

Abstract
Altered expression of the INT6 gene encoding the e subunit of the translational initiation factor eIF3 occurs in human breast cancers, but how INT6 relates to carcinogenesis remains unestablished. Here we show that INT6 is involved in the DNA damage response. INT6 was required for cell survival following γ-irradiation and G2/M checkpoint control. RNAi-mediated silencing of INT6 reduced phosphorylation of the checkpoint kinases CHK1 and CHK2 after DNA damage. Additionally, INT6 silencing prevented sustained accumulation of ATM at DNA damage sites in cells treated with γ-radiation or the radiomimetic drug neocarzinostatin. Mechanistically, this result could be explained by interaction of INT6 with ATM, which together with INT6 was recruited to sites of DNA damage. Lastly, INT6 silencing also reduced ubiquitylation events that promote retention of repair proteins at DNA lesions. Accordingly, accumulation of the repair factor BRCA1 was defective in the absence of INT6. Our findings reveal unexpected and striking connections of INT6 with ATM and BRCA1 and suggest that the protective action of INT6 in the onset of breast cancers relies on its involvement in the DNA damage response.
Keywords: ATM, BRCA1, breast cancer, DNA damage, INT6-EIF3E
Introduction
In response to DNA damage, eukaryotic cells initiate a complex signaling pathway called DNA damage response (DDR) (1, 2). At the core of DDR is the kinase ataxia-telangiectasia mutated (ATM) (3). As part of its activation process, ATM undergoes autophosphorylation, re-localizes rapidly to sites of DNA double-strand breaks (DSBs) through its association with the MRE11-RAD50-NBS1 (MRN) complex (4, 5), and phosphorylates numerous substrates including histone H2AX. Chromatin marked with phosphorylated H2AX (referred to as γ-H2AX) establishes a chromatin domain onto which DDR proteins accumulate, among them ATM, MRN components, and MDC1. MDC1 phosphorylated by ATM then recruits the RNF8 ubiquitin ligase, which in turn catalyzes local ubiquitylation of H2A-type histones, thereby facilitating accumulation of repair complexes, including BRCA1 (6–8).
Most of the known breast cancer susceptibility genes reported to date code for proteins involved in maintaining genome stability by engaging in DDR pathway. These genes include the prototypic BRCA1 and BRCA2 genes, of which inherited mutations were found to confer a ~15-fold increased risk of breast cancer (9). ATM mutations confer a moderate risk (doubling) of breast cancer in relative heterozygous carriers; however the risk is elevated for certain missense variants of ATM (T7271G and L1420F) since these alleles are sufficiently penetrant to generate multiple-case breast cancer families (10–13). Inherited mutations in the gene encoding checkpoint kinase 2 (CHK2), a major signal transducer of DDR, are associated with a ~2-fold increased risk of breast cancer incidence (14, 15). The three MRE11, RAD50, and NBS1 genes are all considered as hereditary breast cancer susceptibility genes as well (16–18). However, ~70% of familial breast cancers remain unexplained by currently known predisposition genes, suggestive of existence of other breast cancer susceptibility genes.
The integration site 6 (INT6) gene, also known as EIF3E, encodes one of the thirteen subunits of human eukaryotic translation initiation factor 3 (eIF3) (19). Several lines of evidence indicate that this gene is critical for preventing breast carcinogenesis in mice and humans. Regarding this matter, various roles were proposed for INT6 either as an oncoprotein (20–22) or a tumor suppressor (23–25). In this study, we have discovered that INT6 is involved in the DDR pathway. Specifically, silencing INT6 decreases cell survival after exposure to γ-irradiation and impairs the G2 DNA damage checkpoint. A fraction of INT6 localizes rapidly to DSB sites and interacts with ATM. Moreover, the ability of ATM to remain at breaks is reduced in INT6-depleted cells. As a consequence, several components operating at different levels of DDR are not recruited efficiently to DSB sites, including the repair factor BRCA1. Since the bulk of breast cancer susceptibility genes discovered to date encode proteins involved in DDR, our results establishing a role for INT6 in this pathway pave the way for large epidemiological and molecular studies to assess the potential clinical outcome of INT6 defects in breast cancers.
Materials and Methods
Cell culture and transfection
HeLa and 293T cells were obtained from ECACC and U2OS cells from ATCC. They were repeatedly screened for mycoplasma, and were negative. HeLa cells were possessed for less than 6 months. Cells were maintained in DMEM containing 10% fetal calf serum. 293T cells were transfected using standard calcium phosphate method. HeLa and U2OS cells were transfected with siRNAs and plasmids using Lipofectamine 2000 (Invitrogen). Unless mentioned, all assays were performed 72 h after transfection. Control and INT6-specific siRNAs (I6.1, I6.3) have been described previously (26). The FLAG-ATM construct (27, 28) was obtained from M. Kastan.
Immunoprecipitations and immunoblots
Transfected 293T cells were lysed in Nonidet P-40-desoxycholate buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1% Nonidet P-40, 0.5% Na desoxycholate, 0.5 mM Tris(2-carboxyethyl)phosphine, and 0.5 mM Pefabloc). For the interaction between endogenous proteins, HeLa cells were lysed in 50 mM Tris pH 7.4, 120 mM NaCl, 0.5% Nonidet P-40, 1 mM EDTA, 50 mM NaF, 1 mM Na3VO4, and protease inhibitors (Roche). Extracts were incubated for 2 h at 4°C with antibodies, then with Protein A agarose for 1 h, washed three times for 10 min before resuspension in 2X SDS sample buffer. Cell extracts used only for immunoblotting were prepared in Laemmli sample buffer. Proteins were separated on SDS-polyacrylamide gels and transferred to PVDF membranes. Blots were developed using ECL or ECL plus reagents (GE Healthcare) or were analyzed with the Odyssey Infrared Imager (LI-COR Biosciences).
In vitro binding assays
In vitro protein interaction between GST-ATM fusion proteins and INT6 were performed as described (29). In brief, extracts from cells untreated orγ-irradiated were mixed with glutathione agarose beads containing GST-ATM fusion proteins. Bound proteins were analyzed by immunoblotting with anti-INT6 antibody and levels of GST-ATM fragments were detected by coomassie staining.
Immunofluorescence and confocal microscopy
Cells were fixed in 4% paraformaldehyde for 10 min, incubated in 100 mM glycine for 10 min, permeabilized with 0.5% Triton X-100 for 5 min, and blocked with 1% BSA for 30 min. Primary antibodies were incubated for 2 h at room temperature or overnight at 4°C and secondary antibodies conjugated with Alexa Fluor 488 or Alexa Fluor 546 (Invitrogen) were incubated for 1 h. Slides were mounted in medium containing Mowiol and observed with a LSM 510 confocal microscope (Carl Zeiss MicroImaging, Inc) mounted on an Axioplan2 (Carl Zeiss MicroImaging, Inc) equipped with a Plan-Apochromat 63x/1.4 NA oil-immersion objective. Acquisitions were performed under constant settings. Co-localization was evaluated by visual inspection of signal overlap on merged images or by using the Co-localization Highlighter plug-in of ImageJ software (National Institutes of Health). Threshold settings were automatically set with the threshold tool and assigned to the input window ofthe Co-localization Highlighter plug-in. The ratio of intensity was set at 50%. Two points are considered as co-localized if their respective intensities are higher than the threshold of their channels, and if their ratio of intensity is higher than 50%.
Live-cell imaging combined with laser micro-irradiation
U2OS cells were micro-irradiated with a pulsed nitrogen laser (365 nm, 10Hz; Spectra-Physics) with output set at 80% of the maximum, as described (30). For live-cell imaging, cells were transfected with a GFP-ATM construct, laser micro-irradiated, time-lapse imaged, and fluorescence intensities of micro-irradiated areas relative to non-irradiated areas calculated as described previously (31).
Antibodies
Antibodies to INT6 (C-169 for immunofluorescence and C-20 for immunoprecipitation and immunoblotting) have been described previously (32) as well as those directed against EIF3D and EIF3L (33). Commercial antibodies are listed in Supplemental Materials.
Results
INT6 is essential for cell survival and G2/M checkpoint following γ-irradiation
To understand how altered expression of INT6 prevents breast cancer onset, we explored if INT6 is involved in the activation of DNA damage signaling pathway. First, the sensitivity of INT6-silenced cells to different doses of γ-irradiation (IR) was assessed using a MTT-based assay. Compared with control siRNA-treated cells, INT6-depleted cells showed a decreased survival rate after IR with a dose-response curve similar to that obtained for ATM-silenced cells (Fig. 1A). Next, we examined whether INT6 was involved in the G2/M DNA damage checkpoint. Flow cytometry analyses using DNA content and phosphorylated-histone H3 to distinguish between G2 and mitotic cells were performed. Control cells showed a clear G2/M block, as evidenced by an 8-fold decrease in the percentage of mitotic cells after IR (Fig. 1B). In contrast, INT6-deficient cells displayed a less robust G2/M block since γ-radiation reduced the percentage of M-phase cells by only 2 to 3-fold. Together, these results indicate that INT6 is critical for proper DNA damage signaling.
Figure 1.
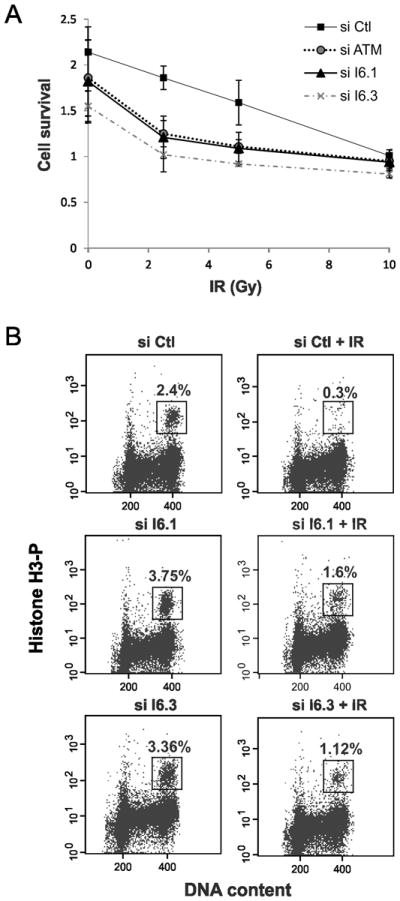
INT6 is required for cell survival and G2/M checkpoint following γ-irradiation. (A) HeLa cells were transfected with control siRNAs, ATM siRNAs or two siRNAs targeting distinct regions of the INT6 mRNA. Fourty hours later, cells were exposed with increasing doses of γ-radiation and allowed to grow for 72 h before cell proliferation was measured by a MTT-based assay. Shown are the averages of triplicate samples. Error bars correspond to standard errors. (B) HeLa cells transfected for 60 h with siRNAs control or targeting INT6 were untreated or γ-irradiated (6 Gy) and harvested 2h later. Cells were labeled with an antibody to phospho-histone H3 and propidium iodide. Percentages of mitotic cells (boxed area) were determined by FACS.
INT6 is required for sustained accumulation of ATM to DNA damage sites
To further understand the involvement of INT6 in DDR, we investigated whether the ATM kinase, essential for cellular signaling in response to DNA damage, was normally activated upon INT6 deficiency. First, the phosphorylation status of its substrate CHK2 was examined. As expected, irradiation of control siRNA-treated cells led to a rapid and optimal phosphorylation of CHK2 within 30 min after irradiation that significantly decreased by 2 h (Fig. 2A). By contrast INT6-depleted cells failed to show similar levels of increase at 30 min or at a later time point. This defect in CHK2 phosphorylation was not caused by a reduction in CHK2 protein levels (Fig. 2A). Phosphorylation of the checkpoint kinase CHK1 was also reduced in these cells (Fig. 2A). Activation of these two kinases was similarly impaired in cells depleted of INT6 by means of a short hairpin RNA (shRNA) and treated with the radiomimetic drug neocarzinostatin (NCS) (Fig. 2B). The rescue of INT6 expression using an INT6 cDNA resistant to degradation by shRNA restored the levels of CHK2- and CHK1-phosphorylation in these cells to control levels (Fig. 2B). Of note, the kinetics of phosphorylation of the two kinases is different; CHK2 showed maximal phosphorylation within 30 min and CHK1 at 2h. Next, ATM activation was assessed by immunoblot using an antibody to Ser1981-autophosphorylated ATM. The ATM active form was detected after, but not before γ-irradiation and silencing INT6 caused a slight decrease in the levels of phosphorylated ATM (Fig. 3A). The impact of INT6 depletion on ATM activation was further studied by immunofluorescence analyses. As expected, γ-irradiation of control cells induced local accumulation of active ATM in nuclear foci (Fig. 3B). Formation of these ATM nuclear foci was impaired in INT6-silenced cells. A signal was also visible in the cytoplasm of these cells. However, specificity of this staining is questionable as it was also observed in non-irradiated cells (Fig. 3B), which showed no signal for phosphorylated ATM by immunoblotting (Fig. 3A). Specificity of the nuclear staining was established by its disappearance when cells were transfected with siRNAs against ATM (Fig. S1A). Formation of ATM foci was also assessed in cells treated with NCS. Again, silencing INT6 impaired the accumulation of ATM to γ-H2AX foci (Fig. S1B). To rule out any effect specific to HeLa cells, localization of ATM was studied in immortalized human fibroblasts treated with NCS and again ATM was not properly retained at DSBs in the absence of INT6 (Fig. S1C). To elucidate this defect, real-time recruitment of ATM tagged with GFP was monitored by live-cell imaging combined with laser micro-irradiation as previously described (30, 31). U2OS cells expressing GFP-ATM were transfected with control or INT6-targeting siRNAs and a small area of the nucleus was laser micro-irradiated to generate DSBs. Time-lapse imaging of control cells showed that GFP-ATM, which was nuclear-diffuse before irradiation, became rapidly recruited to DSBs (within 1 min) and accumulated progressively before reaching a plateau at ~20 min (Fig. 3C–D). In INT6-silenced cells, recruitment of GFP-ATM to DSBs was weaker and reached a plateau at ~5 min after micro-irradiation (Fig. 3C–D). Similarly, recruitment of endogenous phospho-ATM along the laser track was impaired following INT6 knock-down (Fig. S1D). In line with these data, biochemical fractionation of HeLa cells exposed to NCS showed that loss of INT6 led to decreased levels of chromatin-bound ATM (Fig. 3E). Of note, no phospho-ATM signal was detected in cytoplasmic fractions of INT6-depleted cells, thus arguing against localization of the ATM active form in the cytoplasm. Since MDC1 is necessary for ATM retention at DSBs (34), kinetics of MDC1 recruitment to laser tracks was analyzed and was similar with or without INT6 knock-down (Fig. S2), indicating that the defective accumulation of ATM in the absence of INT6 was not due to an impaired localization of MDC1 at DSBs.
Figure 2.

INT6 knock-down impairs activation of CHK2 and CHK1. (A) HeLa cells transfected with siRNAs control or targeting INT6 were untreated or γ-irradiated (6 Gy) and collected after 0.5 h or 2 h. Immunoblots were performed using antibodies to CHK2 and CHK1 unphosphorylated or phosphorylated on Thr68 and Ser345, respectively. RNAi efficacy was controlled by detection of INT6. (B) Hela cells were transfected with the parental vector (pCEP-FGFP), the vector containing the INT6 shRNA (pCEP-SUPI6-FGFP), or the one containing the INT6 shRNA plus an shRNA-resistant INT6 cDNA (pCEP-SUPI6-I6M5). Plasmid description is in Supplemental Materials. Cells were treated with 200 ng/ml NCS for 30 min and collected at indicated time points. Immunoblots were performed with phospho-specific antibodies to CHK2 and CHK1. Efficiency of INT6-depletion and re-expression and equal protein loading were controlled by detection of INT6 and β-actin, respectively.
Figure 3.

Silencing INT6 impairs sustained accumulation of ATM to DNA damage sites. (A) HeLa cells transfected with siRNAs were exposed, or not, to γ-irradiation (10 Gy) and collected after 1 h. Immunoblots were performed using antibodies to unphosphorylated or Ser1981-phosphorylated ATM. RNAi efficacy and protein loading were controlled as in Fig. 2B. (B) HeLa cells transfected with control or INT6-targeting siRNAs were untreated or γ-irradiated (10 Gy). Cells were immunostained 1 h later with an antibody to Ser1981-phosphorylated ATM. Representative confocal images are shown. Scale bar, 10μM. (C) Accumulation of GFP-ATM at laser-induced DSBs was monitored by time-lapse imaging in U2OS cells transfected with control or INT6-targeting siRNAs. White arrows on left images indicate micro-irradiated areas. Scale bar, 10 μM. (D) Kinetics of GFP-ATM relative accumulation at laser-induced DSBs in control and INT6-depleted cells. Mean value of fluorescence intensities for each time point was calculated from at least 30 independent measurements. Error bars represent standard deviations. (E) Biochemical fractionation of HeLa cells transfected with control and INT6-specific siRNAs. Cells were treated with NCS (200 ng/ml, 15 min) and fractionated 3 h later as described in Supplemental Methods. Aliquots of each fraction corresponding to an equal cell number were used for immunoblots using antibodies to ATM, either total or autophosphorylated. Successful fractionation was verified through detection of RRM2 in the cytoplasm and a modified form of histone H3 on chromatin. RNAi efficiency was checked by detecting INT6.
INT6 localizes to sites of DSBs generated by NCS treatment or laser micro-irradiation
In order to elucidate how INT6 might regulate ATM function, we first examined whether INT6 could be localized at DSB sites. In the vast majority of mock-treated cells, no γ-H2AX foci was detected and INT6 was dispersed in the cytoplasm and nucleus (Fig. 4A). Of note, INT6 staining seems only nuclear because the cytoplasm did not lie in this confocal plane. In NCS-treated cells, a substantial fraction of the nuclear pool of INT6 was concentrated into foci co-localized with that of γ-H2AX. Specificity of the INT6 signal was established by its disappearance in cells transfected with a siRNA targeting INT6. Next, kinetics of INT6 recruitment to DSBs was monitored by time-lapse microscopy of cells subjected to micro-irradiation. Localization of INT6 along the laser track was detected within 5 min after micro-irradiation and kinetics of INT6 accumulation was similar to that of γ-H2AX (Fig. 4B). The INT6 signal at laser-induced DSBs was significantly attenuated upon INT6 depletion (Fig. S3A–B). We also found that ATM activity was not required for recruitment of INT6 at DSBs as INT6 was localized to laser tracks normally in cells treated with the ATM inhibitor KU55933 (Fig. 4C and Fig. S3C) or in ATM-deficient cells (Fig. 4D). Together, these findings suggest that INT6 is directly involved in DDR at DSB sites and that it is recruited independently of ATM.
Figure 4.

INT6 is recruited to DSBs induced by NCS treatment or micro-irradiation. (A) HeLa cells transfected with control or INT6-targeting siRNAs were treated, or not, with NCS (200 ng/ml, 15 min), and immunostained 3 h later with antibodies against INT6 and γ-H2AX. Representative confocal images are shown. Co-localized pixels were determined using the Co-localization Highlighter plug-in for ImageJ software. They appear as white dots on right panels. Scale bar, 10 μM. (B and C) U2OS cells were untreated (B) or pre-treated with 5 μM KU55933 (C), micro-irradiated, fixed at the indicated time points, immunostained with antibodies to INT6 and γ-H2AX, and counterstained with DAPI. The merged red and green channels show co-localization in yellow. Bar, 10 μM. (D) Recruitment of INT6 was assessed in ATM-deficient cells (AT5) processed as above.
INT6 interacts physically with ATM
The above results together with our observation that INT6 co-localized with ATM at DNA damage sites (Fig. S4) suggested that a fraction of INT6 might interact physically with ATM. This was tested by immunoprecipitation assays. Co-purification of exogenous FLAG-tagged ATM was detected with INT6, either ectopically-expressed or endogenous (Fig. 5A). This result was confirmed in reciprocal immunoprecipitation experiment (Fig. 5B). Of note, the extent of INT6 overexpression in 293T cells was rather weak. We next investigated whether endogenous ATM could also interact with INT6 in response to DNA damage. The kinase was indeed detected in INT6 immunoprecipitates and the extent of ATM recovery was comparable with or without prior exposure of cells to NCS (Fig. 5C). It should be added that although the signal for co-precipitated ATM was weak, this result was reproducible. Consistent with that, a recent study reported that the ATM average copy number per cell is ~100-fold lower than that of INT6 (35).
Figure 5.

INT6 interacts physically with ATM. (A) Association of ectopically-expressed ATM and INT6. 293T cells were transiently transfected with vectors expressing FLAG-ATM and INT6. Cell extracts were blotted directly (lanes 1–3) or after immunoprecipitation with pre-immune serum (lane 7) or an antibody to INT6 (lanes 4–6). An antibody against FLAG was used to detect FLAG-ATM. (B) Reverse pull-down. Extracts as above were immunoprecipitated using an antibody to FLAG (lanes 4–6) or a control antibody (lane 7) and immunoblotted with an antibody against INT6. (C) Association of the endogenous proteins. HeLa cells were non-treated (lane 1) or treated with NCS (200 ng/ml, 30 min) and collected after 1 h (lane 2). Whole cell lysates were immunoprecipitated with pre-immune serum (lanes 3, 5) or an antibody to INT6 (lanes 4, 6) and immunoblotted using an antibody against ATM. Recovery of INT6 is shown on bottom panel. Uncropped images of blots showing the co-precipitation of ATM and INT6 are in Fig. S7. (D) Mapping of interacting regions between ATM and INT6. A schematic representation of ATM is shown at top of the panel. Structural domains within ATM include the FRAP/ATM/TRRAP (FAT) domain, the kinase domain (PI3K), and the FAT c-terminal (FAT-C) domain. Various GST-ATM fusion proteins that cover the full ATM protein sequence were incubated with lysates from 293T cells untreated or γ-irradiated (6 Gy) and bound proteins were analyzed by immunoblotting with an antibody to INT6. Purification of GST-ATM proteins on glutathione agarose beads was controlled on a gel stained with coomassie blue.
Next, we sought to define the region of the kinase involved in binding INT6 using in vitro assays. INT6 was found to bind strongly to GST-ATM (residues 1764–2138) and GST-ATM (residues 2842–3056) but not the other fusion proteins and binding of INT6 did not respond to DNA damage (Fig. 5D). These two ATM fragments correspond to separate regions, one upstream of the FAT domain and the second encompassing the FAT-C domain.
INT6 activity in DDR does not involve translation
A striking aspect of our findings is that one subunit of the eIF3 translation initiation factor can interact with one crucial kinase of DDR. In this regard, the bulk of eIF3 and translation machinery are mainly cytoplasmic while INT6 shuttles between the cytoplasm and the nucleus in human cells (36). We therefore tested the possibility that INT6 might be released from eIF3 during conditions of DNA damage. Co-purification with INT6 of five other eIF3 subunits was examined and their recovery was similar with or without prior exposure of cells to NCS (Fig. 6A). Additionally, cytoplasmic INT6 levels associated with the EIF3B subunit were comparable with or without NCS treatment (Fig. 6B), thus suggesting that the fraction of INT6 associated with eIF3 is not affected by DNA damage. Next, to determine if INT6 effect on DDR was related to a translational activity, we first tested the ability of ATM to localize at DSBs after inhibition of translation using cycloheximide. Immunofluorescences showed that ATM was normally recruited to NCS-induced DSBs (Fig. 6C). Moreover, silencing INT6 had no significant effect on the separation of ribosomal components and polysomes after DNA damage (Fig. 6D). Together, these findings suggest that the altered functioning of ATM following INT6 suppression is likely not due to an INT6-mediated translational effect. As two recent studies on the impact of INT6 depletion on protein synthesis in human cells pointed that it is not required for global translation but for regulating translation of specific mRNAs (25, 37), we studied whether INT6 knock-down could interfere with translation of mRNAs encoding DDR proteins and ubiquitin under DNA damage conditions. Using the NanoString nCounter technology (38, 39), we found that the relative abundances of the selected DDR transcripts and the four ubiquitin-coding mRNAs varied with a fold-change ranged from 0.6 to 1.5 in INT6-depleted cells treated with NCS, and these minor variations were common to both polysomal and total RNAs (Fig. 6E, Tables S1–S2). However, since these variations mostly impact on transcripts encoding the three components of the MRN complex, which is the DSB sensor that recruits ATM at the break, we analyzed by immunoblot their corresponding protein levels that result from coupling of protein synthesis and degradation. Silencing of INT6 did not modulate overall protein abundance of MRE11, RAD50, and NBS1 regardless of whether cells were treated, or not, with NCS (Fig. S5). From these data, we concluded that INT6 does not significantly influence the translational efficiencies of the transcripts selected here. Together, our results are consistent with a role for INT6 in DDR unrelated to translation.
Figure 6.

INT6 activity in DDR does not rely on a translational effect. (A) HeLa cells were treated, or not, with NCS (200 ng/ml, 30 min) and whole cell extracts were prepared 1 h later. Lysates were immunoprecipitated with preimmune serum (lanes 1, 3) or an antibody to INT6 (lanes 2, 4). Co-immunoprecipitated proteins were analyzed by immunoblotting using antibodies against total eIF3 (only the part of the blot corresponding to the largest eIF3 subunits is shown) or against EIF3L and EIF3D. Recovery of INT6 is shown on bottom panel. (B) Cytoplasmic extracts were prepared from HeLa cells treated as in (A) and were immunoprecipitated with a control antibody (lanes 1, 3) or an antibody to EIF3B (lanes 2, 4). Co-immunoprecipitated proteins were immunoblotted with an antibody to INT6. The same membrane was reprobed with an antibody against EIF3B to verify its pull-down. (C) Hela cells were treated with NCS (200 ng/ml, 15 min) in the presence or absence of 50 μg/ml cycloheximide (CHX). After washing out of NCS, CHX was maintained for 2 h and cells were immunostained using antibodies to Ser1981-phosphorylated ATM and γ-H2AX. Representative confocal images are shown. Scale bar, 10 μM. White squares on merge images delineate regions shown in right panels. These are composite images obtained using the Co-localization Highlighter plug-in for ImageJ. Co-localized pixels appear as white dots. (D) UV-absorbance profiles of cytoplasmic extracts from HeLa cells through a 10–50% sucrose gradient. Cells were transfected with siRNAs control or targeting INT6 for 70 h, treated with NCS (200 ng/ml, 1 h), and collected after 1 h. Positions of 40S and 60S ribosomal subunits, 80S monosomes and polysomes are shown. (E) Transcripts encoding DDR proteins were measured using the NanoString nCounter system from total and polysomal RNAs isolated from cells transfected as in (D). Results are expressed as the mean fold-change of three independent experiments (INT6 knock-down versus control) and error bars correspond to standard deviation. Detailed results and procedures are in Tables S1, S2 and Supplemental Methods.
INT6 facilitates ATM signaling
Because ATM acts very early in DDR and fails to properly re-localize to DSB sites following INT6 depletion, we addressed the consequences of this impairment on the correct assembly of DDR factors. We first examined the phosphorylation of the ATM downstream target NBS1. Phospho-NBS1 was mainly detected after, but not before, exposure to NCS, and its overall abundance did not seem to be modified after INT6 deficiency (Fig. S5). We then tested if it was localized at DSBs. By visual inspection, phospho-NBS1 signal within γ-H2AX foci appeared to be reduced in the vast majority of INT6-deficient cells (Fig. 7A). This was confirmed by measuring fluorescence intensity that showed a 47% decrease of phospho-NBS1 labeling in γ-H2AX foci (Fig. 7D). The signal in the entire nuclear compartment was also reduced by 31%, in agreement with an ATM defect in INT6-depleted cells. We next examined whether INT6 knock-down interfered with the DNA damage-induced ubiquitin signaling pathway which ubiquitylates histones and promotes the recruitment at DSBs of essential repair factors, including BRCA1 (6–8). The formation of ubiquitin conjugates was investigated by immunostaining using the anti-ubiquitin FK2 antibody. Total nuclear ubiquitin signal was normal but ubiquitin intensity in γ-H2AX foci was reduced by 36% when INT6 was silenced (Fig. 7B, D). Consistent with this ubiquitin pathway being compromised, BRCA1 labeling, which remained unchanged in the whole nucleus, was also attenuated by 36% in γ-H2AX foci upon INT6 silencing (Fig. 7C, D). It should be noted that γ-H2AX intensities were normal in these cells. Collectively, these results show that INT6, by stabilizing ATM at DSBs, facilitates ATM downstream signaling upon DNA damage.
Figure 7.

INT6 is required for an efficient DDR. (A–C) HeLa cells transfected with siRNAs were treated with NCS (200 ng/ml, 15 min) and immunostained 3 h post-treatment with antibodies to γ-H2AX and Ser343-phosphorylated NBS1 (panel A), conjugated ubiquitin (FK2 antibody, panel B), and BRCA1 (panel C). ~30 cells were acquired for each cell population and experiments were repeated at least twice. Representative confocal images shown were processed as in Fig. 6C. (D) Fluorescence intensities of γ-H2AX, phospho-NBS1, ubiquitin and BRCA1 labeling were measured within the entire nucleus (Total) and within γ-H2AX foci (Foci). Ten nuclei from control cells and from INT6-deficient cells were analyzed for each experiment. Shown are the mean values of fluorescence intensities. Error bars represent standard errors of the mean.
Discussion
Besides its association with eIF3, INT6 also binds to subunits of the COP9 signalosome (CSN) and the 26S proteasome lid (40), two complexes that regulate proteolysis. These three protein assemblies are referred to as PCI complexes (for Proteasome lid, CSN, eIF3) because a conserved motif was found in subunits of these complexes. Proteins harboring such a PCI domain, including INT6, are thought to serve as structural scaffolds (41). Our results strongly suggest that INT6 does not function in DDR together with eIF3 through regulating translation of mRNAs encoding DDR components. In this regard, a recent study that measured mRNA and protein abundance for more than 5,000 genes in mammalian cells provides valuable information about stoichiometry of protein complexes (35). For INT6, the average protein copy number per cell was estimated to be more than 2-fold that of other subunits of eIF3. This is fully consistent with the notion that INT6, beyond acting with the eIF3 complex, fulfills other functions in the cell. Our results indicate that INT6 is likely to have a direct role in DDR; this is supported by its rapid recruitment at DSBs and by its capacity to interact with ATM. The nature of the signal that triggers this localization of INT6 remains unclear. Phosphorylation of INT6 by ATM is a tempting possibility, as INT6 harbors two PIKK consensus motifs at Ser415 and Thr439. In line with this speculation, a large-scale proteomic analysis aiming at identifying PIKK substrates (42) reported the phosphorylation on Thr439 of the murine protein, but no phosphorylation was found for human INT6 in this work as well as in other studies (42–44). We also searched for such a modification by various experimental approaches but failed to identify INT6 as an ATM substrate. Also, the interaction of INT6 with ATM was not induced by DNA damage, and importantly, recruitment of INT6 at DSBs was normal when ATM kinase activity was inhibited and in the AT5 cell line which lacks ATM. Collectively, these data support the notion that ATM is not required for INT6 recruitment at DNA breaks. Of note, proteasome is recruited at DSBs and is required for efficient DNA repair (45, 46). Future studies should help to determine whether INT6 acts on its own or as part of a macromolecular complex to stabilize ATM at DSBs.
In conclusion, our findings show that INT6 is involved in early stages of DDR and provide a molecular basis to the protective role of INT6 in breast cancer onset. Furthermore, by linking INT6 suppression to a BRCA1 deficiency, these observations might help in the identification of genetic markers that would predict sensitivity to novel PARP-1 inhibitors, which exploit the concept of synthetic lethality and which are so far limited to BRCA1/2-mutated tumors (47, 48).
Supplementary Material
Acknowledgments
We are grateful to P. Descombes and D. Chollet for NanoString experiments, K. Savage for help with GST pull-down, G. Yvert for normalization of Nanostring data, M. Kastan and Y. Shiloh for ATM vectors, J. Hershey for eIF3 antibody, T. Kinsella for RRM2 antibody. We acknowledge the contribution of the Cytometry platform (S. Mouradian-Garcia) and PLATIM platform (C. Lionnet, C. Chamot) of SFR Biosciences Gerland-Lyon Sud (UMS344/US8).
Grant Support
This work was supported by grants from Association pour la Recherche sur le Cancer to PJ, National Institutes of Health [RO1 CA149461], National Aeronautics and Space Administration [NNX10AE08G] and Cancer Prevention and Research Institute of Texas [RP100644] to SB, and National Health and Medical Research Council Program Grant to KKK.
Footnotes
Conflicts of Interest: No potential conflicts of interest were disclosed.
Note: Supplemental Materials and Methods are available at Cancer Research Online.
References
- 1.Lukas J, Lukas C, Bartek J. Mammalian cell cycle checkpoints: signalling pathways and their organization in space and time. DNA Repair (Amst) 2004;3:997–1007. doi: 10.1016/j.dnarep.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 2.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lavin M. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol. 2008;9:759–69. doi: 10.1038/nrm2514. [DOI] [PubMed] [Google Scholar]
- 4.Berkovich E, Monnat RJ, Kastan M. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat Cell Biol. 2007;9:683–90. doi: 10.1038/ncb1599. [DOI] [PubMed] [Google Scholar]
- 5.You Z, Bailis JM, Johnson SA, Dilworth SM, Hunter T. Rapid activation of ATM on DNA flanking double-strand breaks. Nat Cell Biol. 2007;9:1311–8. doi: 10.1038/ncb1651. [DOI] [PubMed] [Google Scholar]
- 6.Huen M, Grant R, Manke I, Minn K, Yu X, Yaffe M, et al. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007;131:901–14. doi: 10.1016/j.cell.2007.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kolas N, Chapman J, Nakada S, Ylanko J, Chahwan R, Sweeney F, et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science. 2007;318:1637–40. doi: 10.1126/science.1150034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, et al. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 2007;131:887–900. doi: 10.1016/j.cell.2007.09.040. [DOI] [PubMed] [Google Scholar]
- 9.Nathanson K, Wooster R, Weber B, Nathanson K. Breast cancer genetics: what we know and what we need. Nat Med. 2001;7:552–6. doi: 10.1038/87876. [DOI] [PubMed] [Google Scholar]
- 10.Swift M, Morrell D, Massey R, Chase C. Incidence of cancer in 161 families affected by ataxia-telangiectasia. N Engl J Med. 1991;325:1831–6. doi: 10.1056/NEJM199112263252602. [DOI] [PubMed] [Google Scholar]
- 11.Thompson D, Duedal S, Kirner J, McGuffog L, Last J, Reiman A, et al. Cancer risks and mortality in heterozygous ATM mutation carriers. J Natl Cancer Inst. 2005;97:813–22. doi: 10.1093/jnci/dji141. [DOI] [PubMed] [Google Scholar]
- 12.Renwick A, Thompson D, Seal S, Kelly P, Chagtai T, Ahmed M, et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet. 2006;38:873–5. doi: 10.1038/ng1837. [DOI] [PubMed] [Google Scholar]
- 13.Chenevix-Trench G, Spurdle AB, Gatei M, Kelly H, Marsh A, Chen X, et al. Dominant negative ATM mutations in breast cancer families. J Natl Cancer Inst. 2002;94:205–15. doi: 10.1093/jnci/94.3.205. [DOI] [PubMed] [Google Scholar]
- 14.Meijers-Heijboer H, van den Ouweland A, Klijn J, Wasielewski M, de Snoo A, Oldenburg R, et al. Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat Genet. 2002;31:55–9. doi: 10.1038/ng879. [DOI] [PubMed] [Google Scholar]
- 15.Walsh T, Casadei S, Coats K, Swisher E, Stray S, Higgins J, et al. Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in families at high risk of breast cancer. JAMA. 2006;295:1379–88. doi: 10.1001/jama.295.12.1379. [DOI] [PubMed] [Google Scholar]
- 16.Górski B, Debniak T, Masojć B, Mierzejewski M, Medrek K, Cybulski C, et al. Germline 657del5 mutation in the NBS1 gene in breast cancer patients. Int J Cancer. 2003;106:379–81. doi: 10.1002/ijc.11231. [DOI] [PubMed] [Google Scholar]
- 17.Heikkinen K, Rapakko K, Karppinen S, Erkko H, Knuutila S, Lundán T, et al. RAD50 and NBS1 are breast cancer susceptibility genes associated with genomic instability. Carcinogenesis. 2006;27:1593–9. doi: 10.1093/carcin/bgi360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bartkova J, Tommiska J, Oplustilova L, Aaltonen K, Tamminen A, Heikkinen T, et al. Aberrations of the MRE11-RAD50-NBS1 DNA damage sensor complex in human breast cancer: MRE11 as a candidate familial cancer-predisposing gene. Mol Oncol. 2008;2:296–316. doi: 10.1016/j.molonc.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Asano K, Merrick W, Hershey J. The translation initiation factor eIF3-p48 subunit is encoded by int-6, a site of frequent integration by the mouse mammary tumor virus genome. J Biol Chem. 1997;272:23477–80. doi: 10.1074/jbc.272.38.23477. [DOI] [PubMed] [Google Scholar]
- 20.Marchetti A, Buttitta F, Miyazaki S, Gallahan D, Smith G, Callahan R. Int-6, a highly conserved, widely expressed gene, is mutated by mouse mammary tumor virus in mammary preneoplasia. J Virol. 1995;69:1932–8. doi: 10.1128/jvi.69.3.1932-1938.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rasmussen S, Kordon E, Callahan R, Smith G. Evidence for the transforming activity of a truncated Int6 gene, in vitro. Oncogene. 2001;20:5291–301. doi: 10.1038/sj.onc.1204624. [DOI] [PubMed] [Google Scholar]
- 22.Mack D, Boulanger C, Callahan R, Smith G. Expression of truncated Int6/eIF3e in mammary alveolar epithelium leads to persistent hyperplasia and tumorigenesis. Breast Cancer Res. 2007;9:R42. doi: 10.1186/bcr1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marchetti A, Buttitta F, Pellegrini S, Bertacca G, Callahan R. Reduced expression of INT-6/eIF3-p48 in human tumors. Int J Oncol. 2001;18:175–9. doi: 10.3892/ijo.18.1.175. [DOI] [PubMed] [Google Scholar]
- 24.Umar A, Kang H, Timmermans A, Look M, Meijer-van Gelder M, den Bakker M, et al. Identification of a putative protein profile associated with tamoxifen therapy resistance in breast cancer. Mol Cell Proteomics. 2009;8:1278–94. doi: 10.1074/mcp.M800493-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suo J, Snider SJ, Mills GB, Creighton CJ, Chen AC, Schiff R, et al. Int6 regulates both proteasomal degradation and translation initiation and is critical for proper formation of acini by human mammary epithelium. Oncogene. 2011;30:724–36. doi: 10.1038/onc.2010.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morris C, Wittmann J, Jack HM, Jalinot P. Human INT6/eIF3e is required for nonsense-mediated mRNA decay. Embo Reports. 2007;8:596–602. doi: 10.1038/sj.embor.7400955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ziv Y, Bar-Shira A, Pecker I, Russell P, Jorgensen T, Tsarfati I, et al. Recombinant ATM protein complements the cellular A-T phenotype. Oncogene. 1997;15:159–67. doi: 10.1038/sj.onc.1201319. [DOI] [PubMed] [Google Scholar]
- 28.Canman C, Lim D, Cimprich K, Taya Y, Tamai K, Sakaguchi K, et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–9. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- 29.Khanna KK, Keating KE, Kozlov S, Scott S, Gatei M, Hobson K, et al. ATM associates with and phosphorylates p53: mapping the region of interaction. Nat Genet. 1998;20:398–400. doi: 10.1038/3882. [DOI] [PubMed] [Google Scholar]
- 30.Tomimatsu N, Mukherjee B, Burma S. Distinct roles of ATR and DNA-PKcs in triggering DNA damage responses in ATM-deficient cells. EMBO Rep. 2009;10:629–35. doi: 10.1038/embor.2009.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bolderson E, Tomimatsu N, Richard DJ, Boucher D, Kumar R, Pandita TK, et al. Phosphorylation of Exo1 modulates homologous recombination repair of DNA double-strand breaks. Nucleic Acids Res. 2010;38:1821–31. doi: 10.1093/nar/gkp1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morris-Desbois C, Bochard V, Reynaud C, Jalinot P. Interaction between the Ret finger protein and the int-6 gene product and colocalisation into nuclear bodies. Journal of Cell Science. 1999;112:3331–42. doi: 10.1242/jcs.112.19.3331. [DOI] [PubMed] [Google Scholar]
- 33.Morris-Desbois C, Rety S, Ferro M, Garin J, Jalinot P. The human protein HSPC021 interacts with Int-6 and is associated with eukaryotic translation initiation factor 3. Journal of Biological Chemistry. 2001;276:45988–95. doi: 10.1074/jbc.M104966200. [DOI] [PubMed] [Google Scholar]
- 34.So S, Davis AJ, Chen DJ. Autophosphorylation at serine 1981 stabilizes ATM at DNA damage sites. J Cell Biol. 2009;187:977–90. doi: 10.1083/jcb.200906064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, et al. Global quantification of mammalian gene expression control. Nature. 2011;473:337–42. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 36.Watkins SJ, Norbury CJ. Cell cycle-related variation in subcellular localization of eIF3e/INT6 in human fibroblasts. Cell Prolif. 2004;37:149–60. doi: 10.1111/j.1365-2184.2004.00305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grzmil M, Rzymski T, Milani M, Harris AL, Capper RG, Saunders NJ, et al. An oncogenic role of eIF3e/INT6 in human breast cancer. Oncogene. 2010;29:4080–9. doi: 10.1038/onc.2010.152. [DOI] [PubMed] [Google Scholar]
- 38.Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol. 2008;26:317–25. doi: 10.1038/nbt1385. [DOI] [PubMed] [Google Scholar]
- 39.Beaume M, Hernandez D, Docquier M, Delucinge-Vivier C, Descombes P, François P. Orientation and expression of methicillin-resistant Staphylococcus aureus small RNAs by direct multiplexed measurements using the nCounter of NanoString technology. J Microbiol Methods. 2011;84:327–34. doi: 10.1016/j.mimet.2010.12.025. [DOI] [PubMed] [Google Scholar]
- 40.Hoareau Alves K, Bochard V, Réty S, Jalinot P. Association of the mammalian proto-oncoprotein Int-6 with the three protein complexes eIF3, COP9 signalosome and 26S proteasome. FEBS Lett. 2002;527:15–21. doi: 10.1016/s0014-5793(02)03147-2. [DOI] [PubMed] [Google Scholar]
- 41.Pick E, Hofmann K, Glickman M. PCI complexes: Beyond the proteasome, CSN, and eIF3 Troika. Mol Cell. 2009;35:260–4. doi: 10.1016/j.molcel.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 42.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, Hurov KE, Luo J, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–6. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 43.Mu JJ, Wang Y, Luo H, Leng M, Zhang J, Yang T, et al. A proteomic analysis of ataxia telangiectasia-mutated (ATM)/ATM-Rad3-related (ATR) substrates identifies the ubiquitin-proteasome system as a regulator for DNA damage checkpoints. J Biol Chem. 2007;282:17330–4. doi: 10.1074/jbc.C700079200. [DOI] [PubMed] [Google Scholar]
- 44.Bensimon A, Schmidt A, Ziv Y, Elkon R, Wang SY, Chen DJ, et al. ATM-dependent and -independent dynamics of the nuclear phosphoproteome after DNA damage. Sci Signal. 2010;3:rs3. doi: 10.1126/scisignal.2001034. [DOI] [PubMed] [Google Scholar]
- 45.Krogan NJ, Lam MH, Fillingham J, Keogh MC, Gebbia M, Li J, et al. Proteasome involvement in the repair of DNA double-strand breaks. Mol Cell. 2004;16:1027–34. doi: 10.1016/j.molcel.2004.11.033. [DOI] [PubMed] [Google Scholar]
- 46.Jacquemont C, Taniguchi T. Proteasome function is required for DNA damage response and fanconi anemia pathway activation. Cancer Res. 2007;67:7395–405. doi: 10.1158/0008-5472.CAN-07-1015. [DOI] [PubMed] [Google Scholar]
- 47.Bryant H, Schultz N, Thomas H, Parker K, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 48.Farmer H, McCabe N, Lord C, Tutt A, Johnson D, Richardson T, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.