

Abstract
The RDX gene anchors cytoskeletal actin of stereocilia to hair cell transmembrane and is responsible for autosomal recessive nonsyndromic hearing impairment (ARNSHI) due to DFNB24. A genome scan was performed using DNA samples from a consanguineous Pakistani family with ARNSHI. A significant maximum two-point LOD score of 4.5 (θ = 0) and multipoint LOD score of 5.8 were achieved at marker D11S1998 (chr11 : 117.20 Mb). The region of homozygosity is bounded by markers D11S2000 (105.06 Mb) and D11S4464 (123.13 Mb) and contains the NSHI genes TECTA and RDX. Although no potentially causal variants were identified in the TECTA gene, within the RDX gene a novel deletion c.1076_1079delTTAA (p.Ile359Lysfs∗6) was identified. The RDX deletion segregates with ARNSHI within the family and was not observed in 500 control chromosomes. It is predicted to cause premature truncation of radixin at the α-helical domain and to result in nonfunctional transcripts within the cochlea. RDX isoforms which encode the coiled-coil region of the α-helical domain are deemed necessary for proper function of hair cell stereocilia.
1. Introduction
Within the mammalian cochlea, the most specialized cells for hearing that mediate conversion of mechanical vibration from sound energy to neural impulses in the brain (mechanotransduction) are the hair cells, so-named because of the hair-like projections at their apical surfaces, which are called stereocilia. Of the 54 nonsyndromic hearing impairment (NSHI) genes that are currently known, ∼20 genes are associated with stereocilia structure or assembly (Hereditary Hearing Loss Homepage). Variants in RDX (MIM 179410) cause autosomal recessive (AR) NSHI (DFNB24) and can be found in all three domains of the radixin protein [1, 2]. Radixin is a member of the ERM family of highly homologous proteins including ezrin and moesin. ERM proteins link cell membrane proteins to actin, which is the basic component of the cytoskeleton of microvillar structures such as stereocilia [3]. There are three well-characterized ERM protein domains: (1) the FERM domain, which binds to transmembrane proteins [4]; (2) the α-helical domain, which masks interaction sites of the FERM domain to regulate activation [5]; (3) the C-terminal tail or ERM association domain (C-ERMAD) which has an F-actin-binding site [6]. Consistent with its anchoring function in the hair cell, radixin is mainly located at the stereociliary base [7]. This paper describes the fifth family known to date to have ARNSHI due to an RDX mutation.
2. Materials and Methods
The study was approved by the Institutional Review Boards of Quaid-I-Azam University and the Baylor College of Medicine and Affiliated Hospitals. Informed consent was obtained from all members of family 4208 who participated in the study.
The members of family 4208 are from the Muzafar Ghar district of Punjab province. Venous blood was obtained from 13 members of family 4208, five of whom have HI (Figure 1). Genomic DNA was extracted from whole blood following a standard protocol [8]. All 13 samples underwent a whole genome linkage scan at the Center for Inherited Disease Research (CIDR) using 405 short tandem repeat markers with average spacing of 9 cM. Data quality control was performed on the resulting genotype data using PedCheck [9] in order to identify Mendelian inconsistencies and MERLIN [10] to detect occurrences of double recombination events over short genetic distances, which are most likely due to genotyping error. Two-point linkage analysis was performed with MLINK of the FASTLINK package [11]. Multipoint linkage analysis was carried out using Allegro1.2c [12]. An AR mode of inheritance with complete penetrance and a disease allele frequency of 0.001 were used in the parametric linkage analysis. Marker allele frequencies were estimated from observed and reconstructed genotypes of founders from family 4208 and 35 other families who underwent genome scan at the same time at CIDR. For multipoint linkage analysis, genetic map positions were based on the Rutgers combined linkage-physical map of the human genome Build 36 version [13]. For markers used in the analysis which are not included on the Rutgers map, the physical map position from the human reference sequence (Build 36) was used to interpolate the genetic map position on the Rutgers map. Haplotypes were reconstructed using SimWalk2 [14].
Figure 1.
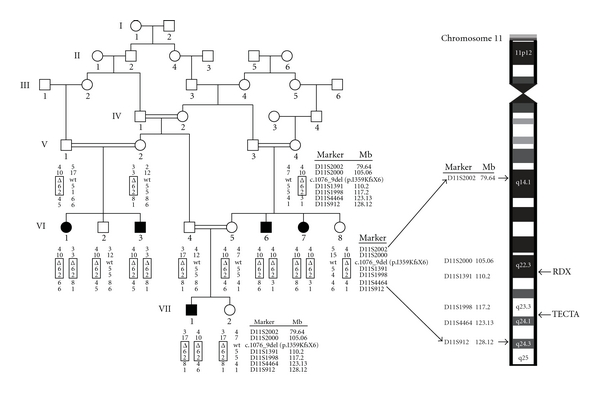
Pedigree drawing and haplotype of family 4208 with the genetic interval on chromosome 11. Filled symbols denote individuals with ARNSHI, while clear symbols represent hearing individuals. The haplotype segregating with ARNSHI is shown in a box and includes short tandem repeat markers and the RDX deletion c.1076_1079delTTAA (p.Ile359Lysfs*6) as a triangle. The positions of the TECTA and RDX genes relative to the genome scan markers are labelled. The genetic interval with genotyped marker loci and locations of RDX and TECTA genes were indicated on the cartoon of chromosome 11.
All exons of the RDX (MIM 179410; NM_002906.3) and α-tectorin (TECTA [MIM 602574; NM_005422.2]) genes were sequenced in HI individuals VI-1 and VII-1 and hearing individual VI-5 who are members of family 4208 (Figure 1). After PCR amplification and purification, sequencing was performed using the BigDye Terminator v3.1 Cycle Sequencing Kit and the ABI 3730 DNA Analyzer (Applied Biosystems Inc., Foster City, Calif, USA). The resulting sequences were assembled and analysed using the Sequencher software (Gene Codes Corp., Ann Arbor Mizh, USA). After identification of the RDX deletion, exon 10 was sequenced using DNA samples from additional family members and 250 unrelated hearing control individuals from Pakistan.
3. Results and Discussion
Family 4208 (Figure 1) is a consanguineous kindred with ARNSHI from the Punjab region of Pakistan and the family members speak Saraiki. The hearing impairment (HI) was described by family members as prelingual in onset, bilateral, and profound. No possible environmental causes of HI such as perinatal, ototoxic, traumatic, and infectious factors were elicited from the clinical history. Evidence of syndromic and vestibular phenotypes was not found after physical examination that included balance and gait testing.
A significant maximum two-point LOD score of 4.5 was obtained at marker D11S1998 (chr11 : 117.20 Mb) at θ = 0. At the same marker, a maximum multipoint LOD score of 5.8 was obtained. The 3-unit support interval (Table 1) and the region of homozygosity (Figure 1) completely overlap, with the proximal limit at D11S2000 (105.06 Mb) and the distal limit at D11S4464 (123.13 Mb). The linkage interval extends over a 25.28 cM region at 11q22.3-q24.1 and contains 18.07 Mb of sequence.
Table 1.
Two-point and multipoint LOD scores for family 4208 at chromosome 11q22.3-q24.1.
| Marker name1 | Physical map position2 | Genetic map position3 | Multipoint LOD score | Two-point LOD score at θ = | |||||
|---|---|---|---|---|---|---|---|---|---|
| 0.0 | 0.01 | 0.05 | 0.10 | 0.20 | 0.30 | ||||
| D11S2371 | 73,182,778 | 84.41 | −∞ | −∞ | −0.35 | 0.24 | 0.37 | 0.31 | 0.15 |
| D11S2002 | 79,643,050 | 91.48 | −13.66 | −∞ | −1.41 | −0.18 | 0.20 | 0.33 | 0.22 |
| D11S2000 | 105,063,887 | 111.71 | −6.18 | −6.08 | −0.15 | 0.90 | 1.07 | 0.83 | 0.42 |
| D11S1391 | 110,200,327 | 115.14 | 5.20 | 3.18 | 3.10 | 2.74 | 2.29 | 1.43 | 0.68 |
| D11S1998 | 117,202,969 | 126.24 | 5.78 | 4.47 | 4.36 | 3.95 | 3.41 | 2.33 | 1.26 |
| D11S4464 | 123,131,592 | 136.99 | −∞ | −∞ | −2.84 | −0.93 | −0.25 | 0.16 | 0.17 |
| D11S912 | 128,129,301 | 145.13 | −∞ | −∞ | −1.52 | −0.62 | −0.25 | 0.01 | 0.06 |
| D11S968 | 133,323,584 | 160.10 | −4.03 | −1.95 | −0.06 | 0.54 | 0.64 | 0.45 | 0.18 |
1Markers in bold denote marker limits based on the 3-unit support interval and the homozygous region.
2Physical map positions in base pairs from Build 36 of the human reference sequence.
3Genetic map positions in cM from Rutgers combined linkage-physical map of the human genome Build 36 version.
Within the linkage interval there are 167 RefSeq genes, including two NSHI genes, RDX and TECTA. Both genes were sequenced. No sequence variants were identified in the TECTA gene. On the other hand, a novel RDX deletion c.1076_1079delTTAA (p.Ile359Lysfs*6), which segregates with ARNSHI in family 4208 (Figure 2), was identified. This deletion was not found in 500 control chromosomes, in either the homozygous or in the heterozygous state. The deletion results in a frameshift and premature truncation of the radixin protein to 363 residues and, subsequently, in loss of most of the α-helical domain and the whole F-actin-binding domain (Figure 3(a)). According to the PROSITE database [15], the deleted domains contain multiple phosphorylation sites for protein kinase C and casein kinase II, which are receptor specific for the ERM protein family [16].
Figure 2.

Chromatogram displaying the novel RDX deletion c.1076_1079delTTAA (p.Ile359Lysfs*6). Chromatograms shown are from sequences of the following: (a) wild type, (b) hearing-impaired homozygous individual VII-1, and (c) unaffected heterozygous individual VI-5. The deleted nucleotides TTAA are indicated with a red triangle.
Figure 3.
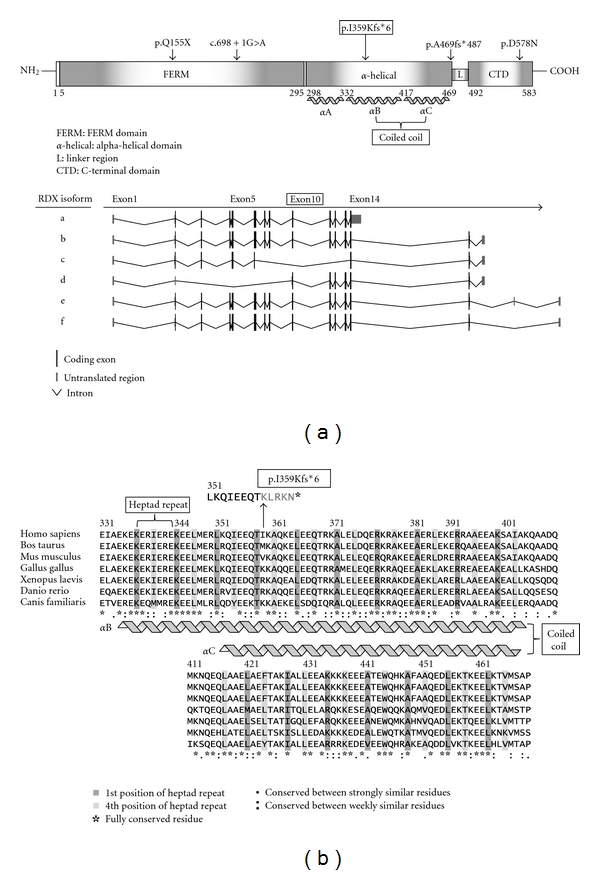
(a) Schematic representation of protein domains and isoforms of RDX gene. The positions of known pathogenic mutations were indicated with arrow and the newly identified mutation was boxed. The protein domain structure is based on the reference sequence NP_002897 (transcript isoform a). (b) Clustal W multiple sequence alignment of alpha-helical domain of RDX protein sequences of seven different species. The 1st and 4th amino acid residues of heptad repeat sequences are highly conserved through the species.
The TECTA gene causes both autosomal dominant (DFNA8/12) and AR (DFNB21) NSHI [17]. When inherited recessively, TECTA mutations cause moderate-to-severe hearing impairment with a flat or U-shaped audiogram pattern [18]. In contrast, the HI in family 4208 is profound, which is similar to the previously reported HI pattern for RDX [2]. In Rdx −/− mice, degeneration of outer hair cell stereocilia began at onset of hearing [19], and this is consistent with the prelingual onset of HI in humans with RDX mutations, as was observed in family 4208. Congenital jaundice was also noted in Rdx −/− mice [20]. However there is no evidence of hyperbilirubinemia in family 4208. Additionally in the previously reported four families that segregate RDX mutations there is no evidence of hyperbilirubinemia [1, 2].
The mutation c.1076_1079delTTAA is located in exon 10 of RDX. Of the six isoforms of RDX (Figure 3(a)), isoform c does not include exons 7 to 13 of isoform a. In the original DFNB24 family, the p.Gln155* stop codon mutation can be found in exon 5, which is not included in RDX isoform d [2] (Figure 3(a)). In particular, exon 10 marks the beginning of a series of highly conserved heptad repeats in the αB helix which interacts with the corresponding highly conserved heptad repeats in the αC helix (Figure 3(b)) as the α-helical domain folds on itself to form an antiparallel coiled coil in the dormant state [5] (Figures 3(a) and 3(b)). When active, the coiled coil is fully extended, unmasking ligand-binding sites [5]. This may indicate that the αB/αC coiled coil is necessary for regulation of radixin activity within hair cell stereocilia.
4. Conclusion
A novel deletion c.1076_1079delTTAA (p.Ile359Lysfs*6) in the RDX gene was identified in a large Pakistani consanguineous pedigree that segregates ARNSHI. This novel RDX deletion is predicted to cause premature truncation of radixin at the α-helical domain and to result in nonfunctional transcripts within the cochlea. Identification of the c.1076_1079delTTAA (p.Ile359Lysfs*6) deletion gives a better understanding of the role the RDX gene plays in hearing impairment.
Electronic Database Information
The following URLs were accessed for data in this paper: Hereditary Hearing Loss Homepage (http://hereditaryhearingloss.org/); UCSC Genome Browser (http://genome.ucsc.edu/); OMIM (http://www.omim.org/). ClustalW (http://www.ebi.ac.uk/Tools/msa/clustalw2/).
Acknowledgments
The authors thank the family members who participated in this study. The study was supported by the Higher Education Commission, Islamabad, Pakistan (to W. Ahmed) and the National Institutes of Health (NIH)-National Institute of Deafness and other Communication Disorders Grants R01-DC03594 and R01-DC011651 (to S. M. Leal). Genotyping services were provided by CIDR and were funded through a federal contract from the NIH to the Johns Hopkins University (Contract no. N01-HG-65403).
References
- 1.Khan SY, Ahmed ZM, Shabbir MI, et al. Mutations of the RDX gene cause nonsyndromic hearing loss at the DFNB24 locus. Human Mutation. 2007;28(5):417–423. doi: 10.1002/humu.20469. [DOI] [PubMed] [Google Scholar]
- 2.Shearer AE, Hildebrand MS, Bromhead CJ, et al. A novel splice site mutation in the RDX gene causes DFNB24 hearing loss in an Iranian family. The American Journal of Medical Genetics, Part A. 2009;149(3):555–558. doi: 10.1002/ajmg.a.32670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jankovics F, Sinka R, Lukácsovich T, Erdélyi M. MOESIN crosslinks actin and cell membrane in Drosophila oocytes and is required for OSKAR anchoring. Current Biology. 2002;12(23):2060–2065. doi: 10.1016/s0960-9822(02)01256-3. [DOI] [PubMed] [Google Scholar]
- 4.Chishti AH, Kim AC, Marfatia SM, et al. The FERM domain: a unique module involved in the linkage of cytoplasmic proteins to the membrane. Trends in Biochemical Sciences. 1998;23(8):281–282. doi: 10.1016/s0968-0004(98)01237-7. [DOI] [PubMed] [Google Scholar]
- 5.Li Q, Nance MR, Kulikauskas R, et al. Self-masking in an intact ERM-merlin protein: an active role for the central α-helical domain. Journal of Molecular Biology. 2007;365(5):1446–1459. doi: 10.1016/j.jmb.2006.10.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turunen O, Wahlström T, Vaheri A. Ezrin has a COOH-terminal actin-binding site that is conserved in the ezrin protein family. Journal of Cell Biology. 1994;126(6):1445–1453. doi: 10.1083/jcb.126.6.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pataky F, Pironkova R, Hudspeth AJ. Radixin is a constituent of stereocilia in hair cells. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(8):2601–2606. doi: 10.1073/pnas.0308620100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grimberg A, Nawoschik S, Belluscio L, McKee R, Turck A, Eisenberg A. A simple and efficient non-organic procedure for the isolation of genomic DNA from blood. Nucleic Acids Research. 1989;17(20):83–90. doi: 10.1093/nar/17.20.8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Connell JR, Weeks DE. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. The American Journal of Human Genetics. 1998;63(1):259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nature Genetics. 2002;30(1):97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 11.Cottingham RW, Jr., Idury RM, Schäffer AA. Faster sequential genetic linkage computations. The American Journal of Human Genetics. 1993;53(1):252–263. [PMC free article] [PubMed] [Google Scholar]
- 12.Gudbjartsson DF, Jonasson K, Frigge ML, Kong A. Allegro, a new computer program for multipoint linkage analysis. Nature Genetics. 2000;25(1):12–13. doi: 10.1038/75514. [DOI] [PubMed] [Google Scholar]
- 13.Matise TC, Chen F, Chen W, et al. A second-generation combined linkage-physical map of the human genome. Genome Research. 2007;17(12):1783–1786. doi: 10.1101/gr.7156307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sobel E, Lange K. Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. The American Journal of Human Genetics. 1996;58(6):1323–1337. [PMC free article] [PubMed] [Google Scholar]
- 15.Sigrist CJA, Cerutti L, de Castro E, et al. PROSITE, a protein domain database for functional characterization and annotation. Nucleic Acids Research. 2010;38:161–166. doi: 10.1093/nar/gkp885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fazioli F, Wong WT, Ullrich SJ, Sakaguchi K, Appella E, Di Fiore PP. The ezrin-like family of tyrosine kinase substrates: receptor-specific pattern of tyrosine phosphorylation and relationship to malignant transformation. Oncogene. 1993;8(5):1335–1345. [PubMed] [Google Scholar]
- 17.Mustapha M, Weil D, Chardenoux S, et al. An α-tectorin gene defect causes a newly identified autosomal recessive form of sensorineural pre-lingual non-syndromic deafness, DFNB21 . Human Molecular Genetics. 1999;8(3):409–412. doi: 10.1093/hmg/8.3.409. [DOI] [PubMed] [Google Scholar]
- 18.Naz S, Alasti F, Mowjoodi A, et al. Distinctive audiometric profile associated with DFNB21 alleles of TECTA . Journal of Medical Genetics. 2003;40(5):360–363. doi: 10.1136/jmg.40.5.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kitajiri SI, Fukumoto K, Hata M, et al. Radixin deficiency causes deafness associated with progressive degeneration of cochlear stereocilia. Journal of Cell Biology. 2004;166(4):559–570. doi: 10.1083/jcb.200402007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kikuchi S, Hata M, Fukumoto K, et al. Radixin deficiency causes conjugated hyperbilirubinemia with loss of Mrp2 from bile canalicular membranes. Nature Genetics. 2002;31(3):320–325. doi: 10.1038/ng905. [DOI] [PubMed] [Google Scholar]