

Abstract
Tissue fusion is an essential morphogenetic mechanism in development, playing a fundamental role in developing neural tube, palate and the optic fissure. Disruption of genes associated with the tissue fusion can lead to congenital malformations, such as spina bifida, cleft lip/palate and ocular coloboma. For instance, the Pax2 transcription factor is required for optic fissure closure, although the mechanism of Pax2 action leading to tissue fusion remains elusive. This lack of information defining how transcription factors drive tissue morphogenesis at the cellular level is hampering new treatments options. Through loss- and gain-of-function analysis, we now establish that pax2 in combination with vax2 directly regulate the fas-associated death domain (fadd) gene. In the presence of fadd, cell proliferation is restricted in the developing eye through a caspase-dependent pathway. However, the loss of fadd results in a proliferation defect and concomitant activation of the necroptosis pathway through RIP1/RIP3 activity, leading to an abnormal open fissure. Inhibition of RIP1 with the small molecule drug necrostatin-1 rescues the pax2 eye fusion defect, thereby overcoming the underlying genetic defect. Thus, fadd has an essential physiological function in protecting the developing optic fissure neuroepithelium from RIP3-dependent necroptosis. This study demonstrates the molecular hierarchies that regulate a cellular switch between proliferation and the apoptotic and necroptotic cell death pathways, which in combination drive tissue morphogenesis. Furthermore, our data suggest that future therapeutic strategies may be based on small molecule drugs that can bypass the gene defects causing common congenital tissue fusion defects.
INTRODUCTION
During tissue morphogenesis, the coordination of cell migration, proliferation and cell death are under tight spatiotemporal molecular control requiring cooperative action of distinct signalling pathways. Genetic screens have identified molecularly diverse genes that mediate localized epithelial fusion processes in the developing neural tube, palate and eye, yet how these genes exert their influence at the cellular level is still poorly understood (1,2). During eye development, failure of the optic fissure to fuse leads to congenital ocular coloboma (3). Closure of the optic fissure requires the coordination of at least three distinct steps: alignment of the apposed edges, contact-induced adhesion and dissolution of the basal lamina (4), so that a continuous epithelial sheet is formed (5). Mutations in developmentally important genes are implicated in optic fissure closure defects, including PAX2, CHD7, SOX2, PAX6, GDF6, OTX2, SHH, SIX3, FADD, MAF, ZFHX1B, RX and GDF3, as well as many chromosomal aberrations (6–13). However, network interactions between these genes and their downstream targets underlying the cellular mechanism of tissue fusion have yet to be established.
As a starting point for understanding how alteration in gene function leads to defective optic fissure morphogenesis, we have focused on the PAX2 transcription factor as it is one of the most common causes of ocular coloboma (3) and plays a crucial evolutionary role in the development of the eye (4,14,15). To identify downstream targets of Pax2 in optic fissure closure, we have taken advantage of the pax2.1/noi zebrafish coloboma mutant (16). We conducted gene expression studies of known coloboma genes in this mutant, and identified one which was down-regulated, the fas-associated death domain (FADD) gene. Recently, we showed that hemizygous deletion of the FADD gene was the underlying genetic basis of inherited ocular coloboma (9). The principal role of the FADD protein is the induction of apoptotic cell death (17), implying that fine tuning of cell death may be required during optic fissure closure. FADD recruits pro-caspase-8 and c-FLIP to create a death-inducing signalling complex (known as DISC), which in turn activates downstream effector caspases leading to cell death (18). Recent investigations have, however, also uncovered the seminal finding that FADD has an apoptosis-independent role in embryogenesis, whereby it suppresses RIP1/RIP3 kinase-mediated necroptosis pathways (19,20), essential for the maintenance of the vasculature, haematopoiesis, innate immunity and T-cell proliferation during lymphocyte development (21–24). The raises a number of possible mechanisms by which loss of FADD function may cause ocular coloboma.
In this study, we demonstrate that pax2 and vax2 transcription factors co-regulate fadd transcriptional activation. Loss-of-function analysis of fadd confirmed that it is required for optic fissure closure. In the absence of fadd, cell proliferation is increased and RIP1/RIP3-mediated necroptosis is activated. Fadd over-expression in pax2.1-deficient embryos is sufficient to rescue the proliferation defect and close the optic fissure. Inhibition of RIP1 kinase activity by necrostatin-1 inhibits necroptotic cell death and also rescues the optic fissure closure defect in the pax2.1-deficient zebrafish. Taken together, our results demonstrate for the first time that fadd has an essential physiological function in protecting the developing optic fissure neuroepithelium from RIP3-dependent necroptosis, thus preventing ocular coloboma. Abnormal necroptosis may therefore be an important molecular trigger leading to failure of epithelial fusion due to mutation of fadd and pax2. Targeting the cellular mechanisms that drive necroptosis may be a promising paradigm for preventing congenital tissue fusion abnormalities such as ocular coloboma.
RESULTS
Fadd gene expression analysis in mutant zebrafish lines
To determine where the fadd gene is positioned in the transcriptional cascade regulating optic fissure closure (8), the spatiotemporal expression profile of fadd was tested in two ocular coloboma mutant zebrafish lines. The no isthmus (noitu29a) line exhibiting ocular coloboma (15) has a R139X non-sense mutation in the pax2.1 gene (genotyping for this mutation can be found in Supplementary Material, Fig. S1A). The grumpy (gupm189) line is homozygous for Q524X non-sense mutation in the lamb1 gene (25) displaying a severe ocular coloboma defect (26,27).
In wild-type embryos at 24 h post-fertilization (hpf), high levels of fadd expression were restricted to the developing brain, otic vesicles, the lens, retina and at the site of the optic fissure (Fig. 1A). However, in the noi/pax2.1 mutant, fadd expression was absent except in the floorplate (Fig. 1B), a structure that guides neuronal differentiation along the dorsoventral axis of the neural tube. At 48 hpf, the optic fissure was closed and there was strong expression in the mandibular mesenchyme, otic vesicles and lens (Fig. 1C), whereas in the noi/pax2.1 mutant there was only minimal fadd expression in the floorplate (Fig. 1D). This demonstrates that the fadd gene is downstream of pax2.1 in the signalling cascade in vivo. In contrast, fadd gene expression patterns in the gup/lamb1 zebrafish mutant resembles wild-type expression at 24 hpf (compare Fig. 1A and E) and 48 hpf (Fig. 1C and F). Furthermore, the expression of pax2.1 was not disrupted in the gup/lamb1 mutant (Fig. 1G and H). This establishes that fadd and pax2.1 are in a different signalling pathway to lamb1.
Figure 1.
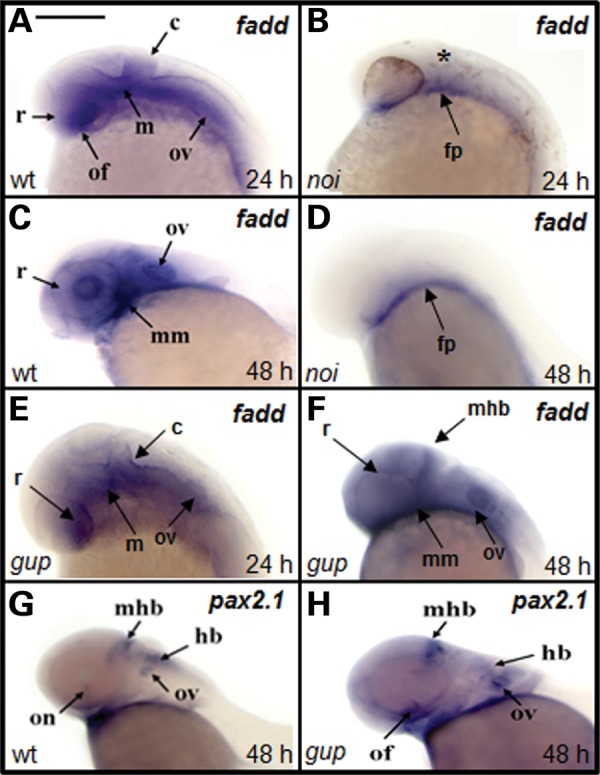
Expression of fadd is down-regulated in noi mutant zebrafish. (A–F) fadd expression in wild-type (wt), pax2.1−/− coloboma mutant (noi) and lamb1−/− coloboma mutant (gup). Gene expression tested at either 24 (24 h) or 48 hpf (48 h). Asterisk (*) denotes the absence of the tectum and cerebellum in noi mutants at 24 hpf. c, cerebellum; fp, floorplate; hb, hindbrain; m, midbrain; mhb, mid-hindbrain boundary; mm, mandibular mesenchyme; of, optic fissure; on, optic nerve; ov, otic vesicle; r, retina. Scale bar for all images shown in (A) 300 µm. (G and H) pax2.1 expression in wt and gup embryos is the same at 48 h.
Fadd promoter characterization
To determine whether fadd is a direct or indirect downstream target of pax2.1, we carried out a number of in vitro and in vivo assays. Using in silico motif analysis, we searched the genomic DNA sequence immediately upstream of the fadd transcription start site for pax2-binding sites using MatInspector program (www.genomatix.de). This analysis revealed five potential pax2 consensus-binding sites (Supplementary Material, Fig. S2) based on mouse (28,29) and medaki fish (Oryzias latipes) sequences (30). A number of other putative transcription factor-binding sites were also identified in the fadd upstream sequence, including barx2, oct1, rxr, ap-1, vax2, cdx1 and irf2. Of specific interest were the vax2- and rxr-binding sites as mutations in these genes cause ocular coloboma (31,32).
To verify pax2 responsive elements in the fadd promoter, deletion constructs were tested in luciferase reporter assays. The construct containing DNA sequence from −417 to +97 bp of the fadd gene had two pax2-binding sites and one vax2 site and was able to drive the highest level of expression in Madin-Derby canine kidney (MDCK) cells (Fig. 2A). Site-directed mutagenesis of the pax2 site nearest to the ATG start codon reduced luciferase expression by 74 ± 3%, whereas mutation of the second pax2 site reduced reporter expression by 85 ± 5%. When the vax2 site was mutated, luciferase activity was reduced by 74 ± 4%, suggesting that it contributed to driving high-level fadd expression. This was confirmed by mutation of pax2- and vax2-binding sites which completely abolished gene transcription. Longer constructs containing more potential pax2 sites had decreased luciferase activity compared with the −417 bp construct. In these constructs, there was an rxr-binding site which could be mediating the decreased transcription. However, when this site was mutated, there was no increase in reporter activity, suggesting that rxr did not have a negative regulatory effect on the fadd promoter. Reverse transcription–polymerase chain reaction (RT–PCR) for Rxr (Fig. 2B) and western blot analysis for Pax2 and Vax2 (Fig. 2C) showed that these transcription factors were expressed endogenously in MDCK cells. Specificity of transcriptional activation was verified for the −417 bp construct when transfected into COS-7 cells that do not express Pax2 (Fig. 2D).
Figure 2.

pax2.1 responsive elements in the fadd promoter. (A) Diagram at top depicts symbols used for transcription factor-binding sites. p, pax2.1 site; v, vax2 site; rxr, rxr site. Hatched box, exon 1 of fadd. L, luciferase gene. Reporter constructs transfected into Madin-Derby canine kidney (MDCK) cells. In lower part of (A), the filled symbols represent mutation of the site so it is no longer functional. The activity of the last construct relates to the −417 bp fragment transfected into COS-7 cells. The relative luciferase activity for the longest construct (−767 bp) was set at 100%. Activity (± SEM, n= 6) of all other constructs measured against this. (B) Confirmation that MDCK cells express Rxr. Lane 1, no RT control for Rxr-α; lane 2, Rxr-α (495 bp); lane 3, no RT control Rxr-β; lane 4, Rxr-β (520 bp). M, 100 bp marker. (C) Western blot of MDCK cells with Pax2 or Vax2 antibodies. (D) Western blot showing the Pax2 protein is absent in COS-7 cells.
To further validate pax2.1 regulation of fadd, electrophoretic mobility shift assays (EMSAs) were performed using in vitro translated zebrafish pax2.1 protein. We used probes containing the predicted pax2-binding sites flanked between 3–22 bases up and downstream of the binding site (Supplementary Material, Table S1). Pax2.1 bound with highest affinity to the second pax2-binding site upstream of the ATG (Supplementary Material, Fig. S2), which could be displaced by an excess of unlabelled competitor and the DNA–protein complex could be shifted to a higher migrating band by a Pax2 antibody (Fig. 3A). To determine whether endogenous pax2.1 could bind to the zebrafish fadd promoter in vivo, we used chromatin immunoprecipitation (ChIP) assays. We performed ChIP-qPCR on zebrafish embryos at 36 hpf during the window of optic fissure morphogenesis, when pax2.1 is expressed. A 423 bp fragment containing the second pax2 site was specifically amplified from pax2.1 immunoprecipitates (Fig. 3B). Similarly, the same fadd fragment was amplified from vax2 immunoprecipitates (Fig. 3C), confirming that vax2 is a co-activator of fadd expression in vivo.
Figure 3.

Pax2.1 binds to the fadd promoter. (A) EMSA using fadd oligonucleotide probe containing the second pax2-binding site. Fadd probe bound to the pax2.1 protein (lane 2), which was displaced by unlabelled competitor (lane 3) and in the presence of pax2 antibody resulted in a supershifted (SS) complex. FP, free probe. Inset, western blot of in vitro translated full-length pax2.1 protein. (B) ChIP-PCR using Pax2 antibody (44 kDa protein detected by western blot of 36 hpf embryos). A fadd band (arrow) was specifically amplified from pax2 immunoprecipitates. IgG, negative control. Input corresponds to PCR from chromatin suspension without immunoprecipitation. (C) ChIP-PCR for Vax2 antibody (31 kDa protein detected by western blot of 36 hpf embryos). A fadd band (arrow) was specifically amplified from vax2 immunoprecipitates.
Fadd loss-of-function analysis
To provide functional evidence that fadd is important in optic fissure morphogenesis, we utilized morpholino antisense knockdown technology (33). At 72 hpf, a time when the fissure is closed in wild-type embryos (Fig. 4A–C), fadd morphants displayed an open fissure (Fig. 4E–G), demonstrating that fadd is necessary for optic fissure closure. In addition, there were systemic defects including pericardial oedema and a shortened curved spine. Since one of the functions of fadd is to activate caspase-3-dependent cell death, we hypothesized that if cell death was important during optic fissure closure there would be a reduction in cell death and/or increased proliferation in fadd morphants. The level of phosphohistone H-3 (PH3) labelling as a marker for cell proliferation was found to be highly up-regulated throughout the eye of fadd morphants compared with mismatch controls (compare Fig. 4I with J and K), whereas there was no change in activated caspase-3 activity (compare Fig. 4L with M and N). The fadd morpholino was distributed throughout the embryo as shown by imaging of the fluorescent tag on the morpholino (Fig. 4D and H). The specificity of the ATG-blocking morpholino was confirmed by western blot (Fig. 4O), which showed that the 27 kDa fadd band was markedly reduced in fadd morphant embryos.
Figure 4.

Fadd knockdown and analysis of zebrafish morphants at 72 hpf. (A–C) Representative images of control fadd mismatch morpholino (mm) injection on eye development. (D) Distribution of fadd morpholino detected by 3′-carboxyfluorescein modification of antisense morpholino. (E–G) Representative images of fadd antisense morpholino (mo) injection on eye development. (H) Magnified image of the eye showing distribution of the fadd antisense morpholino in the lens and retina. (I–K) Phosphohistone-H3 (PH3) labelling (green) in fadd mm and fadd mo knockdown eyes. (L–N) Activated caspase-3 (casp3) labelling (green) in fadd mm and fadd mo knockdown eyes. (O) Western blot from fadd mm and fadd mo embryos. GAPDH used as loading control. (A–C and E–G) Red fluorescence derived from ath5-Gal4 x UAS-NR:mcherry transgene driving reporter expression in the developing eye. (I–N) embryos counterstained with Bodipy Texas-Red. All embryo images represent a projection through a confocal stack. (E–N) optic fissure (of) is indicated by a white arrow.
Fadd gain-of-function analysis
To further demonstrate that fadd is a direct downstream target of pax2.1, we reasoned that over-expression of fadd mRNA in pax2.1-deficient embryos should at least partially or fully rescue the mutant eye phenotype. To confirm that the pax2.1 mutant phenotype could be rescued, we injected pax2.1 mRNA into pax2.1-deficient embryos. Injection of pax2.1 mRNA into wild-type embryos had no obvious detrimental effect at 72 hpf (Fig. 5A and B). Since PH3 labelling was elevated in fadd morphant fish, we also found it was elevated in pax2.1-deficient embryos (Fig. 5F) and therefore PH3 labelling could be used as a phenotypic measure. When pax2.1 mRNA was injected into pax2.1-deficient embryos, it resulted in normal closure of the optic fissure (Fig. 5K) and also greatly reduced the number of PH3-positive cells (12% ± 2 of mutant PH3-positive cells, n= 5). The cerebellum and otic vesicle defects were normalized and the pericardial oedema greatly reduced, although there was evidence of a small haemorrhage around the developing heart (Fig. 5L). When fadd mRNA was injected into pax2.1-deficient embryos, the optic fissure closure defect was also rescued (Fig. 5P); however, there were still some PH3-positive cells still present (38 ± 4% of mutant PH3-positive cells, n= 5). The pericardial oedema was reduced and haemorrhaging was more obvious than with pax2.1 injection (Fig. 5Q). Although the systemic defects were not fully rescued by fadd mRNA injection, the optic fissure was closed in all cases.
Figure 5.

Rescue of the pax2.1-deficient phenotype by mRNA injection. (A–E) Wild-type embryos (wt) were injected with pax2.1 mRNA. (F–J) Uninjected pax2.1-deficient (mt) embryos (K–O) pax2.1-deficient embryos injected with pax2.1 mRNA. (P–T) pax2.1-deficient embryos injected with fadd mRNA. (A, F, K, P) Proliferation status detected PH3 immunohistochemistry. (B, G, L, Q) Systemic phenotype of control and injected embryos. c, cerebellum; ov, otic vesicle; po, pericardial oedema. Punctate red staining in the 12 right-hand panels represents RIP1 localization with DAPI counterstain. White arrows denote an open optic fissure.
Activation of the necroptosis pathway
Since fadd also has a role in repressing RIP1/RIP3 kinase-mediated necroptosis, we examined wild-type and pax2.1-deficient embryos for evidence of RIP1 kinase activity. In wild-type embryos, there were minimal RIP1-positive cells present at 24 hpf, but by 72 hpf RIP1 labelling was extinguished (Fig. 5D and E). In pax2.1 mutant embryos, the number of RIP-positive cells was greatly increased at 48 and 72 hpf, particularly in the margins of the open optic fissure (Fig. 5I and J). In mutant embryos injected with either pax2.1 mRNA (Fig. 5M–O) or fadd mRNA (Fig. 5R–T), the RIP1 labelling corresponded to the wild-type pattern. These data confirm that in the absence of pax2.1 and consequently fadd, RIP1 localization is greatly increased. Since fadd is also an endogenous inhibitor of RIP3-mediated necroptosis (34), we investigated the distribution of RIP3 kinase in the mutant pax2.1 embryos. In wild-type embryos, RIP3 labelling was very sparse (Fig. 6A–C); however, in mutant embryos, RIP3 labelling was greatly increased at all time points, especially in the region of the optic fissure (Fig. 6D–F). To determine whether RIP1/RIP3 labelling was evidence that necroptosis was contributing to the disease pathogenesis in vivo, we tested the effect of necrostatin-1, a specific inhibitor of RIP1 kinase activity that has been validated for use in fish (35). Using dose–response analysis in wild-type embryos, we established that a concentration of 6 µm necrostain-1 was the highest dose we could use without causing any detrimental harm to the developing embryos (Supplementary Material, Fig. S3). In treated mutants, only a few RIP1-positive cells were present and the optic fissure was fully closed by 72 hpf (compare Fig. 6G–I with Fig. 5H–J), showing that the drug had rescued the eye phenotype. Furthermore, RIP3 labelling was also greatly decreased at all time points with the optic fissure fully closed at 72 hpf (Fig. 6J–L). Interestingly, we found that mutants treated with necrostatin-1 had more than double the expected lifespan compared with untreated mutants (Fig. 6M–P), suggesting there was a systemic effect of necrostatin-1 affecting survival. Necrostatin-1 had no effect on the increased proliferation in the eye in the absence of pax2.1 (Supplementary Material, Fig. S4).
Figure 6.

Spatiotemporal RIP3 localization and rescue of eye phenotype using necrostatin-1. (A–C) RIP3 immunohistochemistry (red) in wild-type (wt) eyes counterstained with DAPI. (D and E) High-level RIP3 labelling in pax2.1-deficient eyes (mt). White arrow denotes open optic fissure. (G–I) RIP1 labelling in pax2.1-deficient eyes treated with 6 µm necrostatin-1. Optic fissure completely closed at 72 hpf. (J–L) RIP3 labelling in pax2.1-deficient eyes treated with 6 µm necrostatin-1. Optic fissure completely closed at 72 hpf. (M) Lifespan of pax2-deficient embryos (mt) is increased when treated with necrostatin-1 (+N). Data presented as mean ± SEM, n= 30. (N) The systemic phenotype of untreated pax2.1-deficient embryos at 3 dpf. (O) pax2.1-deficient embryos treated with necrostatin-1 (+N) at 3 dpf. Heart bleeding and pericardial oedema are reduced (black arrow). At 9 dpf, there is extreme oedema (the mt would not be alive at this age without treatment).
Cell death during normal optic fissure morphogenesis
Since cell proliferation appeared to be up-regulated in the pax2.1-deficent embryos, we hypothesized whether this was due to loss of fadd activity that would normally stimulate caspase-dependent apoptosis, keeping the proliferation in check. We reasoned that caspase inhibitors would mimic this loss of fadd activity and may therefore disrupt the morphogenesis process. Whole wild-type zebrafish embryos were incubated with zDEVD-fmk (caspase-3-specific inhibitor) from 10 to 72 hpf as previously described (25) and pax2 immunohistochemistry was used as a marker for optic fissure morphogenesis. At 24 hpf, pax2 was highly restricted ventrally to the opposed edges of the open fissure (Fig. 7A) and between 36 and 48 hpf the intensity of labelling decreased as the fissure began to close (Fig. 7B and C). By 72 hpf, the fissure was fully closed and pax2 labelling was extinguished (Fig. 7D). However, in embryos treated with zDEVD-fmk, the expression of pax2 was expanded throughout the ventral eyecup at 24 hpf (Fig. 7E). At 36–48 hpf, the optic fissure was still clearly visible and labelling of pax2 was expanded at the edges of the fissure compared with wild-type eyes (Fig. 7F and G). Pax2 expression persisted until 72 hpf coincident with an open optic fissure (Fig. 7H). The delayed closure of the optic fissure was observed at doses of zDEVD-fmk that had no embryotoxic effects as indicated by yolk sac circulation, growth parameters and morphological appearance (25). In the presence of zDEVD-fmk, the levels of PH3 labelling throughout the eye at 24 hpf were higher than untreated embryos (compare Fig. 7M and I). Although decreasing with developmental time, there were still more PH3-positive cells present in the zVAD-fmk treated embryos (Fig. 7N and O), than in wild-type embryos (Fig. 7J and K). By 72 hpf, there was no PH3 labelling in wild-type or treated embryos (Fig. 7L and P). These experiments indicate that the increase in cell proliferation occurs when there is loss of the caspase-dependent cell death and this is associated with a delay in optic fissure closure.
Figure 7.
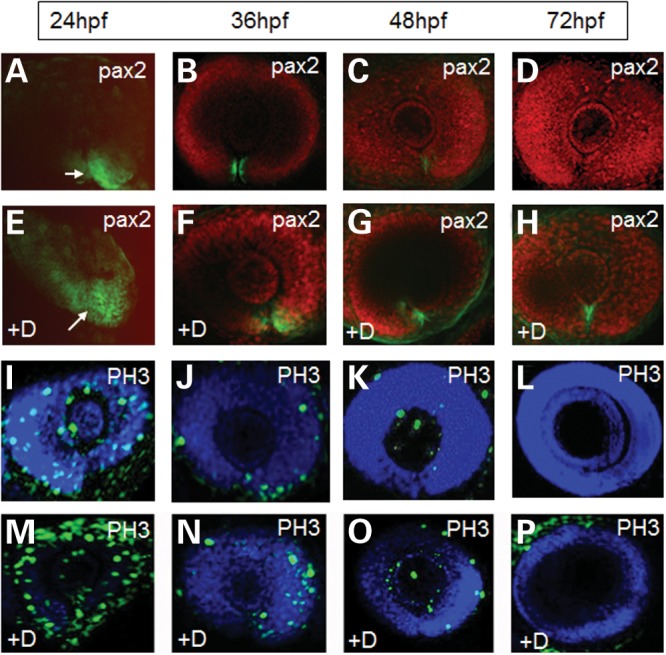
Effect of DEVD-fmk (caspase-3 inhibitor) on optic fissure closure in wild-type eyes. (A–D) Localization of the pax2 protein (green labelling) in embryos treated with 300 µm control inhibitor zFA-fmk continuously from 10 hpf. (E–D) pax2 localization in embryos exposed to 300 µm DEVD-fmk (+D) continuously from 10 hpf. (A and E) No BOBO-3 counterstain in these panels to better show the expanded Pax2 localization in the treated embryos. White arrows indicate the position of the optic fissure. (I–L) PH3 staining (green) from 24–72 h in embryos treated with control zFA-fmk. (M–P) PH3 staining from 24 to 72 h in embryos treated with 300 µm DEVD-fmk (+D).
DISCUSSION
Although it has been known for more than 15 years that PAX2 deficiency causes ocular coloboma (36), the direct targets of Pax2 regulating optic fissure closure have remained elusive. In this study, we demonstrate by both in vivo and in vitro experiments that the fadd gene is a direct downstream target of the pax2.1 transcription factor in the developing eye. Furthermore, we show that the vax2 is required to co-activate fadd transcription. This is significant because mutations in both pax2 and vax2 (31) cause ocular coloboma, implying that fadd is the common downstream target of these transcription factors, where its loss of action leads to failure of epithelial fusion and an open optic fissure. In other aspects of embryonic development, several Pax2 targets have been identified. In the kidney, Pax2 negatively regulates Wnt5a during nephron development (37) and activates Naip in the ureteric bud (38). In the developing medaka otic vesicle, Pax2 activates stm-l to promote otolith formation (30). In the developing mid-hindbrain boundary, Brn1 is a direct target of Pax2 (39). Thus, Pax2 has a range of regulatory activities in developing tissues that account for other phenotypes associated with PAX2 mutations such as kidney dysfunction, hearing deficits and brain abnormalities (3).
Our investigations have uncovered two functions for fadd in the developing eye. First, loss of fadd activity through absence of pax2 (as in the noi mutant zebrafish), by direct loss of fadd itself (morpholino knockdown) or by inhibition of downstream effector caspases in wild-type embryos, results in highly up-regulated proliferation throughout the eye. This suggests that fadd is keeping proliferation in check by inducing cell death through a caspase-dependent mechanism. Loss of fadd leading to an increase in proliferation has also been observed in the intestinal epithelium of conditional Fadd (40) or conditional Casp8 knockout mice (41). In these studies, the chronic inflammation of the colon was associated with hyperproliferation resulting in epithelial dysplasia. The critical nature of controlling cell proliferation in the developing eye is exemplified in N-cadherin zebrafish mutants (ncad) which exhibit ocular coloboma (42). Increased proliferation in the retinal neuroepithelium leads to loss of apico-basal cell polarity, collapse of the adherens junctions, detachment of cells undergoing mitosis, severe retinal disorganization and patterning defects (43). Furthermore, abrogation of vax1 and vax2 activity in zebrafish or mice leads to progressive expansion of retinal tissue along the optic nerve and results in an open optic fissure (31,44,45). Similarly, reduced Tfap2a activity results in ocular coloboma due to expansion of retinal pigment epithelium out of the back of the eye towards the midline brain (46). These observations provide evidence that deficiencies in the control of cellular proliferation are common to optic fissure closure defects and may be a target for therapeutic strategies.
However, in the absence of fadd, we also found that RIP1/RIP3-mediated necroptosis is switched on, especially in the cells lining the margins of the optic fissure, indicating a second role for fadd in eye development in repressing necroptotic cell death. When pax2-deficient embryos were treated with the specific RIP1 inhibitor necrostatin-1 (47), RIP1 and consequently RIP3 were down-regulated resulting in optic fissure closure. This is in contrast to zVAD-fmk, which is ineffective in rescuing optic fissure closure in these mutants (48), providing supportive evidence that cell death in the optic fissure is a not driven by a caspase-dependent process. Activation of necroptosis is similarly observed in Fadd and Casp8 knockout mice studies, where RIP1 and RIP3 are highly elevated and could be inhibited by necrostatin-1 (40,41). The mechanism by which fadd represses necroptosis in development involves a caspase-8-dependent activity that is post-translational. Necroptosis and apoptosis that are induced by death receptor activation share the same pathway from initiation to the formation of the DISC complex, composed of Fadd, RIP1, RIP3 and caspase-8. Under normal circumstances, caspase-8 cleaves RIP3 (49) and RIP1 (50) and consequently induces apoptosis through downstream effector caspases. However, if caspase-8 activity is blocked directly (e.g. by zVAD-fmk) or fadd activity is missing (fadd dimerizes with and subsequently activates caspase-8), phosphorylation of RIP1 and RIP3 occurs resulting in the formation of the necrosome and initiation of necroptosis. Our data provide compelling evidence that inhibiting cell death occurring through the necroptosis pathway is the fundamental element enabling optic fissure closure in pax2-deficient embryos.
Our data suggest that targeting the necroptosis pathway might be beneficial for preventing optic fissure closure caused by failure of epithelial fusion due to Pax2, Vax2 or Fadd mutation. Several in vitro and in vivo studies have suggested a beneficial effect of necrostatin-1 treatment to other types of tissue damage. For example, animal models of hypoxic injury, ischaemic reperfusion damage to the brain and retina, neurologic deficits of traumatic brain injury and myocardial infarction have all benefited to various extents from necrostain-1 treatment (51–54). Furthermore, necrostain-1 treatment may be more broadly applicable to other types of tissue fusion defects that may be associated with necroptotic cell death. For example, during neural tube closure acridine orange labelling in the hinge region of the neural tube suggested that cell death was important to neurulation (55) and in mice, excessive terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) positive labelling was associated with failure of neural tube closure in several genetic mutants (56). However, caspase-3-deficient mice did exhibit neural tube closure defects (57) providing evidence against apoptotic cell death associated with neural tube closure. Furthermore, Fbxl10-deficient mice exhibit neural tube defects and ocular coloboma associated with increased TUNEL staining in these tissues (58). However, since acridine orange and TUNEL are not exclusively markers of apoptotic cell death (59,60), it would be worthwhile re-investigating neural tube mutants for activation of the necroptosis pathway.
Here, our data demonstrate for the first time the cellular consequence of gene defects that cause the congenital abnormality ocular coloboma. Fadd is the common downstream target of Pax2 and Vax2 ocular coloboma genes and that abrogation of Fadd activity leads to activation of the necroptosis cell death pathway, preventing closure of the optic fissure (Fig. 8). A broader study investigating these findings in the mammalian system and ocular coloboma associated with other epithelial fusion defects is warranted. However, since the mechanism of optic fissure closure is well conserved between human and zebrafish, this model system provides the ideal test bed for new therapeutic strategies. The downstream effectors of RIP3 activation have yet to be fully elucidated, but it may involve catastrophic disruption to energy metabolism (61), production of reactive oxygen species (62,63), phospholipase activity (64,65) or endoplasmic reticulum stress/autophagy (40,66). Targeting the necroptotic cellular mechanisms such as inhibition of RIP3 activity itself or its downstream effectors could reveal new preventative strategies for congenital epithelial fusion defects.
Figure 8.
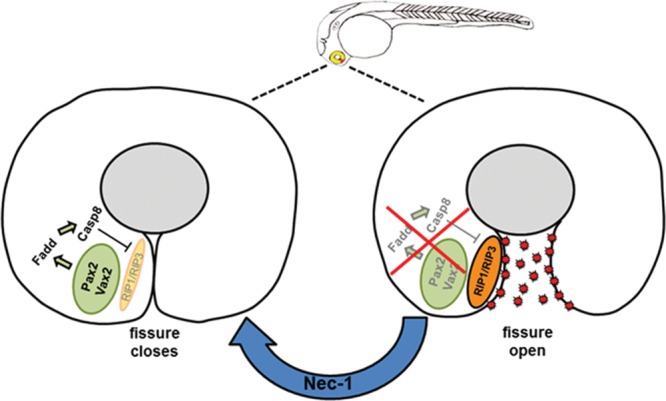
Mechanism of cell death in the pax2-deficient optic fissure. Schematic representation of normal optic fissure closure (left) compared with necroptosis activation in the developing mutant eye (right image). Activation of Fadd by Pax2 and Vax2 transcription factors leads to up-regulation of caspase-8 which inhibits RIP1/RIP3 complex allowing the optic fissure to close. The absence of Pax2 or Fadd relieves the repression on RIP1/RIP3 due to the lack of caspase-8 activity causing excessive cell death through necroptosis. Treatment with necrostatin-1 (Nec-1) inhibits RIP1 and results in closure of the optic fissure.
MATERIALS AND METHODS
Zebrafish strains and maintenance
Zebrafish strains [wild-type AB (67), gupm189 (68), noitu29a (69) and Ath5-Gal4/UAS:mcherry line (70)] were staged according to the morphological criteria (71). Research was carried in accordance with protocols compliant to the Canadian Council on Animal Care with the approval of the Animal Care Committee at the University of British Columbia and with the Association for Research in Vision and Ophthalmology (ARVO) statement for the use of animals in vision research. Embryos were raised at 28.5°C on a 14 h light/10 h dark cycle in 100 mm2 petri dishes containing aquaria water. To aid image analysis, 0.2 mm phenylthiourea (PTU, Sigma-Aldrich, St Louis, MO, USA) was added to the embryos at 10 hpf to inhibit pigment formation. To confirm the genotype in experimental embryos, DNA was extracted by the NaOH method as described previously (72). Extracted DNA was amplified by PCR using the specific primers listed in Supplementary Material, Table S1. Amplified products were digested with TaqI enzyme (Invitrogen, Carlsbad, CA, USA) and separated by 2% agarose gel electrophoresis (Supplementary Material, Fig. S1).
RT–PCR for MDCK cells
Total RNA was extracted from MDCK cells using Absolutely RNA Miniprep Kit (Stratagene, La Jolla, CA, USA) according to the manufacturer's instruction, and purified RNA was treated with DNase (Qiagen, Crawley, UK). The QuantiTect Reverse Transcription System (Qiagen) was used to synthesize first-strand cDNA from 250 ng total RNA. Primers were synthesized based on the cDNA sequence of either canine RXR alpha (RXR α) or RXR beta (RXR β) and sequences are shown in Supplementary Material, Table S1. RT–PCR was performed at 95°C for 30 s, 55°C for 30 s and 72°C for 40 s, for 35 cycles. Products of RT–PCR were analysed using 2% agarose gel electrophoresis in 1× tris-borate-EDTA (TBE) buffer in the presence of ethidium bromide for UV visualization.
In situ hybridization
A pax.2.1 cDNA clone (pGEM®-T Easy) was generated by RT–PCR amplification and cloning of a 635 bp product from zebrafish RNA obtained from whole embryos (Absolutely RNA Miniprep Kit, Stratagene). For PCR amplification, a forward primer, 5′-TCTAACAGGCACATCCCATTC-3′, and reverse primer, 5′-TGTGCGAGCTCCACGATTC-3′, were used with the Superscript III RT-PCR System (Invitrogen). Digoxigenin-labelled pax2.1 riboprobe was generated using DIG RNA Labeling Kit (SP6/T7) according to the manufacturer's instructions (Roche, Indianapolis, IN, USA). Whole-mount RNA in situ hybridization was carried out using the pax2.1 riboprobe or a riboprobe to fadd as previously described (9). Embryos were mounted in 70% glycerol and images captured using a DFC300 FX Digital Color Camera attached to a Leica MZ16F stereomicroscope.
Electrophoretic mobility shift assays (EMSAs)
Full-length zebrafish pax2.1 protein was synthesized by in vitro translation from a pax2.1 expression vector (pGEM-T Easy) using the TNT T7-coupled reticulocyte lysate system (Promega). The translated protein was verified using the Transcend™ non-radioactive translation detection system (Promega) according to the manufacturer's instructions. Single-stranded complementary oligonucleotides containing the Pax2-binding sites (Supplementary Material, Table S1) were biotinylated using the Biotin 3′-end DNA-labelling kit (Pierce) and then annealed overnight at room temperature (RT) to produce double-stranded probes. EMSA was performed using the LightShift Chemiluminescent EMSA kit according to the manufacturer's instructions (Pierce). For each EMSA reaction, 20 fmol biotinylated oligonucleotide, 8 µl of in vitro translation product and 50 ng/µl poly (dI-dC) in binding buffer (10 mm Tris–HCl pH 7.5, 50 mm KCl, 1 mm dithiothreitol) were incubated for 20 min at 25°C. For competition assays, unlabelled double-stranded probes were added to the reaction mix prior to addition of labelled probes in 100-fold excess molar concentrations. For supershift assays, 1 µg Pax2 antibody (Abcam) was added to the EMSA reaction 20 min before the biotinylated probes were added. DNA–protein complexes were resolved by native 6% polyacrylamide gel electrophoresis, transferred to positively charged nylon membrane (Amersham) by electroblotting and UV cross-linked to the membrane for 15 min. Biotinylated DNAs were visualized with streptavidin-bound horseradish peroxidase (HRP) and Luminol/Enhancer substrate (Pierce). All EMSAs were repeated at least twice to ensure reproducibility.
Luciferase reporter assays
Genomic DNA isolated from whole embryos using an Illustra DNA Extraction Kit (GE Healthcare, London, UK). Deletion constructs for the upstream region of fadd (up to −700 bp) were directionally cloned into the luciferase reporter plasmid pGL3 (Promega). Forward primer sequences for PCR amplification of each construct are listed in Supplementary Material, Table S1, as well as the reverse primer sequence which was common to all PCR products. Sequencing was used to ensure clone integrity. These pGL3 plasmid constructs were purified with a standard protocol (Midiprep; Qiagen) and quantified using a NanoDrop® ND-1000 spectrophotometer. Lipofectamine™-mediated transient transfections (Invitrogen) were performed in COS-7 or MDCK cells (which constitutively express Pax2) using 1 μg of each construct as previously described (73). For each construct, three independent transfections were performed with Renilla luciferase (pRL-CMV) co-transfection, as an internal control for efficiency of transfection. The Dual-Luciferase Reporter Assay System (Promega) and a GloMax 20/20 luminometer were used to measure luciferase activity in cell lysates.
Chromatin immunoprecipitation assay (ChIP-PCR)
Approximately 300 embryos were washed once with sterile water, placed in phosphate buffered saline (PBS) 20 mm Na-butyrate and transferred to a 5 ml syringe. The embryos were forced through a 20-gauge needle directly into a 6 cm Petri dish containing 5 ml of PBS/20 mm Na-butyrate. The empty floating chorions were removed. The embryos were transferred into 1.5 ml tube and were homogenized in 1% formaldehyde in PBS/butyrate and incubated at 10 min, prior to the addition of 125 mm glycine followed by incubation at RT for 5 min, cells were washed twice with PBS/butyrate, pelleted and snap-frozen in ethanol-dry ice (74). Cells were lysed in 300 µl lysis buffer and sonicated in ice water (Bioruptor Sonicator, Diagenode) for 20, 25 and 30 cycles of 30 s ON, 30 s OFF for 24, 36 and 48 hpf embryos, respectively. Fragmented chromatin sizes (300–500 bp) were confirmed by agarose gel electrophoresis, and sheared chromatin was diluted in radioimmunoprecipitation assay (RIPA) ChIP buffer to achieve 0.2 U A260 before immunoprecipitation.
Protein A-coated paramagnetic beads were pre-cleared with 100 µl RIPA ChIP buffer (10 mm Tris–HCl, pH 7.5, 140 mm NaCl, 1 mm ethylene diamine tetraacetic acid (EDTA) 0.5 mm ethylene glycol tetraacetic acid (EGTA) 1% Triton X-100, 0.1% sodium dodecyl sulphate (SDS) 0.1% Na-deoxycholate, protease inhibitors, 20 mm Na-butyrate) and then were mixed with 10 µl antibody-Protein A coated magnetic (Diagenode) complexes overnight at 4°C. To generate these complexes, 3 µg of either ChIP-grade Pax2 antibody (Abcam, cat. no. ab23799), Vax2 (Santa Cruz, cat. no. sc-81422; Supplementary Material, Fig. S1B) antibody or negative control IgG (Diagenode) were pre-mixed with protein A coated magnetic for 2 h at 4°C in RIPA buffer. The immunoprecipitated ChIP material was washed thrice with RIPA buffer and one time with 10 mm Tris–HCl, pH 8.0, 10 mm EDTA buffer. The cross-link was reversed and DNA eluted in a single step for 2 h at 68°C in 150 µl ChIP elution buffer (20 mm Tris–HCl, pH 7.5, 5 mm EDTA, 20 mm Na-butyrate, 50 mm NaCl, 1% SDS and 50 mg/ml proteinase K) containing 5 µg RNase (Roche). Eluted DNA was purified by phenol-chloroform isoamylalcohol extraction method and precipitated with ethanol. The DNA pellet was resuspended in nuclease-free water. An aliquot of the extracted DNA (1 µl) was used for PCR analysis. For PCR, different primers to the fadd proximal promoter were generated to produce the amplicon sizes between (∼150 and 199 bp) covering the putative pax2-binding sites within the promoter region (Supplementary Material, Table S1). The intensity of PCR products was confirmed using a 2% Agarose gel and visualized with ethidium bromide staining; DNA marker: 100 bp Hyperladder IV.
Western blot analysis
Briefly, 25 wild-type or mutant embryos at various stages of development were snap frozen in liquid nitrogen and homogenized by sonication in lysis buffer [10 mm Tris, pH 7.5, 10 mm NaCl, 1% SDS, 1 × Protease Inhibitor Cocktail (Roche)]. Insoluble material was removed by a 10 min centrifugation (25 000g). Protein concentration was determined by the DC protein assay (Bio-Rad). Zebrafish proteins (80 μg) were separated on a 12% SDS–polyacrylamide gel and transferred to Immobilon-FL membrane (Millipore, Bedford, MA, USA). For MDCK or COS-7 cells, only 40 µg protein/lane was used. The membrane was incubated in 5% non-fat milk powder in PBS/0.1% Tween-20 (PBST) for 2 h at RT and incubated overnight at 4°C with primary antibody. Following three washes in PBST, the membrane was incubated in the dark for 1 h with a Li-COR secondary antibody (Mandel Scientific, Guelph, ON, Canada). After the membrane was washed three times in PBST in the dark, protein bands were visualized using a Li-COR Odyssey detector.
Specific primary antibodies were obtained from commercial sources and used as follows: 1: 500 Pax2 rabbit polyclonal (Covance, Montreal, Canada; cat. no. PRB-276P-200); 1:500 Vax2 mouse monoclonal antibody (Santa Cruz; cat. no. sc-81422); 1:700 fadd rabbit polyclonal (AnaSpec, Fremont, CA, USA; cat. no. 55462); and 1:2000 GAPDH mouse monoclonal (Abcam, cat. no. 9485). The secondary antibody was IRDye 680LT goat anti-rabbit (Mandel Scientific; cat. no. 827-08364) or IRDye 700DX goat anti-mouse (Rockland; cat No. 610-130-121).
Whole-mount immunohistochemistry
Specific primary and secondary antibodies were obtained from commercial sources and used as follows: 1:100 cleaved caspase-3 rabbit polyclonal (Cell Signalling Technology; Cat No. 9661) (75,76); 1:100 Pax2 rabbit polyclonal (Covance, cat. no. PRB-276P-200); 1:50 RIP1 mouse monoclonal (Abcam, cat. no. ab56815; Supplementary Material, Fig. S1C) (77); 1:200 PH3 rabbit polyclonal (Abcam, cat. no. ab5176); 1:100 RIP3 rabbit polyclonal (Abgent, San Diego, CA, USA; cat. no. AP7184a); 1:200 goat anti-rabbit IgG conjugated to Alexa 488, 1:200 goat anti-rabbit IgG conjugated to Alexa-556 or 1:200 goat anti-mouse IgG conjugated to Alexa-546 (Invitrogen; cat. no. A11008, A11034 and A11010, respectively).
Embryos were dechorionated and fixed in 4% paraformaldehyde (PFA) washed three times for 15 min in PBS containing 0.1% Tween-20 PBS-Tw and then incubated with 10 µg/ml proteinase K (Sigma-Aldrich) for 5 min. Embryos were washed three times in PBS-Tw before re-fixing in 4% PFA for 10 min at RT. Non-specific antibody-binding sites were inhibited in blocking buffer (PBS-Tw containing 10% heat-inactivated normal goat serum and 1% dimethyl sulphoxide (DMSO)) for 2 h at RT.
Immunohistochemical staining was carried out as previous described (78). Images were obtained by scanning confocal microscopy. Optical sections were acquired at 3 µm intervals and merged into a maximum projection image for analysis.
Morpholino injections
Morpholino sequence directed against the transcriptional start site of zebrafish fadd was designed by GeneTools: 5′-TTGCTCGGAATCCTCTCTTGTCCAT (fadd MO) and mismatch (fadd MM) 5′-TTGCTCCGAATGCTGTCTTCTCGAT-3′ and were modified at the 3′-end with carboxy fluorescein. In addition to fadd morpholinos, a p53 morpholino was used to suppress off-target morpholino-induced apoptosis (79). Morpholino oligonucleotides were injected into the yolk of 1–2 cell-stage embryos as follows: 0.75 ng each of fadd MO or fadd MM plus 0.75 ng of p53 MO or with p53 MO alone. Morpholino oligonucleotides were injected in 1× Danieau buffer [58 mm NaCl, 0.7 mm KCl, 0.4 mm MgSO4, 0.6 mm Ca (NO3)2, 5 mm hydroxyethyl piperazineethanesulfonic acid, pH 7.6] with 0.1% phenol red using a Nanoject II variable injector (Drummond Scientific, Broomall, PA, USA). The morphant embryos were grown at 28.5°C and observed for morphological changes under a stereoscopic microscope and fixed in 4% PFA at specific developmental time points.
RNA injection into embryos
Zebrafish fadd and pax2.1, 5′-capped mRNAs were synthesized using a T7 mMESSAGE mMACHINE Kit (Ambion). A full-length fragment of pax2.1 or fadd was cloned into the pGEM T-easy vector and then linearized with NdeI restriction enzyme. For in vitro transcription, 1 µg of linear plasmid was used as template according to the manufacturer's instruction. The mRNA was precipitated with 7.5 m lithium chloride and the capped mRNA diluted in nuclease-free water prior to injection. One–2 cell-stage embryos were injected with fadd (5–25 pg) or pax2.1 (25–100 pg) mRNA, respectively. Embryos were collected at different developmental stages 24, 48 and 72 hpf and fixed overnight in 4% PFA prior to processing for immunohistochemistry.
Drug treatments
Stock solutions of necrostatin-1, caspase inhibitor DEVD-fmk and its control zFA-fmk (BD Biosciences, San Jose, CA, USA) were prepared in DMSO and added to aquaria water. Wild-type embryos were dechorionated at 10 hpf and treated with 300 µm zVAD-fmk or zFA-fmk for up to 72 hpf as previously described (25,80). Pax2.1-deficient embryos were treated with 6 µm necrostatin-1 for up to 13 dpf. Previous experiments demonstrated that these doses of drugs are not embryotoxic during this developmental period (25; Supplementary Material, Fig. S3). For each treatment, 30 embryos were used and 3 independent experiments were performed.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by The Wellcome Trust (084346 to C.Y.G.-E.); Canadian Institutes of Health Research (222728 to C.Y.G.-E. and K.G.-E.) and an unrestricted grant from the Department of Ophthalmology, University of British Columbia (to C.Y.G.-E.).
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr Derek Stemple at the Wellcome Trust Sanger Institute, UK for providing us with the gup zebrafish line, Dr Michael Brand, Dresden University of Technology, Germany for the noi zebrafish line and Dr Herwig Baier, University of California at San Francisco, USA for the ath5-Gal4 driver zebrafish line. The authors also thank Dr Orson Moritz for advice on image analysis and Dr Jiang Ping Zhang for technical advice.
Conflict of Interest statement. None declared.
References
- 1.Martin P., Wood W. Epithelial fusions in the embryo. Curr. Opin. Cell Biol. 2002;14:569–574. doi: 10.1016/s0955-0674(02)00369-1. [DOI] [PubMed] [Google Scholar]
- 2.Yu H., Smallwood P.M., Wang Y., Vidaltamayo R., Reed R., Nathans J. Frizzled 1 and frizzled 2 genes function in palate, ventricular septum and neural tube closure: general implications for tissue fusion processes. Development. 2010;137:3707–3717. doi: 10.1242/dev.052001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bower M., Salomon R., Allanson J., Antignac C., Benedicenti F., Benetti E., Binenbaum G., Birk-Jensen U., Cochat P., et al. Update of PAX2 mutations in renal coloboma syndrome and establishment of a locus specific database. Hum. Mutat. 2011 doi: 10.1002/humu.22020. [Epub ahead of print]. doi: 10.1002/humu.22020. [DOI] [PubMed] [Google Scholar]
- 4.Torres M., Gómez-Pardo E., Gruss P. Pax2 contributes to inner ear patterning and optic nerve trajectory. Development. 1996;122:3381–3391. doi: 10.1242/dev.122.11.3381. [DOI] [PubMed] [Google Scholar]
- 5.Remington L.A., McGill E.C. Ocular Embryology. In: McGill E.D., editor. Clinical Anatomy of the Visual System. Oxford, UK: Butterworth-Heinemann; 1997. pp. 103–121. [Google Scholar]
- 6.Asai-Coakwell M., French C.R., Berry K.M., Ye M., Koss R., Somerville M., Mueller R., van Heyningen V., Waskiewicz A.J., Lehmann O.J. GDF6, a novel locus for a spectrum of ocular developmental anomalies. Am. J. Hum. Genet. 2007;80:306–315. doi: 10.1086/511280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gregory-Evans C.Y., Vieira H., Dalton R., Adams G.G., Salt A., Gregory-Evans K. Ocular coloboma and high myopia with Hirschsprung disease associated with a novel ZFHX1B missense mutation and trisomy 21. Am. J. Med. Genet. A. 2004a;131:86–90. doi: 10.1002/ajmg.a.30312. [DOI] [PubMed] [Google Scholar]
- 8.Gregory-Evans C.Y., Williams M.H., Halford S., Gregory-Evans K. Ocular coloboma: a reassessment in the age of molecular neuroscience. J. Med. Genet. 2004b;41:881–891. doi: 10.1136/jmg.2004.025494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gregory-Evans C.Y., Moosajee M., Hodges M.D., Mackay D.S., Game L., Vargesson N., Bloch-Zupan A., Rüschendorf F., Santos-Pinto L., Wackens G., et al. SNP genome scanning localizes oto-dental syndrome to chromosome 11q13 and microdeletions at this locus implicate FGF3 in dental and inner-ear disease and FADD in ocular coloboma. Hum. Mol. Genet. 2007;16:2482–2493. doi: 10.1093/hmg/ddm204. [DOI] [PubMed] [Google Scholar]
- 10.London N.J.S., Kessler P., Williams B., Pauer G.J., Hagstrom S.A., Traboulsi E.I. Sequence alterations in RX in patients with microphthalmia, anophthalmia, and coloboma. Mol. Vis. 2009;15:162–167. [PMC free article] [PubMed] [Google Scholar]
- 11.Ragge N.K., Brown A.G., Poloschek C.M., Lorenz B., Henderson R.A., Clarke M.P., Russell-Eggitt I., Fielder A., Gerrelli G., Martinez-Barbera J.P., et al. Heterozygous mutations of OTX2 cause severe ocular malformations. Am. J. Hum. Genet. 2005;76:1008–1022. doi: 10.1086/430721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang P., Liang X., Yi J., Zhang Q. Novel SOX2 mutation associated with ocular coloboma in a Chinese family. Arch. Ophthalmol. 2008;126:709–713. doi: 10.1001/archopht.126.5.709. [DOI] [PubMed] [Google Scholar]
- 13.Ye M., Berry-Wynne K.M., Asai-Coakwell M., Sundaresan P., Footz T., French C.R., Abitbol M., Fleisch V.C., Corbett N., Allison W.T., et al. Mutation of the bone morphogenetic protein GDF3 causes ocular and skeletal anomalies. Hum. Mol. Genet. 2010;19:287–298. doi: 10.1093/hmg/ddp496. [DOI] [PubMed] [Google Scholar]
- 14.Cross S.H., McKie L., West K., Coghill E.L., Favor J., Bhattacharya S., Brown A.D., Jackson I.J. The Opdc missense mutation of Pax2 has a milder than loss-of-function phenotype. Hum. Mol. Genet. 2011;20:223–234. doi: 10.1093/hmg/ddq457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Macdonald R., Scholes J., Strähle U., Brennan C., Holder N., Brand M., Wilson S.W. The Pax protein Noi is required for commissural axon pathway formation in the rostral forebrain. Development. 1997;124:2397–2408. doi: 10.1242/dev.124.12.2397. [DOI] [PubMed] [Google Scholar]
- 16.Lun K., Brand M. A series of no isthmus (noi) alleles of the zebrafish pax2.1 gene reveals multiple signaling events in development of the midbrain-hindbrain boundary. Development. 1998;125:3049–3062. doi: 10.1242/dev.125.16.3049. [DOI] [PubMed] [Google Scholar]
- 17.Chinnaiyan A.M., O'Rourke K., Tewari M., Dixit V.M. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81:505–512. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- 18.Tourneur L., Chiocchia G. FADD: a regulator of life and death. Trends Immunol. 2010;31:260–269. doi: 10.1016/j.it.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 19.Zhang H., Zhou X., McQuade T., Li J., Chan F.K-M., Zhang J. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature. 2011;471:373–376. doi: 10.1038/nature09878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaiser W.J., Upton J.W., Long A.B., Livingston-Rosanoff D., Daley-Bauer L.P., Hakem R., Caspary T., Mocarski E.S. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011;471:368–372. doi: 10.1038/nature09857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Imtiyaz H.Z., Rosenberg S., Rahman Z.S., Hou Y.J., Manser T., Zhang J. The Fas-associated death domain protein is required in apoptosis and TLR-induced proliferative responses in B cells. J. Immunol. 2006;176:6852–6861. doi: 10.4049/jimmunol.176.11.6852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosenberg S., Zhang H., Zhang J. FADD deficiency impairs early hematopoiesis in the bone marrow. J. Immunol. 2011;186:203–213. doi: 10.4049/jimmunol.1000648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yeh W-C., Pompa J.L., McCurrach M.E., Shu H-B., Elia A.J., Shahinian A., Ng M., Wakeham A., Khoo W., Mitchell K., et al. FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science. 1998;279:1954–1958. doi: 10.1126/science.279.5358.1954. [DOI] [PubMed] [Google Scholar]
- 24.Zhang J., Cado D., Chen A., Kabra N.H., Winoto A. Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort1. Nature. 1998;392:296–300. doi: 10.1038/32681. [DOI] [PubMed] [Google Scholar]
- 25.Moosajee M., Gregory-Evans K., Ellis C.D., Seabra M.C., Gregory-Evans C.Y. Translational bypass of nonsense mutations in zebrafish rep1, pax2.1 and lamb1 highlights a viable therapeutic option for untreatable genetic eye disease. Hum. Mol. Genet. 2008;17:3987–4000. doi: 10.1093/hmg/ddn302. [DOI] [PubMed] [Google Scholar]
- 26.Biehlmaier O., Makhankov Y., Neuhauss S.C. Impaired retinal differentiation and maintenance in zebrafish laminin mutants. Invest. Ophthalmol. Vis. Sci. 2007;48:2887–2894. doi: 10.1167/iovs.06-1212. [DOI] [PubMed] [Google Scholar]
- 27.Parsons M.J., Pollard S.M., Saude L., Feldman B., Coutinho P., Hirst E.M., Stemple D.L. Zebrafish mutants identify an essential role for laminins in notochord formation. Development. 2002;129:3137–3146. doi: 10.1242/dev.129.13.3137. [DOI] [PubMed] [Google Scholar]
- 28.Czery T., Schaffner G., Busslinger M. DNA sequence recognition by Pax proteins: bipartite structure of the paired domain and its binding site. Genes Dev. 1993;7:2048–2061. doi: 10.1101/gad.7.10.2048. [DOI] [PubMed] [Google Scholar]
- 29.Epstein J., Cai J., Glaser T., Jepeal L., Maas R. Identification of a Pax paired domain recognition sequence and evidence for DNA-dependent conformational changes. J. Biol. Chem. 1994;269:8355–8361. [PubMed] [Google Scholar]
- 30.Bajoghli B., Ramialison M., Aghaallaei N., Czerny T., Wittbrodt J. Identification of starmaker-like in medaka as a putative target gene of Pax2 in the otic vesicle. Dev. Dyn. 2009;238:2860–2866. doi: 10.1002/dvdy.22093. [DOI] [PubMed] [Google Scholar]
- 31.Barbieri A.M., Lupo G., Bulfone A., Andreazzoli M., Mariani M., Fougerousse F., Consalez G.G., Borsani G., Beckmann J.S., Barsacchi G., et al. A homeobox gene, vax2, controls the patterning of the eye dorsoventral axis. Proc. Natl Acad. Sci. USA. 1999;96:10729–10734. doi: 10.1073/pnas.96.19.10729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mascrez B., Ghyselinck N.B., Chambon P., Mark M. A transcriptionally silent RXRalpha supports early embryonic morphogenesis and heart development. Proc. Natl Acad. Sci. USA. 2009;106:4272–4277. doi: 10.1073/pnas.0813143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nasevicius A., Ekker S.C. Effective targeted gene ‘knockdown’ in zebrafish. Nat. Genet. 2000;26:216–220. doi: 10.1038/79951. [DOI] [PubMed] [Google Scholar]
- 34.Galluzzi L., Kepp O., Kroemer G. FADD: an endogenous inhibitor of RIP-3 driven regulated necrosis. Cell Res. 2011;21:1383–1385. doi: 10.1038/cr.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krumschnabel G., Ebner H.L., Hess M.W., Villunger A. Apoptosis and necroptosis are induced in rainbow trout cell lines exposed to cadmium. Aquat. Toxicol. 2010;99:73–85. doi: 10.1016/j.aquatox.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 36.Sanyanusin P., Schimmenti L.A., McNoe L.A., Ward T.A., Pierpont M.E., Sullivan M.J., Dobyns W.B., Eccles M.R. Mutation of the PAX2 gene in a family with optic nerve colobomas, renal anomalies and vesicoureteral reflux. Nat. Genet. 1995;9:358–364. doi: 10.1038/ng0495-358. [DOI] [PubMed] [Google Scholar]
- 37.Tamimi Y., Ekuere U., Laughton N., Grundy P. WNT5A is regulated by PAX2 and may be involved in blastemal predominant Wilms tumorigenesis. Neoplasia. 2008;10:1470–1480. doi: 10.1593/neo.08442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dziarmaga A., Hueber P.A., Iglesias D., Hache N., Jeffs A., Gendron N., Mackenzie A., Eccles M., Goodyer P. Neuronal apoptosis inhibitory protein is expressed in developing kidney and is regulated by PAX2. Am. J. Physiol. Renal. Physiol. 2006;291:F913–F920. doi: 10.1152/ajprenal.00004.2006. [DOI] [PubMed] [Google Scholar]
- 39.Bouchard M., Grote D., Craven S.E., Sun Q., Steinlein P., Busslinger M. Identification of Pax2-regulated genes by expression profiling of the mid-hindbrain organizer region. Development. 2005;132:2633–2643. doi: 10.1242/dev.01833. [DOI] [PubMed] [Google Scholar]
- 40.Welz P-S., Wullaert A., Vlantis K., Kondylis V., Fernández-Majada V., Ermolaeva M., Kirsh P., Sterner-Kock A., van Loo G., Pasparakis M. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature. 2011;477:300–334. doi: 10.1038/nature10273. [DOI] [PubMed] [Google Scholar]
- 41.Günther C., Martini E., Wittkopf N., Amann K., Weigmann B., Neumann H., Waldner M.J., Hedrick S.M., Neurath M.F., Becker C. Caspase-8 regulates TNF-α-induced epithelial necroptosis and terminal ileitis. Nature. 2011;477:335–339. doi: 10.1038/nature10400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Masai I., Lele Z., Yamaguchi M., Komori A., Nishiwaki Y., Wada H., Tanaka H., Nojima Y., Hammerschmidt M., Wilson S.W., et al. N-cadherin mediates retinal lamination, maintenance of forebrain compartments and patterning of retinal neuritis. Development. 2003;130:2479–2494. doi: 10.1242/dev.00465. [DOI] [PubMed] [Google Scholar]
- 43.Yamaguchi M., Imai F., Tonou-Fujimori N., Masai I. Mutations in N-cadherin and a Stardust homolog, Nagie oko, affect cell-cycle exit in zebrafish retina. Mech. Dev. 2010;127:247–264. doi: 10.1016/j.mod.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 44.Take-uchi M., Clarke J.D., Wilson S.W. Hedgehog signalling maintains the optic stalk-retinal interface through the regulation of Vax gene activity. Development. 2003;130:955–968. doi: 10.1242/dev.00305. [DOI] [PubMed] [Google Scholar]
- 45.Hallonet M., Hollemann T., Pieler T., Gruss P. Vax1, a novel homeobox-containing gene, directs development of the basal forebrain and visual system. Genes Dev. 1999;13:3106–3114. doi: 10.1101/gad.13.23.3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gestri G., Osborne R.J., Wyatt A.W., Gerrelli D., Gribble S., Stewart H., Fryer A., Bunyan D.J., Prescott K., Collin J.R., et al. Reduced TFAP2A function causes variable optic fissure closure and retinal defects and sensitizes eye development to mutations in other morphogenetic regulators. Hum. Genet. 2009;126:791–803. doi: 10.1007/s00439-009-0730-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Degterev A., Hitomi J., Germscheid M., Ch'en I.L., Korkina O., Teng X., Abbott D., Cuny G.D., Yuan C., Wagner G., et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gregory-Evans C.Y., Moosajee M., Shan X., Gregory-Evans K. Gene-specific differential response to anti-apoptotic therapies in zebrafish models of ocular coloboma. Mol. Vis. 2011;17:1473–1484. [PMC free article] [PubMed] [Google Scholar]
- 49.Feng S., Yang Y., Mei Y., Ma L., Zhu D.E., Hoti N., Castanares M., Wu M. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal. 2007;19:2056–2067. doi: 10.1016/j.cellsig.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 50.Lin Y., Devin A., Rodriguez Y., Liu Z.G. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 1999;1:2514–2526. doi: 10.1101/gad.13.19.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu X., Chua K.W., Chua C.C., Liu C.F., Hamdy R.C., Chua B.H. Synergistic protective effects of humanin and necrostatin-1 on hypoxia and ischaemia/reperfusion injury. Brain Res. 2010;1355:189–194. doi: 10.1016/j.brainres.2010.07.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rosenbaum D.M., Degterev A., David J., Rosenbaum P.S., Roth S., Grotta J.C., Cuny G.D., Yuan J., Savitz S.I. Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J. Neurosci. Res. 2010;88:1569–1576. doi: 10.1002/jnr.22314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li Y., Yang X., Ma C., Qiao J., Zhang C. Necroptosis contributes to the NMDA-induced excitotoxicity in rat's cultured cortical neurons. Neurosci. Lett. 2008;447:120–123. doi: 10.1016/j.neulet.2008.08.037. [DOI] [PubMed] [Google Scholar]
- 54.Smith C.C., Davidson S.M., Lim S.Y., Simpkin J.C., Hothersall J.S., Yellon D.M. Necrostatin: a potentially novel cardioprotective agent? Cardiovasc. Drugs Ther. 2007;21:227–233. doi: 10.1007/s10557-007-6035-1. [DOI] [PubMed] [Google Scholar]
- 55.Weil M., Jacobson M.D., Raff M.C. Is programmed cell death required for neural tube closure? Curr. Biol. 1997;7:281–284. doi: 10.1016/s0960-9822(06)00125-4. [DOI] [PubMed] [Google Scholar]
- 56.Mirkes P.E. Warkany lecture: to die or not to doe, the role of apoptosis in normal and abnormal mammalian development. Teratology. 2002;65:228–239. doi: 10.1002/tera.10049. [DOI] [PubMed] [Google Scholar]
- 57.Massa V., Savery D., Ybot-Gonzalez P., Ferraro E., Rongvaux A., Cecconi F., Flavell R., Green N.D., Copp A.J. Apoptosis is not required for mammalian neural tube closure. Proc. Natl Acad. Sci. USA. 2009;106:8233–8238. doi: 10.1073/pnas.0900333106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fukuda T., Tokunaga A., Sakamoto R., Yoshida N. Fbxl10/Kdm2b deficiency accelerates neural progenitor cell death and leads to exencephaly. Mol. Cell Neurosci. 2011;46:614–624. doi: 10.1016/j.mcn.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 59.Delic J., Coppey J., Magdelenat H., Coppey-Moisan M. Impossibility of acridine orange intercalation in nuclear DNA of the living cell. Exp. Cell Res. 1991;194:147–153. doi: 10.1016/0014-4827(91)90144-j. [DOI] [PubMed] [Google Scholar]
- 60.Grasl-Kraupp B., Ruttkay-Nedecky B., Koudelka H., Bukowska K., Bursch W., Schulte-Hermann R. In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology. 1995;21:1465–1468. doi: 10.1002/hep.1840210534. [DOI] [PubMed] [Google Scholar]
- 61.Zhang D-W., Shao J., Lin J., Zhang N., Lu B-J., Lin S-C., Dong M-Q., Han J. RIP3, an energy metabolism regulator that stiches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 62.Lin Y., Choksi S., Shen H.M., Yang Q.F., Hur G.M., Kim Y.S., Tran J.H., Nedospasov S.A., Liu Z.G. Tumor necrosis factor-induced nonapoptotic cell death requires receptor-interacting protein-mediated cellular reactive oxygen species accumulation. J. Biol. Chem. 2004;279:10822–10828. doi: 10.1074/jbc.M313141200. [DOI] [PubMed] [Google Scholar]
- 63.Cho Y.S., Challa S., Moquin D., Genga R., Ray T.D., Guildford M., Chan F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.De Valck D., Vercammen D., Fiers W., Beyaert R. Differential activation of phospholipases during necrosis or apoptosis: a comparative study using tumor necrosis factor and anti-Fas antibodies. J. Cell Biochem. 1998;71:392–399. [PubMed] [Google Scholar]
- 65.Berghe T.V., Vanlangenakker N., Parthoens E., Deckers W., Devos M., Festjens N., Guerin C.J., Brunk U.T., Declercq W., Vandenabeele P. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2010;17:922–930. doi: 10.1038/cdd.2009.184. [DOI] [PubMed] [Google Scholar]
- 66.Ch'en I.L., Tsau J.S., Molkentin J.D., Komatsu M., Hedrick S.M. Mechanisms of necroptosis in T cells. J. Exp. Med. 2011;208:633–641. doi: 10.1084/jem.20110251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Johnson S.L., Midson C.N., Ballinger E.W., Postlethwait J.H. Identification of RAPD primers that reveal extensive polymorphisms between laboratory strains of zebrafish. Genomics. 1994;19:152–156. doi: 10.1006/geno.1994.1026. [DOI] [PubMed] [Google Scholar]
- 68.Stemple D.L., Solnica-Krezel L., Zwartkruis F., Neuhauss S.C., Schier A.F., Malicki J., Stainier D.Y., Abdelilah S., Rangini Z., Mountcastle-Shah E., et al. Mutations affecting development of the notochord in zebrafish. Development. 1996;123:117–128. doi: 10.1242/dev.123.1.117. [DOI] [PubMed] [Google Scholar]
- 69.Brand M., Heisenberg C.P., Jiang Y.J., Beuchle D., Lun K., Furutani-Seiki M., Granato M., Haffter P., Hammerschmidt M., Kane D.A., et al. Mutations in zebrafish genes affecting the formation of the boundary between midbrain and hindbrain. Development. 1996;123:179–190. doi: 10.1242/dev.123.1.179. [DOI] [PubMed] [Google Scholar]
- 70.Maddison L.A., Lu J., Victoroff T., Scott E., Baier H., Chen W. A gain-of-function screen in zebrafish identifies a guanylate cyclase with a role in neuronal degeneration. Mol. Genet. Genomics. 2009;281:551–563. doi: 10.1007/s00438-009-0428-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kimmel C.B., Ballard W.W., Kimmel S.R., Ullmann B., Schilling T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- 72.Truett G.E., Heeger P., Mynatt R.L., Truett A.A., Walker J.A., Warman M.L. Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT) Biotechniques. 2000;29:552–554. doi: 10.2144/00291bm09. [DOI] [PubMed] [Google Scholar]
- 73.Hodges M.D., Vieira H., Gregory-Evans K., Gregory-Evans C.Y. Characterisation of the genomic and transcriptional structure of the CRX gene: substantial differences between human and mouse. Genomics. 2002;80:531–542. doi: 10.1006/geno.2002.6854. [DOI] [PubMed] [Google Scholar]
- 74.Lindeman L.C., Vogt-Kielland L.T., Aleström P., Collas P. Fish'n ChIPs: chromatin immunoprecipitation in the zebrafish embryo. Methods Mol. Biol. 2009;567:75–86. doi: 10.1007/978-1-60327-414-2_5. [DOI] [PubMed] [Google Scholar]
- 75.Moro E., Gnügge L., Braghetta P., Bortolussi M., Argenton F. Analysis of beta cell proliferation dynamics in zebrafish. Dev. Biol. 2009;332:299–308. doi: 10.1016/j.ydbio.2009.05.576. [DOI] [PubMed] [Google Scholar]
- 76.Veth K.N., Willer J.R., Collery R.F., Gray M.P., Willer G.B., Wagner D.S., Mullins M.C., Udvadia A.J., Smith R.S., John S.W., et al. Mutations in zebrafish lrp2 result in adult-onset ocular pathogenesis that models myopia and other risk factors for glaucoma. PLoS Genet. 2011;7:e1001310. doi: 10.1371/journal.pgen.1001310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang W.L., Hong J.R., Lin G.H., Liu W., Gong H.Y., Lu M.W., Lin C.C., Wu J.L. Stage-specific expression of TNFα regulates Bad/Bid-mediated apoptosis and RIP1/ROS-mediated secondary necrosis in birnavirus-infected fish cells. PLoS ONE. 2011;6:e16740. doi: 10.1371/journal.pone.0016740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Duggan C.D., DeMaria S., Baudhuin A., Stafford D., Ngai J. Foxg1 is required for development of the vertebrate olfactory system. J. Neurosci. 2008;28:5229–5239. doi: 10.1523/JNEUROSCI.1134-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Robu M.E., Larson J.D., Nasevicius A., Beiraghi S., Brenner C., Farber S.A., Ekker S.C. p53 activation by knockdown technologies. PLoS Genet. 2007;3:e78. doi: 10.1371/journal.pgen.0030078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yabu T., Kishi S., Okazaki T., Yamashita M. Characterization of zebrafish caspase-3 and induction of apoptosis through ceramide generation in fish fathead minnow tailbud cells and zebrafish embryo. Biochem. J. 2001;360:39–47. doi: 10.1042/0264-6021:3600039. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.