

Abstract
The understanding of the molecular genetics in sensorineural hearing loss (SNHL) has advanced rapidly during the last decade, but the molecular etiology of hearing impairment in the Portuguese population has not been investigated thoroughly. To provide appropriate genetic testing and counseling to families, we analyzed the whole mitochondrial genome in 95 unrelated children with SNHL (53 nonsyndromic and 42 syndromic) and searched for variations in two frequent genes, GJB2 and GJB6, in the non-syndromic patients. Mutations in mtDNA were detected in 4.2% of the cases, including a hitherto undescribed change in the mtDNA-tRNATrp gene (namely, m.5558A>G). We also identified mono- or biallelic GJB2 mutations in 20 of 53 non-syndromic cases and also detected two novel mutations (p.P70R and p.R127QfsX84). Our data further reinforce the notion that genetic heterogeneity is paramount in children with SNHL.
1. Introduction
Sensorineural hearing loss (SNHL) is one of the most common disabilities in human, and genetics is an important aspect in research and clinical practice for SNHL. One in 1000 children is born with bilateral SNHL, and 50–70% of them have monogenic causes for their deafness [1]. In addition, 10% of the people over 65 years have SNHL that limits considerably speech communication. Although the etiology is polygenic in most of the cases, with different contribution of ageing and environment, discovering monogenic causes has important clinical implications in terms of better counseling and management.
Hereditary hearing loss can be classified into syndromic and nonsyndromic depending on the associated features. Whilst over 400 genetic syndromes have been described in association with mono- or bilateral deafness, syndromic conditions account for about 30% of hereditary congenital hearing loss, whereas the relative contribution to all deaf people is much higher (>70%) for nonsyndromic subtypes [1].
Nonsyndromic sensorineural hearing loss (NSSNHL) is predominantly inherited in an autosomal recessive patterns (DFNB loci) (80%) but can be also autosomal dominantly (DFNA) (15–20%), X-linked (DFN) (2-3%), or maternally (1%) transmitted. A polygenic or multifactorial pattern of inheritance should be postulated for late onset cases of hearing impairment [2, 3]. To date, 134 deafness loci (77 DFNB and 57 DFNA) have been reported, with more than 40 genes cloned [4]. Further heterogeneity is, however, expected to emerge.
Mutations in the GJB2 and GJB6 genes on the DFNB1 locus at chromosome 13q11-q12 are responsible for up to 50% of autosomal recessive (AR) NSSNHL [5, 6]. GJB2 and GJB6 encode gap junction proteins connexin 26 (Cx26) and connexin 30 (Cx30), respectively, which are expressed in the cochlea where they colocalize, form heteromeric gap junctions [7], and play a role in cochlear homeostasis [8]. The list of allelic variants in GJB2 is wide with more than 100 variants being detected, mainly in congenital AR deafness, but also in dominant forms. The deletion of a single guanine (c.35delG) individually account for up to 50% of cases of NSSNHL among populations in Europe, North America, and Asia [9, 10]. The common 342-Kb genomic deletion in GJB6 (termed GJB6-D13S1830) occurs in up to 20% of the hearing-impaired American population and may account for ~10% of all DFNB1 alleles with an extremely wide range based on ethnic origin, oftentimes in dysgenic association with the c.35delG/GJB2 variant [11, 12].
Maternally inherited hearing loss account for approximately 1% of cases, but accurate diagnosis is important because of its unique implications for affected individuals and their family members. Hearing loss may be an early manifestation of a more complex mitochondrial disorder such as the Kearns-Sayre (KSS) [13–15], the MERRF (myoclonic epilepsy with ragged-red fibers) [16, 17], or the MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes) [18, 19] syndromes. In addition, SNHL can be the main, or sole, presenting feature of a mtDNA defect. In particular, mutations in the mtDNA-encoded 12S rRNA and tRNASer(UCN) genes are frequent. The m.1555A>G mutation in 12S rRNA has a worldwide distribution, and it is localized in a highly conserved region which is involved in decoding small ribosomal subunit [20]. The new G–C pair in 12S rRNA created by the A>G transition facilitates the binding of aminoglycosides antibiotics such as gentamycin and streptomycin, causing aminoglycoside-induced NSSNHL or worsening hearing loss in individuals harboring this mutation. The prevalence of the m.1555A>G mutation has been shown to be between 20–30% in deaf individuals in Spain and Asia of which 15% of them had the history of aminoglycoside ototoxicity [21]. Additional mutations in 12S rRNA (m.961delT, m.961insC(n), m.1005T>C, m.1095T>C, and m.1494 C>T) have been associated with impaired hearing [22–25], although their pathogenic significance remains controversial. All this information indicates that the mitochondrial 12S rRNA gene is a hotspot for mutations causing nonsyndromic hearing loss as well as increased sensitivity to aminoglycoside ototoxicity [26]. Mutations in the tRNASer(UCN) gene are additional genetic hotspot in Caucasian and Asian pedigrees with nonsyndromic and aminoglycoside-induced hearing loss. In the vast majority of patients, mtDNA mutations are nearly homoplasmic, indicating the requirement of a high mutation load to exert cochlear dysfunction [27].
We performed a comprehensive analysis of GJB2 and GJB6 in 53 non-syndromic patients and scanned the whole mitochondrial genome for mutations in a cohort of 95 paediatric Portuguese SNHL patients recruited in a hearing clinic to investigate the relative contribution of common genetic etiologies to their disorder, provide effective assessment of genetic risk, and to better counsel their relatives.
2. Patients and Methods
2.1. Subjects
Ninety-five Portuguese SNHL children from unrelated families were included in this study. A diagnosis of hearing deficit was performed in a Hearing Clinic according to international guidelines [28]. Fifty-three cases (56%) showed a non-syndromic hearing loss, profound in 80% and mild to moderate in 20%, and 42 (44%) patients had a syndromic disorder. Frequent accompanying manifestations involved the nervous system, eye, external ear, and musculoskeletal system. Parents were interviewed with regard to age of onset, family history, mother's health during pregnancy, and patient's past clinical history, including infection, possible head or brain injury, and the use of aminoglycoside antibiotics. Written informed consent was obtained from all patients or their parents prior to blood sampling and all clinical and molecular studies accomplished with the ethical issues of Declaration of Helsinki.
2.2. DNA Extraction
Total genomic DNA was extracted from peripheral blood (B) or skeletal muscle (M) using commercially available procedures which employed the EZ1 DNA Blood 350 μL Kit (QIAGEN) and Puregene Tissue kit (Gentra) kits, respectively.
2.3. Mutational Analyses
We performed polymerase chain reaction (PCR) amplifications and direct gene sequencing of the coding exon and flanking regions of GJB2 in 53 non-syndromic patients, as described elsewhere [5]. The common GJB6-D13S1830 deletion [29] was searched in patients harbouring a heterozygous sequence variation in GJB2. All 95 patients were screened for mutations in the entire mtDNA by using a commercially available kit, mitoSEQr Resequencing System for the Human Mitochondrial Genome (Applied Biosystems, Foster City, Calif), according to the procedure recommended by the manufacturer.
For the description of the mutations, we used the latest conventions of the Human Genome Variation Society nomenclature. Synonymous, missense, and splice site variations were systematically evaluated for modifications of exonic splicing enhancers (Polyphen analysis, http://genetics.bwh.harvard.edu/pph/; ESEfinder, http://rulai.cshl.edu/cgi-bin/tools/ESE/esefinder.cgi) or consensus splicing sequences in order to determine the splice site score (http://www.cbs.dtu.dk/services/NetGene2/ and http://www.fruitfly.org/seq_tools/splice.html). Multiple alignments with GJB2 orthologs were performed using ClustalW (http://www.ebi.ac.uk/Tools/msa/clustalw2/) to evaluate the degree of conservation of missense variants. MtDNA mutations and polymorphisms were searched in MITOMAP data base (http://mitomap.org/MITOMAP).
3. Results
Table 1 summarizes molecular data detected in children with SNHL. Sequence analysis of GJB2 indicated that eight patients carried two mutated alleles with already described pathogenic mutations and one presented the p.M34T mutation in heterozogosity, which has been reported to cause AD-NSSNHL [30]. The frequent c.35delG was homozygous in four children and was detected on a single allele in eight patients, three of whom harbored a wild-type sequence on the other allele and two children harbored the heterozygous common (GJB6-D13S1830) deletion. Two additional patients presented novel variations in GJB2: a homozygous p.R127QfsX84 mutation was found in one case whereas another patient presented the p.P70R on a single allele. Four patients presented with heteroplasmic (two individuals) or homoplasmic (two cases) mutations in mtDNA, including the hitherto unreported m.5558A>G in the mtDNA-encoded tRNATrp gene.
Table 1.
Genotype of patients with mutations in nuclear genes (GJB2 and GJB6) and in mitochondrial genome.
| Number of patients | GJB2 | Reference | GJB6 | Reference | mtDNA | Reference |
|---|---|---|---|---|---|---|
| 4 | c.35delG/c.35delG | [9] | — | — | ||
| 1 | p.W24X/p.W24X | [30] | — | — | ||
| 1 | c.35delG/p.E47X | [9, 31] | — | — | ||
| 1 | c.35delG/p.L76P | [9, 32] | — | — | ||
| 1 | c.35delG/p.V95M | [9, 33] | — | — | ||
| 1 | p.M34T/wt | [30] | — | — | ||
| 1 | p.R127QfsX84/p.R127QfsX84 | This study | — | — | ||
| 3 | c.35delG/wt | [9] | — | — | ||
| 1 | p.P70R/wt | This study | — | — | ||
| 2 | c.35delG | [9] | del(GJB6-D13S1830) | [29] | — | |
| 1 | — | — | m.7445A>G | [34] | ||
| 1 | — | — | m.1555A>G | [20] | ||
| 1 | — | — | m.3243 A>G | [19] | ||
| 1 | — | — | m.5558 A>G | This study |
Collectively, about 17% (16/95) of the Portuguese children analyzed in the present study were molecularly characterized, whereas a single variant was identified in four patients. The c.35delG accounted for 40% (16/40) of all mutant alleles, and up to half of mutations occurring in connexin-related genes. Additionally, 4.2% of studied patients (4/95) presented mutations in mtDNA, two of them (m.5558A>G and m.7445A>G) were identified in non-syndromic patients and the remaining mutations (m.1555A>G and m.3243A>G) in syndromic cases.
4. Discussion
Despite increasing evidence of mutations associated with SNHL, there have been thus far no studies reporting on the relative frequency of mutations in the hearing-impaired Portuguese pediatric population. Although limited to a single center and to a relatively small number of cases, our study demonstrated that screening of two common etiologies (GJB2 and GJB6) can characterize one in five patients with NSSNHL of unknown etiology. The relative frequency of the recurrent c.35delG mutation—64% of all mutated GJB2 alleles—is in agreement with data reported by others [9, 10], as well as, the whole mtDNA in all SNHL patients.
In the present investigation, mutations in the GJB2 gene were very frequent among NSSNHL patients, accounted for 23.6% (25/106) of mutated alleles and appear particularly frequent in South European patients [9, 10]. Among the DFNB-causing GJB2 gene mutations reported so far, the c.35delG mutation accounts for most of mutant alleles (60–85%) in Caucasians, from which 10–50% was present in only one allele. Our data confirms the presence of the common GJB6-D13S1830 deletion in two patients, showing a digenic inheritance in keeping with the severity of their hearing deficit.
We also identified two possibly pathogenic novel variants in GJB2. The new p.P70R that substitutes a proline (iminoacid) at codon 70 to an arginine (basic aminoacid) was predicted to be probably damaging by PolyPhen (The score is 2.108), and arginine was not tolerated when analyzed with the prediction software SIFT. Whether the mutation results in NSSNHL inherited in a dominant fashion remains a possibility. Conversely, the novel p.R127QfsX84 predicts a frameshift with a shorter, prematurely truncated connexin-26.
We also detected four mutations in mtDNA, corresponding to 4.2% of the all cohort. The m.1555A>G mutation has been reported in families whose members presented with aminoglycoside-induced deafness as the sole pathologic feature, but it may cause hearing loss even without aminoglycoside exposure, as in our case, which represents ~1% of this cohort. In our study, this mutation was found in a patient with ataxia and failure to thrive. The m.3243A>G mutation determines not only nonsyndromic, but also syndromic SNHL, such as in MELAS and MIDD (maternally inherited diabetes and deafness) [1]. The case identified in the present work was a 12-year-old boy diagnosed at the age of 8 years with syndromic SNHL; he developed hypertrophic cardiomyopathy and generalized muscle atrophy four years later. The m.7445A>G mutation was detected in a 13-year-old boy who presented a history of moderate progressive hearing loss. This mutation is believed to have led to failure in the processing of the L-strand RNA precursor, thereby reducing the steady-state levels of tRNASer(UCN) and ND6 mRNA [27].
We also found a novel mutation (m.5558A>G) in the tRNATrp in a 10-year-old boy with NSSNHL. The mutation affects the conserved A49 nucleotide in the T-stem of the tRNA (Figure 1) leading to a possible disruption of its secondary structure after the loss of the T-stem. This modification could also affect the tertiary structure of the tRNATrp and has probably a consequence on the aminoacylation. Another mutation (m.5559A>G) has been reported in the T-stem of the tRNATrp [35], and two additional tRNATrp mutations (namely, m.5540A>G and m.5568A>G) have been associated with SNHL [36, 37].
Figure 1.
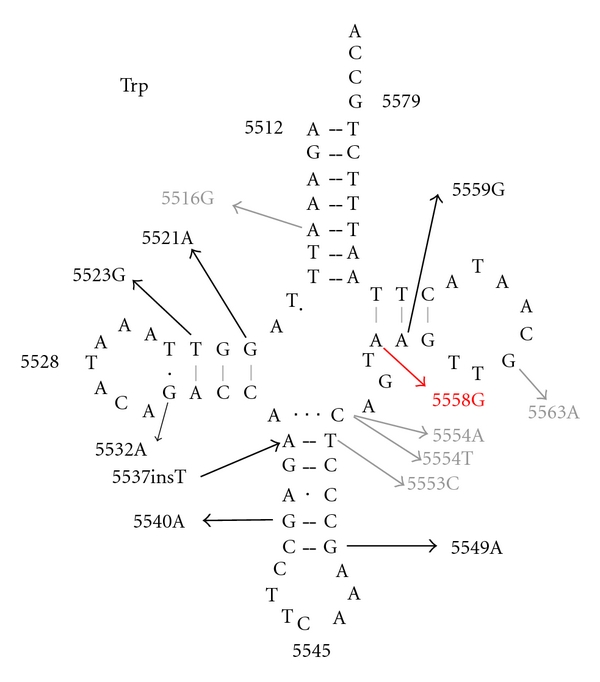
Schematic secondary structure of the wild-type human mitochondrial tRNATrp gene showing the reported variants found in this gene. Black stands for pathogenic mutations, gray stands for polymorphic mutations, and red stands for the novel mutation described in our study.
5. Conclusions
Although the contribution of less common genes remains to be determined, our results suggest that analysis of the GJB2 gene may have clinical implications in the diagnosis of deaf Portuguese children. Also, it would make feasible early rehabilitation and prevention in affected families. The relatively higher incidence of mtDNA mutation also suggests that screening for variations in the mitochondrial genome should always be considered unless mitochondrial inheritance can be excluded for certain. The molecular diagnosis will permit more accurate genetic counseling for family members, monitor possible multisystem complications, and avoid usage of aminoglycosides if infections occur.
Acknowledgment
This study was supported by the grant of the Comissão de Fomento da Investigação em Cuidados de Saúde do Ministério da Saúde, Project 254/2001.
References
- 1.Matsunaga T. Value of genetic testing in the otological approach for sensorineural hearing loss. Keio Journal of Medicine. 2009;58(4):216–222. doi: 10.2302/kjm.58.216. [DOI] [PubMed] [Google Scholar]
- 2.Utrera R, Ridaura V, Rodríguez Y, et al. Detection of the 35delG/GJB2 and del(GJB6-D13S1830) mutations in Venezuelan patients with autosomal recessive nonsyndromic hearing loss. Genetic Testing. 2007;11(4):347–352. doi: 10.1089/gte.2006.0526. [DOI] [PubMed] [Google Scholar]
- 3.Vele O, Schrijver I. Inherited hearing loss: molecular genetics and diagnostic testing. Expert Opinion on Medical Diagnostics. 2008;2(3):231–248. doi: 10.1517/17530059.2.3.231. [DOI] [PubMed] [Google Scholar]
- 4.Van Camp G, Smith R. Hereditary Hearing Loss. 2010, http://hereditaryhearingloss.org/
- 5.Kenneson A, Van Naarden Braun K, Boyle C. GJB2 (connexin 26) variants and nonsyndromic sensorineural hearing loss: a HuGE review. Genetics in Medicine. 2002;4(4):258–274. doi: 10.1097/00125817-200207000-00004. [DOI] [PubMed] [Google Scholar]
- 6.Putcha GV, Bejjani BA, Bleoo S, et al. A multicenter study of the frequency and distribution of GJB2 and GJB6 mutations in a large North American cohort. Genetics in Medicine. 2007;9(7):413–426. doi: 10.1097GIM.0b013e3180a03276. [DOI] [PubMed] [Google Scholar]
- 7.Ahmad S, Chen S, Sun J, Lin X. Connexins 26 and 30 are co-assembled to form gap junctions in the cochlea of mice. Biochemical and Biophysical Research Communications. 2003;307(2):362–368. doi: 10.1016/s0006-291x(03)01166-5. [DOI] [PubMed] [Google Scholar]
- 8.Zhao HB, Kikuchi T, Ngezahayo A, White TW. Gap junctions and cochlear homeostasis. Journal of Membrane Biology. 2006;209(2-3):177–186. doi: 10.1007/s00232-005-0832-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zelante L, Gasparini P, Estivill X, et al. Connexin26 mutations associated with the most common form of non-syndromic neurosensory autosomal recessive deafness (DFNB1) in Mediterraneans. Human Molecular Genetics. 1997;6(9):1605–1609. doi: 10.1093/hmg/6.9.1605. [DOI] [PubMed] [Google Scholar]
- 10.Pandya A, Arnos KS, Xia XJ, et al. Frequency and distribution of GJB2 (connexin 26) and GJBG (connexin 30) mutations in a large North American repository of deaf probands. Genetics in Medicine. 2003;5(4):295–303. doi: 10.1097/01.GIM.0000078026.01140.68. [DOI] [PubMed] [Google Scholar]
- 11.Grifa A, A Wagner C, D’Ambrosio L, et al. Mutations in GJB6 cause nonsyndromic autosomal dominant deafness at DFNA3 locus. Nature Genetics. 1999;23(1):16–18. doi: 10.1038/12612. [DOI] [PubMed] [Google Scholar]
- 12.Fuse Y, Doi K, Hasegawa T, Sugii A, Hibino H, Kubo T. Three novel connexin26 gene mutations in autosomal recessive nonsyndromic deafness. NeuroReport. 1999;10(9):1853–1857. doi: 10.1097/00001756-199906230-00010. [DOI] [PubMed] [Google Scholar]
- 13.Holt IJ, Harding AE, Morgan-Hughes JA. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature. 1988;331(6158):717–719. doi: 10.1038/331717a0. [DOI] [PubMed] [Google Scholar]
- 14.Zeviani M, Moraes CT, DiMauro S, et al. Deletions of mitochondrial DNA in Kearns-Sayre syndrome. Neurology. 1988;38(9):1339–1346. doi: 10.1212/wnl.38.9.1339. [DOI] [PubMed] [Google Scholar]
- 15.Swift AC, Singh SD. Hearing impairment and the Kearns-Sayre syndrome. Journal of Laryngology and Otology. 1988;102(7):626–627. doi: 10.1017/s0022215100105912. [DOI] [PubMed] [Google Scholar]
- 16.Shoffner JM, Lott MT, Lezza AMS, Seibel P, Ballinger SW, Wallace DC. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNALys mutation. Cell. 1990;61(6):931–937. doi: 10.1016/0092-8674(90)90059-n. [DOI] [PubMed] [Google Scholar]
- 17.Silvestri G, Ciafaloni E, Santorelli FM, et al. Clinical features associated with the A→G transition at nucleotide 8344 of mtDNA (‘MERRF mutation’) Neurology. 1993;43(6):1200–1206. doi: 10.1212/wnl.43.6.1200. [DOI] [PubMed] [Google Scholar]
- 18.Goto YI, Nonaka I, Horai S. A mutation in the tRNA(LeuUUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990;348(6302):651–653. doi: 10.1038/348651a0. [DOI] [PubMed] [Google Scholar]
- 19.Van Den Ouweland JMW, Lemkes HHPJ, Ruitenbeek W, et al. Mutation in mitochondrial tRMALeu(UUR) gene in a large pedigree with maternally transmitted type II diabetes mellitus and deafness. Nature Genetics. 1992;1(5):368–371. doi: 10.1038/ng0892-368. [DOI] [PubMed] [Google Scholar]
- 20.Fischel-Ghodsian N. Mitochondrial deafness mutations reviewed. Human Mutation. 1999;13(4):261–270. doi: 10.1002/(SICI)1098-1004(1999)13:4<261::AID-HUMU1>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 21.Lingala HB, Sankarathi, Penagaluru PR. Role of connexin 26 (GJB2) & mitochondrial small ribosomal RNA (mt 12S rRNA) genes in sporadic & aminoglycoside-induced non syndromic hearing impairment. Indian Journal of Medical Research. 2009;130(4):369–378. [PubMed] [Google Scholar]
- 22.Li R, Xing G, Yan M, et al. Cosegregation of C-insertion at position 961 with the A1555G mutation of the mitochondrial 12S rRNA gene in a large Chinese family with maternally inherited hearing loss. American Journal of Medical Genetics. 2004;124(2):113–117. doi: 10.1002/ajmg.a.20305. [DOI] [PubMed] [Google Scholar]
- 23.Li Z, Li R, Chen J, et al. Mutational analysis of the mitochondrial 12S rRNA gene in Chinese pediatric subjects with aminoglycoside-induced and non-syndromic hearing loss. Human Genetics. 2005;117(1):9–15. doi: 10.1007/s00439-005-1276-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tessa A, Giannotti A, Tieri L, Vilarinho L, Marotta G, Santorelli FM. Maternally inherited deafness associated with a T1095C mutation in the mDNA. European Journal of Human Genetics. 2001;9(2):147–149. doi: 10.1038/sj.ejhg.5200601. [DOI] [PubMed] [Google Scholar]
- 25.Zhao H, Li R, Wang Q, et al. Maternally inherited aminoglycoside-induced and nonsyndromic deafness is associated with the novel C1494T mutation in the mitochondrial 12S rRNa gene in a large Chinese family. American Journal of Human Genetics. 2004;74(1):139–152. doi: 10.1086/381133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mkaouar-Rebai E, Fendri-Kriaa N, Louhichi N, et al. Whole mitochondrial genome screening in two families with hearing loss: detection of a novel mutation in the 12S rRNA gene. Bioscience Reports. 2010;30(6):405–411. doi: 10.1042/BSR20090120. [DOI] [PubMed] [Google Scholar]
- 27.Chen J, Yuan H, Lu J, et al. Mutations at position 7445 in the precursor of mitochondrial tRNASer(UCN) gene in three maternal Chinese pedigrees with sensorineural hearing loss. Mitochondrion. 2008;8(4):285–292. doi: 10.1016/j.mito.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 28.Ghirri P, Liumbruno A, Lunardi S, et al. Universal neonatal audiological screening: experience of the University Hospital of Pisa. Italian Journal of Pediatrics. 2011;37, article 16 doi: 10.1186/1824-7288-37-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Del Castillo I, Villamar M, Moreno-Pelayo MA, et al. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. New England Journal of Medicine. 2002;346(4):243–249. doi: 10.1056/NEJMoa012052. [DOI] [PubMed] [Google Scholar]
- 30.Kelsell DP, Dunlop J, Stevens HP, et al. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature. 1997;387(6628):80–83. doi: 10.1038/387080a0. [DOI] [PubMed] [Google Scholar]
- 31.Denoyelle F, Weil D, Maw MA, et al. Prelingual deafness: high prevalence of a 30delG mutation in the connexin 26 gene. Human Molecular Genetics. 1997;6(12):2173–2177. doi: 10.1093/hmg/6.12.2173. [DOI] [PubMed] [Google Scholar]
- 32.Batissoco AC, Auricchio MTBM, Kimura L, Tabith-Junior A, Mingroni-Netto RC. A novel missense mutation p.L76P in the GJB2 gene causing nonsyndromic recessive deafness in a Brazilian family. Brazilian Journal of Medical and Biological Research. 2009;42(2):168–171. doi: 10.1590/s0100-879x2009000200004. [DOI] [PubMed] [Google Scholar]
- 33.Kelley PM, Harris DJ, Comer BC, et al. Novel mutations in the connexin 26 gene (GJB2) that cause autosomal recessive (DFNB1) hearing loss. American Journal of Human Genetics. 1998;62(4):792–799. doi: 10.1086/301807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reid FM, Vernham GA, Jacobs HT. A novel mitochondrial point mutation in a maternal pedigree with sensorineural deafness. Human Mutation. 1994;3(3):243–247. doi: 10.1002/humu.1380030311. [DOI] [PubMed] [Google Scholar]
- 35.Mkaouar-Rebai E, Chamkha I, Kammoun F, et al. Two new mutations in the MT-TW gene leading to the disruption of the secondary structure of the tRNATrp in patients with Leigh syndrome. Molecular Genetics and Metabolism. 2009;97(3):179–184. doi: 10.1016/j.ymgme.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 36.Silvestri G, Mongini T, Odoardi F, et al. A new mtDNA mutation associated with a progressive encephalopathy and cytochrome c oxidase deficiency. Neurology. 2000;54(8):1693–1696. doi: 10.1212/wnl.54.8.1693. [DOI] [PubMed] [Google Scholar]
- 37.Jacobs HT, Hutchin TP, Käppi T, et al. Mitochondrial DNA mutations in patients with postlingual, nonsyndromic hearing impairment. European Journal of Human Genetics. 2005;13(1):26–33. doi: 10.1038/sj.ejhg.5201250. [DOI] [PubMed] [Google Scholar]