed] [Google Scholar]
- 18.Sandri M., Lin J., Handschin C., Yang W., Arany Z.P., Lecker S.H., Goldberg A.L., Spiegelman B.M. PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc. Natl Acad. Sci. USA. 2006;103:16260–16265. doi: 10.1073/pnas.0607795103. doi:10.1073/pnas.0607795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brault J.J., Jespersen J.G., Goldberg A.L. Peroxisome proliferator-activated receptor gamma coactivator 1alpha or 1beta overexpression inhibits muscle protein degradation, induction of ubiquitin ligases, and disuse atrophy. J. Biol. Chem. 2010;285:19460–19471. doi: 10.1074/jbc.M110.113092. doi:10.1074/jbc.M110.113092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arany Z., He H., Lin J., Hoyer K., Handschin C., Toka O., Ahmad F., Matsui T., Chin S., Wu P.H., et al. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005;1:259–271. doi: 10.1016/j.cmet.2005.03.002. doi:10.1016/j.cmet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 21.Handschin C., Chin S., Li P., Liu F., Maratos-Flier E., Lebrasseur N.K., Yan Z., Spiegelman B.M. Skeletal muscle fiber-type switching, exercise intolerance, and myopathy in PGC-1alpha muscle-specific knock-out animals. J. Biol. Chem. 2007;282:30014–30021. doi: 10.1074/jbc.M704817200. doi:10.1074/jbc.M704817200. [DOI] [PubMed] [Google Scholar]
- 22.Biernacka A., Frangogiannis N.G. Aging and cardiac fibrosis. Aging Dis. 2011;2:158–173. [PMC free article] [PubMed] [Google Scholar]
- 23.de Souza R.R. Aging of myocardial collagen. Biogerontology. 2002;3:325–335. doi: 10.1023/a:1021312027486. doi:10.1023/A:1021312027486. [DOI] [PubMed] [Google Scholar]
- 24.Edgar D., Shabalina I., Camara Y., Wredenberg A., Calvaruso M.A., Nijtmans L., Nedergaard J., Cannon B., Larsson N.G., Trifunovic A. Random point mutations with major effects on protein-coding genes are the driving force behind premature aging in mtDNA mutator mice. Cell Metab. 2009;10:131–138. doi: 10.1016/j.cmet.2009.06.010. doi:10.1016/j.cmet.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 25.Williams S.L., Huang J., Edwards Y.J., Ulloa R.H., Dillon L.M., Prolla T.A., Vance J.M., Moraes C.T., Zuchner S. The mtDNA mutation spectrum of the progeroid Polg mutator mouse includes abundant control region multimers. Cell Metab. 2010;12:675–682. doi: 10.1016/j.cmet.2010.11.012. doi:10.1016/j.cmet.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ameur A., Stewart J.B., Freyer C., Hagstrom E., Ingman M., Larsson N.G., Gyllensten U. Ultra-deep sequencing of mouse mitochondrial DNA: mutational patterns and their origins. PLoS Genet. 2011;7:e1002028. doi: 10.1371/journal.pgen.1002028. doi:10.1371/journal.pgen.1002028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vermulst M., Bielas J.H., Kujoth G.C., Ladiges W.C., Rabinovitch P.S., Prolla T.A., Loeb L.A. Mitochondrial point mutations do not limit the natural lifespan of mice. Nat. Genet. 2007;39:540–543. doi: 10.1038/ng1988. doi:10.1038/ng1988. [DOI] [PubMed] [Google Scholar]
- 28.Rebelo A.P., Dillon L.M., Moraes C.T. Mitochondrial DNA transcription regulation and nucleoid organization. J. Inherit. Metab. Dis. 2011;34:941–951. doi: 10.1007/s10545-011-9330-8. doi:10.1007/s10545-011-9330-8. [DOI] [PubMed] [Google Scholar]
- 29.Zahn J.M., Sonu R., Vogel H., Crane E., Mazan-Mamczarz K., Rabkin R., Davis R.W., Becker K.G., Owen A.B., Kim S.K. Transcriptional profiling of aging in human muscle reveals a common aging signature. PLoS Genet. 2006;2:e115. doi: 10.1371/journal.pgen.0020115. doi:10.1371/journal.pgen.0020115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Conley K.E., Jubrias S.A., Amara C.E., Marcinek D.J. Mitochondrial dysfunction: impact on exercise performance and cellular aging. Exerc. Sport Sci. Rev. 2007;35:43–49. doi: 10.1249/JES.0b013e31803e88e9. doi:10.1249/JES.0b013e31803e88e9. [DOI] [PubMed] [Google Scholar]
- 31.Vina J., Gomez-Cabrera M.C., Borras C., Froio T., Sanchis-Gomar F., Martinez-Bello V.E., Pallardo F.V. Mitochondrial biogenesis in exercise and in ageing. Adv. Drug Deliv. Rev. 2009;61:1369–1374. doi: 10.1016/j.addr.2009.06.006. doi:10.1016/j.addr.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 32.Wenz T., Rossi S.G., Rotundo R.L., Spiegelman B.M., Moraes C.T. Increased muscle PGC-1alpha expression protects from sarcopenia and metabolic disease during aging. Proc. Natl Acad. Sci. USA. 2009;106:20405–20410. doi: 10.1073/pnas.0911570106. doi:10.1073/pnas.0911570106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Safdar A., Bourgeois J.M., Ogborn D.I., Little J.P., Hettinga B.P., Akhtar M., Thompson J.E., Melov S., Mocellin N.J., Kujoth G.C., et al. Endurance exercise rescues progeroid aging and induces systemic mitochondrial rejuvenation in mtDNA mutator mice. Proc. Natl Acad. Sci. USA. 2011;108:4135–4140. doi: 10.1073/pnas.1019581108. doi:10.1073/pnas.1019581108. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Lira V.A., Benton C.R., Yan Z., Bonen A. PGC-1alpha regulation by exercise training and its influences on muscle function and insulin sensitivity. Am. J. Physiol. Endocrinol. Metab. 2010;299:E145–E161. doi: 10.1152/ajpendo.00755.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Safdar A., Little J.P., Stokl A.J., Hettinga B.P., Akhtar M., Tarnopolsky M.A. Exercise increases mitochondrial PGC-1alpha content and promotes nuclear-mitochondrial cross-talk to coordinate mitochondrial biogenesis. J. Biol. Chem. 2011;286:10605–10617. doi: 10.1074/jbc.M110.211466. doi:10.1074/jbc.M110.211466. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36.Russell L.K., Mansfield C.M., Lehman J.J., Kovacs A., Courtois M., Saffitz J.E., Medeiros D.M., Valencik M.L., McDonald J.A., Kelly D.P. Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1alpha promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ. Res. 2004;94:525–533. doi: 10.1161/01.RES.0000117088.36577.EB. doi:10.1161/01.RES.0000117088.36577.EB. [DOI] [PubMed] [Google Scholar]
- 37.Rowe G.C., Jiang A., Arany Z. PGC-1 coactivators in cardiac development and disease. Circ. Res. 2010;107:825–838. doi: 10.1161/CIRCRESAHA.110.223818. doi:10.1161/CIRCRESAHA.110.223818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arnold A.S., Egger A., Handschin C. PGC-1alpha and myokines in the aging muscle—a mini-review. Gerontology. 2011;57:37–43. doi: 10.1159/000281883. doi:10.1159/000281883. [DOI] [PubMed] [Google Scholar]
- 39.Pickrell A.M., Fukui H., Wang X., Pinto M., Moraes C.T. The striatum is highly susceptible to mitochondrial oxidative phosphorylation dysfunctions. J. Neurosci. 2011;31:9895–9904. doi: 10.1523/JNEUROSCI.6223-10.2011. doi:10.1523/JNEUROSCI.6223-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sciacco M., Bonilla E. Cytochemistry and immunocytochemistry of mitochondria in tissue sections. Methods Enzymol. 1996;264:509–521. doi: 10.1016/s0076-6879(96)64045-2. doi:10.1016/S0076-6879(96)64045-2. [DOI] [PubMed] [Google Scholar]
- 41.Barrientos A. In vivo and in organello assessment of OXPHOS activities. Methods. 2002;26:307–316. doi: 10.1016/S1046-2023(02)00036-1. doi:10.1016/S1046-2023(02)00036-1. [DOI] [PubMed] [Google Scholar]
- 42.Martinez B., del Hoyo P., Martin M.A., Arenas J., Perez-Castillo A., Santos A. Thyroid hormone regulates oxidative phosphorylation in the cerebral cortex and striatum of neonatal rats. J. Neurochem. 2001;78:1054–1063. doi: 10.1046/j.1471-4159.2001.00487.x. doi:10.1046/j.1471-4159.2001.00487.x. [DOI] [PubMed] [Google Scholar]
- 43.Peacock J.D., Levay A.K., Gillaspie D.B., Tao G., Lincoln J. Reduced sox9 function promotes heart valve calcification phenotypes in vivo. Circ. Res. 2010;106:712–719. doi: 10.1161/CIRCRESAHA.109.213702. doi:10.1161/CIRCRESAHA.109.213702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheung M.C., Spalding P.B., Gutierrez J.C., Balkan W., Namias N., Koniaris L.G., Zimmers T.A. Body surface area prediction in normal, hypermuscular, and obese mice. J. Surg. Res. 2009;153:326–331. doi: 10.1016/j.jss.2008.05.002. doi:10.1016/j.jss.2008.05.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.