

Abstract
Background
Interaction of Helicobacter pylori with gastric mucosa leads to marked cellular and humoral host immunological responses. The signaling pathways initiated by bacteria-host interaction that result in perturbations in cell structure and function remain unclear. Forkhead transcription factors of class O (FoxO) are implicated in the regulation of apoptosis, cell survival, and pathogenesis. H. pylori infection of gastric epithelial cells induces phosphoinositide-3 kinase (PI3K)-dependent Akt activation and cell survival signaling. We investigated the role of H. pylori-activated PI3K/Akt in the regulation of FoxO1/3a in gastric cells.
Methods
Immunoblot, immunoprecipitation, and fluorescence microscopy were used to assess the effect of infection of gastric epithelial cells with wild-type H. pylori and their isogenic cag pathogenicity island (PAI) or oipA mutants on the FoxO1/3a signaling pathways. Interleukin-8 release was determined by enzyme-linked immunosorbent assays.
Results
H. pylori infection resulted in activation of the PI3K p85 subunit and inactivation of FoxO1 and FoxO3a by their phosphorylation and translocation of from the nucleus to the cytoplasm. Inhibition of PI3K or Akt kinase activity reduced FoxO1/3a phosphorylation. Akt, FoxO1, or FoxO3a siRNA reduced H. pylori-induced interleukin-8 production. Infection with oipA mutants reduced PI3K/Akt activation and inhibited FoxO1/3a phosphorylation, whereas infection with cag PAI mutants reduced PI3K/Akt activity but did not inhibit FoxO1/3a activation.
Conclusions
FoxO1 and FoxO3a are novel nuclear substrates of H. pylori-induced PI3K/Akt cell survival signaling pathways that partially control interleukin-8 production. OipA-regulated interleukin-8 release through PI3K/Akt is dependent on FoxO1/3a inactivation whereas cag PAI-mediated interleukin-8 production employs FoxO1/3-independent signaling.
Introduction
Helicobacter pylori infection is the major risk factor for gastric cancer, the second most common cause of cancer-related death worldwide. H. pylori-induced host immunological responses include inflammation of the gastric mucosa characterized by production of proinflammatory cytokines, especially interleukin (IL)-8 which is thought to play a major role in the pathogenesis of H. pylori-associated diseases (1). Increased levels of mucosal IL-8 in gastric cancer tissues are also thought to enhance angiogenesis and the metastatic potential; IL-8 levels have been shown to be a prognostic factor in patients with gastric cancer (2). Bacterial risk factors for H. pylori-related gastric cancer include the expression of proinflammatory virulence factors such as the cag pathogenicity island (PAI), which encodes a type IV secretion system that injects CagA into host cells, and oipA, which encodes the outer inflammatory protein (OipA), an outer membrane protein. Both factors regulate IL-8 production and are involved in the pathogenesis of gastric diseases (3, 4). However, the signaling pathways initiated by interaction of bacteria with host cell membranes that result in perturbations in cell structure and function remain unclear.
One pathway through which H. pylori influences cellular homeostasis involves phosphoinositide-3 kinase (PI3K) and its downstream effector protein kinase B (PKB/Akt), a serine/threonine protein kinase (5). We previously showed that H. pylori-mediated activation of PI3K and Akt played an important role in the regulation of IL-8 production and suggested that these findings provided a potential mechanism for H. pylori-related inflammatory signaling likely involved in gastric carcinogenesis (6). Other laboratories have also shown that H. pylori-mediated activation of Akt plays roles in cell migration, cell survival, and regulation of apoptosis (7, 8). In addition, we showed that both OipA and cag PAI activate site-specific phosphorylation of PI3K/Akt signaling (6).
FoxO family members, including the functionally related proteins FoxO1, FoxO3a, FoxO4, and FoxO6, are downstream effectors of PI3K/Akt signaling. In the absence of growth factors or stimuli, FoxO resides in the nucleus bound to DNA or other transcription factors (transcriptionally active) to regulate genes such as those involved in cell cycle, apoptosis, and oxidative stress. Akt is known to phosphorylate FoxOs (inactivation) resulting in the suppression of transactivation, dissociation from DNA, and promotion of its translocation from the nucleus to the cytoplasm (the transcriptionally inactive form). Its inactivation and degradation results in inhibition of transcriptional activity that affects diverse cellular functions, including cell cycle arrest, gene expression and DNA damage implicated in apoptosis and cytokine production. These cellular events have been implicated in H. pylori-associated carcinogenesis (8–12). However, there are currently no reports describing the role of H. pylori in the regulation of FoxO family transcriptional regulators in gastric epithelial cells. We hypothesized that FoxO transcription factors are involved in H. pylori-mediated PI3K/Akt-dependent signaling, thereby regulating the induction of gastric inflammatory cytokines, especially IL-8. In the present study, we examined the relationship of FoxO and H. pylori-mediated cellular signaling and found that OipA plays a major role in the regulation of FoxO1/3a.
Methods
Reagents
Phospho-specific antibodies (PI3K p85 [Tyr458], Akt [Ser473], FoxO1/3a [Thr24/Thr32], FoxO1 [Ser256], and FoxO3a [Ser318 and Ser321]), total polyclonal antibodies (PI3K p85, Akt, FoxO1, and FoxO3a), affinity-purified horseradish peroxidase (HRP)-linked goat anti-rabbit IgG (H and L) or goat anti-mouse IgG, and the LumiGLO® reagent chemiluminescent substrate detection system were purchased from Cell Signaling Technology, Inc. (Beverly, MA). Immunoprecipitation reagents were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). The mammalian FoxO1 or FoxO3a small interfering RNA (siRNA) and Lipofectamine 2000 were purchased from Invitrogen Corp. (Carlsbad, CA), and the Akt siRNA SignalSilence® kit was from Cell Signaling Technology, Inc. Mouse monoclonal anti-β-actin antibody, Ponceau S solution, and a protease inhibitor cocktail were purchased from Sigma-Aldrich (St. Louis, MO), mouse monoclonal Histone H1 antibody was from Santa Cruz Biotech. (Santa Cruz, CA) and the selective inhibitor of PI3K (LY294002) was from Calbiochem (San Diego, CA).
Cell Culture
Human gastric epithelial cancer cells (AGS; American Type Culture Collection, Manassas, VA), MKN28, and MKN45 (both from Riken Bank, Tsukuba, Japan) were grown in RPMI 1640 medium supplemented with 1% penicillin/streptomycin solution and 10% heat-inactivated fetal bovine serum (FBS) at 37°C in a 5% CO2 humidified atmosphere. Cells were seeded at a density of 1 × 105 cells/well in 6-well plates and 1 × 106 cells/well in 10-cm dishes. Before treatment, cells were washed, serum-starved overnight at 80% confluence, and left untreated in RPMI 1640 medium, or were co-cultured with H. pylori for the specified times and at the specified multiplicities of infection (MOI) as described in the text and figure legends.
H. pylori
Functional oipA-positive/cag PAI-positive H. pylori strains TN2GF4, ATCC43504, and 26695, and their isogenic oipA mutants with intact type IV secretion system or cag PAI-deleted mutants in which CagA was also deleted, were used in this study(3, 13). The oipA/cag PAI double mutant of strain TN2GF4 was also used (13) in some experiments. The H. pylori strain TN2GF4 was isolated from a Japanese gastric ulcer patient and has been shown to cause gastric cancer in Mongolian gerbils (14). Bacteria were cultured on brain heart infusion agar plates containing 7% horse blood and incubated at 37°C under microaerophilic conditions for 24 to 36 h. The bacteria were suspended in phosphate buffered saline (PBS). The density was estimated by spectrophotometry (A625) and microscopic observation.
Protein Extraction, Immunoblotting, and Immunoprecipitation
Total proteins were extracted using cell lysis buffer (Cell Signaling Technology, Inc. Beverly, MA) containing a protease inhibitor cocktail (Sigma). Immunoblotting was performed using whole-cell extracts prepared from AGS cultures either uninfected or infected with H. pylori at the indicated MOI or time. For preparing cytoplasmic and nuclear fractions, serum starved AGS cells (approximately 5×105 cells/ mL) were left uninfected or infected with H. pylori at an MOI of 100. Cell fractions were prepared using hypotonic/nonionic detergent lysis buffers and cytoplasmic and nuclear proteins were normalized using protein assay (Bio-Rad Lab., Hercules, CA), as described previously(4). Equal amounts of protein were resolved by 8% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes (Bio-Rad Laboratories). The blots were stained with Ponceau S solution to visualize proteins and to further process desired proteins. The blots were incubated with the indicated primary antibodies for overnight at 4°C followed by detection with the respective secondary antibodies and visualization with a chemiluminescent detection system according to the manufacturer’s instructions. Semi-quantitative analyses were performed by quantifying the bands on the scanned radiographic films using the Image J 1.36 software (http://rsbweb.nih.gov/ij/) from the National Institutes of Health. Statistical analyses were performed using the Mann-Whitney Rank Sum test and the paired t test depending on the data set of interest using the statistical software SigmaStat 3.01 (Ashburn, VA). P < 0.05 was established as statistically significant.
For immunoprecipitation, equal amounts of protein from uninfected (control) or infected samples were incubated with the indicated antibodies for 2 h at 4°C and antigen-antibody complexes were precipitated with Protein A/G Sepharose (Amersham Biosciences, Piscataway, NJ). The samples were washed, boiled in Laemmli buffer for 3 min, subjected to 8% SDS-PAGE, and transferred to nitrocellulose membranes, followed by probing with specific antibodies as described in the text.
Immunofluorescence Analysis
FoxO3a phosphorylation and translocation in AGS cells were visualized by fluorescence microscopy (Olympus, America Inc., Melville, NY). Uninfected cells and cells infected with H. pylori for 30 min were fixed in 3.7% formaldehyde for 10 min at room temperature. Cells were permeabilized with 0.1% Triton X-100 in PBS for 5 min at 4°C and blocked with 5% normal goat serum for 30 min at room temperature. To visualize FoxO3a localization, cells were incubated with a phospho-specific FoxO3a Ser318/321 antibody or a total FoxO3a antibody (10 µg/mL) overnight at 4°C, followed by incubation with fluorescein isothiocyanate (FITC)-conjugated anti-rabbit IgG secondary antibody, Alexa Fluor 594 phalloidin, and 4′-6-diamidino-2-phenylindole (DAPI). To prevent photobleaching, the SlowFade Antifade kit (Molecular Probes, Eugene, OR) was used as recommended by the manufacturer.
Small Interfering RNA Transfection and Silencing of Akt and FoxO1/3a Protein Expression
To silence Akt or FoxO1/3 expression in AGS cells, Akt, FoxO1, or FoxO3a siRNAs were transfected according to the manufacturer’s instructions. Briefly, AGS cells were transfected with 100 nM nonspecific siRNA (negative control) or specific siRNA in serum-free culture medium using the Lipofectamine 2000 transfection reagent. The cells were cultured for 72 to 96 h post transfection. Cells were starved overnight in serum-free medium before infection with H. pylori at an MOI of 100 for 1 h for immunoblotting using the Akt siRNA and for 21 h for measuring IL-8 production in the Akt and FoxO1/3a knockdown experiments.
Determination and quantification of IL-8
IL-8 produced from H. pylori-infected AGS cells was detected using an enzyme-linked immunosorbent assay (ELISA) (R&D Systems, Minneapolis, MN) as previously described (4). Briefly, AGS cells were grown in 24-well plates and cultured for 2 days. Serum-starved cells that were 80% confluent were pre-incubated with control siRNA, Akt, FoxO1, or FoxO3a siRNAs as described above, followed by H. pylori infection at an MOI of 100 for 21 h. Culture supernatants were collected and assayed for IL-8 production by using an ELISA. Results were expressed as median from triplicate samples/number of experiments, and statistical significance was determined using the Mann-Whitney U test. P < 0.05 was considered statistically significant.
RESULTS
H. pylori-induced Tyrosine Phosphorylation of the PI3K p85 Subunit
We initially measured the effects of the wild-type H. pylori strain TN2GF4 on the phosphorylation of Tyr458 of the PI3K p85 subunit, a major regulator of PI3K/Akt signaling, using cell lysates from AGS cells infected with H. pylori for 1 h at various MOIs. There was an H. pylori MOI-dependent progressive increase in tyrosine phosphorylation of the PI3K p85 subunit (Figure 1A). PI3K p85 phosphorylation levels adjusted to the total PI3K levels were almost identical to those adjusted to the levels of β-actin. In agreement with our previous study (6), time course experiments demonstrated an increase in phosphorylation of PI3K at 5 min after infection; infection at an MOI of 100 for 1 h resulted in maximum phosphorylation of PI3K (data not shown). Similar patterns were observed using other gastric epithelial cells (MKN28 or MKN45 cells) and different H. pylori strains (data not shown).
Figure 1. H. pylori-induced tyrosine phosphorylation of the PI3K p85 subunit.

(A) Equal amounts of total cell lysates from mock-treated AGS cells or cells infected with wild-type H. pylori at an MOI of 12.5 to 200 for 1 h were analyzed by immunoblot using the indicated phospho-specific PI3K, PI3K p85 subunit, or β-actin antibodies. Mock-treated control cells were incubated for 1 h. The density of phospho-specific PI3K was normalized to that of total PI3K and the levels were expressed as fold increase compared with those of mock-treated control cells. At least 3 independent co-cultures were performed. Data are presented as average values ± SE. * P < 0.05, ** P < 0.01 versus mock-treated control cells.
(B) Equal amounts of total cell lysates from mock-treated AGS cells or cells infected with wild-type H. pylori at an MOI of 100 for 1 h were immunoprecipitated with PI3K antibody and immune complexes were analyzed by immunoblot using phospho-specific PI3K p85 or total PI3K p85 antibodies. Data are representative of 3 separate experiments. IP: immunoprecipitation
The specificity of H. pylori-mediated PI3K activation was confirmed by immunoprecipitation with PI3K antibody, followed by immunoblotting with an antibody to the phospho-PI3K p85 subunit (Figure 1B). PI3K phosphorylation was not detected in cell lysates from either mock-treated or infected cells by using normal rabbit IgG or immunoprecipitated with protein A-sepharose beads alone (data not shown).
Effect of H. pylori on FoxO1/3a Phosphorylation
To determine if FoxO1 and FoxO3a phosphorylation was modulated by H. pylori infection, cell lysates from uninfected AGS cells or from cells infected with H. pylori strain TN2GF4 for 1 h were analyzed using a phospho-FoxO1/3a antibody that recognizes both proteins, phosphorylated FoxO1 (Thr24) and FoxO3a (Thr32). H. pylori infection induced phosphorylation of FoxO1 and FoxO3a with maximum levels at an MOI of 100 (Figure 2A), while total FoxO1 or FoxO3a protein levels were not affected by H. pylori infection at the tested concentrations at 1 h (data not shown). Data for an MOI of 200 were variable possibly because, as we previously showed, cell viability was not changed (compared with uninfected cells) at an MOI of 100 but decreased at higher MOI’s (15). We therefore used an MOI of 100 in subsequent experiments. Time course experiments showed that increased phosphorylation of FoxO1 and FoxO3a was evident within 15 min of H. pylori infection with maximum levels 1 h after infection (Figure 2B). Total FoxO1 or FoxO3a protein levels were not affected by H. pylori infection until 60 min and were decreased at 90 min (data not shown). Similar patterns were observed using other gastric epithelial cells and different H. pylori strains (data not shown). Therefore, we used AGS cells and the H. pylori TN2GF4 in subsequent experiments.
Figure 2. H. pylori-mediated inactivation of FoxO1/3a.

(A) Equal amounts of total cell lysates of mock-infected AGS cells or cells infected with H. pylori at an MOI of 12.5 to 200 for 1 h were analyzed by immunoblot analysis using phospho-specific FoxO1 (Thr32) or Fox3a (Thr24) antibodies.
(B) Equal amounts of total cell lysates of mock-treated AGS cells or cells infected with wild-type H. pylori (MOI of 100) for 5 to 90 min were analyzed by immunoblot analysis using FoxO1 (Thr32) or Fox3a (Thr24) antibodies.
(A)(B) For quantitation, the density of phospho-specific sites was normalized to that of β-actin and the levels were expressed as fold increase compared with those of mock-treated control cells. At least 3 independent co-cultures were performed. Data are presented as average values ± SE. * P < 0.05, ** P < 0.01 versus mock-treated control cells.
Immunofluorescence was used to determine the effect of H. pylori infection on phosphorylation and cytoplasmic translocation of FoxO3a in AGS cells using a phospho-specific FoxO3a Ser318/321 antibody. We cultured AGS cells with H. pylori for 30 min to avoid the effects of FoxO degradation. Mock-treated cells showed low basal levels of phosphorylated FoxO3a that were localized exclusively in the nucleus (Figure 3A). H. pylori infection resulted in nuclear exclusion of FoxO3a and markedly enhanced phosphorylation and translocation of FoxO3a to the cytoplasm (Figure 3A). These data are consistent with the notion that unphosphorylated FoxO3a remains in the nucleus and that H. pylori infection resulted in phosphorylation and cytoplasmic translocation of FoxO3a from the nucleus resulting in inactivation of the transcriptional activity of FoxO3a.
Figure 3. Localization of FoxO1/3a and H. pylori infection.
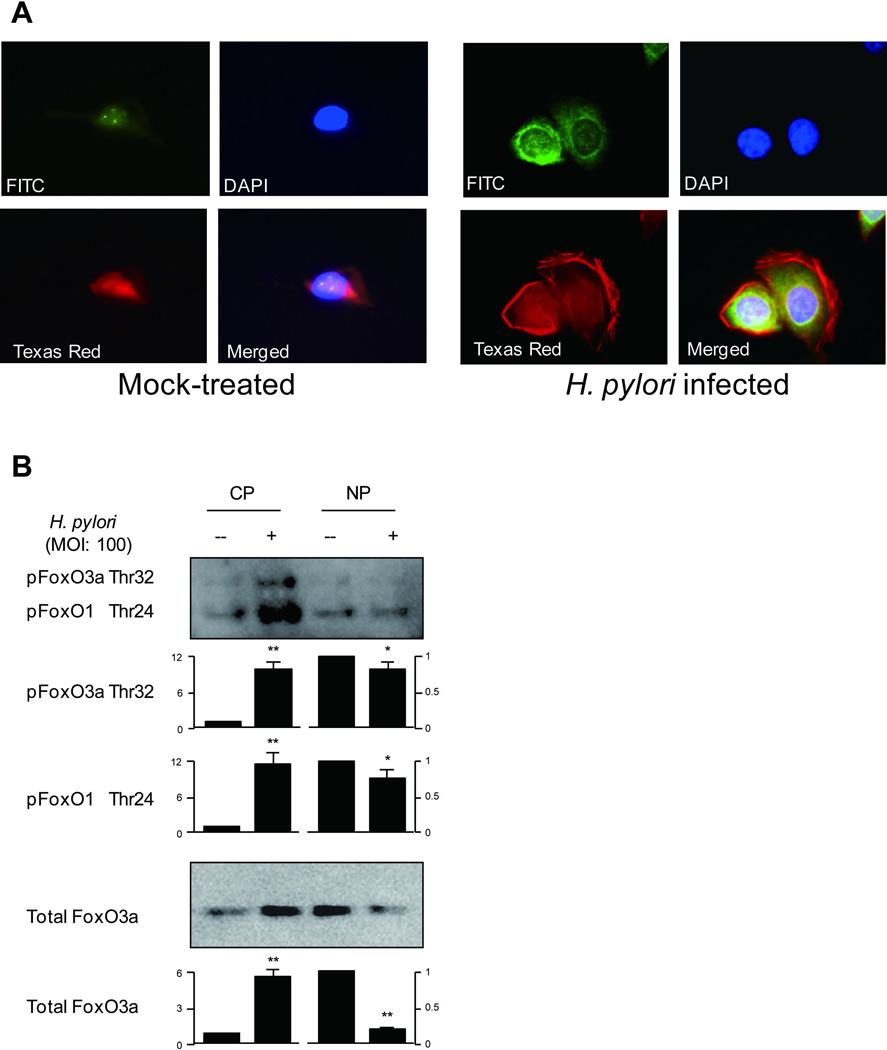
(A) AGS cells were mock-treated or infected with wild-type H. pylori (MOI of 100) for 30 min. Cells were incubated with phospho-specific FoxO3a Ser318/321 antibody followed by FITC-conjugated anti-rabbit IgG secondary antibody, Alexa Fluor 594 phalloidin, and DAPI to localize phosphorylated FoxO3a, F-actin, and the nucleus, respectively. Merged image was also shown. Cells were subjected to identical conditions for fixation, staining, and imaging. Images of each sample were taken in triplicate and we examined approximately 1,000 cells. Representative images are presented. The representative images shown in the right panel with H. pylori infection were in average observed in more than 80% of cells examined, as compared to in less than 10% of mock-treated cells.
(B) For Foxo1/3a subcellular distribution analysis, the cytoplasmic and nuclear proteins from mock-treated AGS cells or H. pylori infected for 1 hour were separated by SDS-PAGE analysis followed by immunoblotting with indicated antibodies. For quantitation, the density was normalized to that of β-actin for cytoplasm and Histone H1 for the nucleus. The levels were expressed as fold increase compared with those of mock-treated control cells. At least 3 independent co-cultures were performed. Data are presented as average values ± SE. * P < 0.05, ** P < 0.01 versus mock-treated control cells.
CP; cytoplasmic proteins, NP; nuclear proteins
We next used subcellular fraction experiments and phosphospecific FoxO1 (Thr24) and FoxO3a (Thr32) or FoxO3a antibodies to determine the status of total FoxO3a or phosphorylated FoxO1/3a in the cytoplasm or in the nucleus following H. pylori infection. Nuclear extracts from mock-treated cells exhibited high level of total FoxO3a and low basal levels of phosphorylated FoxO1/3a (Figure 3B). Interestingly, H. pylori infection resulted in enhancement of phosphorylated FoxO1/3a and total FoxO3a levels in the cytosol whereas the levels of phosphorylated FoxO1/3a and total FoxO3a levels in the nuclear fraction were reduced (Figure 3B).
Role of PI3K and Akt in the Regulation of H. pylori-induced FoxO1/3a Phosphorylation
Akt is a downstream target of PI3K. To examine the role of PI3K in the H. pylori-induced phosphorylation of Akt (Ser473) or FoxO1/3a (Thr24/Thr32), we pre-treated AGS cells with a PI3K chemical inhibitor (LY294002; 20 µM) followed by H. pylori infection (MOI of 100) and immunoblotting using the indicated antibodies (Figure 4A). Pre-incubation with LY294002 reduced H pylori-induced Akt phosphorylation as well as FoxO1/3a phosphorylation, suggesting that both Akt and FoxO1/3a are downstream targets of PI3K. LY294002 had no effect on the total levels of Akt or FoxO1/3a (data not shown).
Figure 4. Role of PI3K/Akt in H. pylori-mediated FoxO1/3a phosphorylation.

(A) AGS cells were left uninfected or pretreated with 20 µM of the PI3K inhibitor LY294002 followed by infection with wild-type H. pylori (MOI of 100) for 1 h. Whole cell lysates were analyzed by immunoblot using phospho-Akt Ser473 or FoxO1/3a (Thr32/Thr24) antibodies. At least 3 independent co-cultures were performed.
(B) Equal amounts of total cell lysates of mock-treated AGS cells, cells transfected with plasmid containing negative control sequence, or cells transfected with Akt-specific siRNA for 72 h followed by a 1-h infection with wild-type H. pylori were analyzed by SDS-PAGE and detected with phospho-specific AktSer473, total Akt, or FoxO1/3a (Thr32/Thr24) antibodies. At least 3 independent co-cultures were performed.
To investigate whether Akt regulates the function of FoxO1/3a, we inhibited the expression of Akt in AGS cells using specific Akt siRNA. The expression of total Akt and phosphorylated Akt was markedly downregulated by Akt-specific siRNA (Figure 4B). Cells transfected with nonspecific siRNA followed by H. pylori infection showed a marked increase in Akt and FoxO1/3a (Thr24/Thr32) phosphorylation compared to uninfected cells. In contrast, the amount of phosphorylated Akt was reduced to basal levels in lysates from cells transfected with Akt siRNA followed by H. pylori infection (Figure 4B). In addition, reduced expression of Akt followed by H. pylori infection resulted in inhibition of FoxO1/3a phosphorylation (Figure 4B), suggesting that Akt is located upstream of FoxO1/3a and plays a role in the inactivation and translocation of FoxO1/3a from the nucleus to the cytoplasm.
Relationship of H. pylori Virulence Factors with PI3K, Akt, and FoxO1/3a Phosphorylation
To understand the roles of the cag PAI and OipA in the regulation of PI3K, Akt, and FoxO1/3a, we used the wild-type H. pylori strain TN2GF4 and its cag PAI and oipA isogenic mutants. In agreement with our previous study (6), and compared to cells infected with wild-type H. pylori, phosphorylation of the PI3K p85 subunit and Akt was reduced in cells infected with the cag PAI and oipA mutants (data not shown) suggesting that both virulent factors are involved in PI3K/Akt signaling.
Next, we examined the effect of these virulence factors on the phosphorylation of FoxO1/3a (Thr24/Thr32), FoxO1a (Ser256), or FoxO3a (Ser318/321) (Figure 5). In wild-type H. pylori-infected cells, the phosphorylation (inactivation) of FoxO1 and FoxO3a was enhanced on both serine and threonine sites until 60 min; however, FoxO phosphorylation was markedly reduced (activated) in cells infected with the oipA mutant compared with those infected with wild-type H. pylori, independent of the infection time. Unexpectedly, at the early infection phase, FoxO1 and FoxO3a phosphorylation levels on both sites were mostly independent of the deletion of cag PAI. The phosphorylation levels at 90 min were even significantly higher in cells infected with the cag PAI mutant compared with those infected with wild-type H. pylori (Figure 5). The phosphorylation levels of FoxO1/3a threonine (Thr24/Thr32) at 60 min were also significantly higher in cells infected with the cag PAI mutant compared with those infected with wild-type H. pylori. Total FoxO1/3a levels were not affected by the deletion or mutation of cag PAI or oipA (data not shown). Overall, OipA appears to be the predominant factor involved in the phosphorylation and cytoplasmic translocation of FoxO1/3a (inactivation), whereas the cag PAI is required to maintain the nuclear retention and unphosphorylated form of FoxO1/3a (activation).
Figure 5. Role of cag PAI and OipA in FoxO1/3a phosphorylation.

AGS cells were mock-treated or infected with wild-type H. pylori, cag PAI mutant, or oipA mutant (MOI of 100) for 5 to 90 min. Equal amounts of total cell lysates were subjected to immunoblot analyses with the indicated antibodies. For quantitation, the density of phospho-specific sites was normalized to that of β-actin and the levels were expressed as fold increase compared with those of mock-treated control cells. Four independent co-cultures were performed. Data are presented as average values ± SE. * P < 0.05, ** P < 0.01 versus wild-type infected cells (corresponding time) when downregulated by the mutants, and # P < 0.05, ## P < 0.01 when upregulated by the mutants.
Pre-incubation with a PI3K inhibitor (LY294002) reduced phosphorylation (inactivation) of wild-type H. pylori-induced FoxO1/3a (Thr24/Thr32) further reduced the phosphorylation of FoxO1/3a in cells infected with the cag PAI mutant (Figure 6). This might suggest that cag PAI-independent factors or a feedback mechanism activated following introduction of CagA into host cells are involved in the PI3K-FoxO pathways. In contrast, FoxO1/3a (Thr24/Thr32) phosphorylation induced by the oipA mutant were very low even in the absence of the inhibitor but were unchanged by the presence of the PI3K inhibitor. These results suggest that OipA regulates FoxO1/3a inactivation through PI3K signaling pathways. Infection with the cag PAI and oipA double mutant resulted in complete inhibition of FoxO1/FoxO3a phosphorylation (Figure 6). Taken together, these data suggest that OipA plays a major role in the FoxO regulation mediated by the PI3K pathway.
Figure 6. Role of cag PAI and OipA in PI3K-FoxO pathways.

AGS cells were left uninfected or pretreated with 20 µM of the PI3K inhibitor LY294002 followed by infection with wild-type H. pylori, cag PAI mutant, oipA mutant or cag PAI/oipA mutants (MOI of 100) for 1 h. Equal amounts of total cell lysates were analyzed by immunoblot using phospho-FoxO1/3a antibodies. The same blots were reprobed with a β-actin antibody to verify equal loading. Four independent co-cultures were performed. Data are presented as average values ± SE. ** P < 0.01 versus infected cells without LY294002.
Reduced Expression of Akt or FoxO1/3a Inhibits H. pylori-induced IL-8 Production
To determine the role of Akt and FoxO1/3a in the regulation of IL-8 production, we abrogated Akt, FoxO1, or FoxO3a expression using the respective siRNAs, followed by H. pylori infection for 21 h. We previously showed that the IL-8 production reached maximum levels at 18 to 21 h after H pylori infection at an MOI of 100 and then plateaued until 30 h (4). Therefore, we co-cultured H. pylori strain TN2GF4 with AGS cells at an MOI of 100 for 21 h. In the presence of nonspecific siRNA, wild-type H. pylori induced IL-8 production; however, knockdown of FoxO1 or FoxO3a with the respective siRNA constructs resulted in an approximately 40% reduction in the H. pylori-induced IL-8 production (Figure 7). Knockdown of Akt resulted in an approximately 50% reduction in the H. pylori-induced IL-8 production (Figure 7). These results suggest that Akt, FoxO1, and FoxO3a are involved in the regulation of the H. pylori-induced IL-8 production.
Figure 7. Role of FoxO1/3a or Akt in H. pylori-induced IL-8 production.

To analyze the IL-8 levels by ELISA, we used equal amounts of culture supernatants of mock-treated AGS cells, cells transfected with a plasmid containing negative control sequence, or cells transfected with a plasmid containing siRNA sequence specific for FoxO1, FoxO3a, or Akt for 72 h followed by 21 h infection with wild-type H. pylori, cag PAI mutant, or oipA mutant (MOI of 100). Results are expressed as box plot from triplicate samples/number of experiments (total of 9 data sets), and statistical significance was determined using the Mann-Whitney U test. The end of the bars indicates the 25th and 75th percentiles. The 50th percentile (median) is indicated with a solid line in the box; the broken line indicates the mean value. The 10th and 90th percentiles are indicated with error bars. *: P < 0.05, **: P < 0.01 compared with wild-type H. pylori-infected cells transfected with control siRNA.
In FoxO1 or FoxO3a silencing experiments, infection of cells with the cag PAI mutant further inhibited IL-8 production to nearly basal levels (Figure 7). In contrast, the oipA mutant reduced IL-8 production to 50% of the levels induced with wild-type H. pylori infection, irrespective of the presence of FoxO1 or FoxO3a siRNA (Figure 7). Collectively, these results suggest that IL-8 induction via inactivation of FoxO1/3a is mainly regulated by OipA, while cag PAI-mediated IL-8 production requires FoxO1/3-independent signaling.
IL-8 levels induced by the cag PAI mutant were slightly, but not significantly reduced in the presence of Akt siRNA (Figure 7). IL-8 levels induced following infection with the oipA mutant were significantly lower (approximately 30% reduction in the presence of Akt siRNA) (Figure 6). These findings suggest that both OipA and cag PAI were partially involved in the IL-8 induction via Akt pathways.
Discussion
Expression of the PI3K/Akt signaling pathway has been linked to gastric carcinogenesis, (16) making this pathway an attractive target for novel gastric cancer therapies. We recently reported that activation of the PI3K/Akt signaling pathway plays an important role in the regulation of H. pylori-related inflammatory signaling related to gastric carcinogenesis (6). The role of H. pylori in the regulation of an Akt substrate, (ie, FoxO family of transcription factors) in gastric epithelial cells remained unknown. We showed that H. pylori induced phosphorylation (activation) of the PI3K p85 subunit and phosphorylation (inactivation) of FoxO1/3a (Thr24/Thr32), FoxO1 (Ser256), and FoxO3a (Ser318 and Ser321) leading to FoxO inactivation and translocation from the nucleus to the cytoplasm. Inhibiting PI3K signaling using a chemical inhibitor or abrogating Akt expression using specific siRNA reduced H. pylori-induced FoxO1/3a phosphorylation. On the basis of these results, we propose that PI3K/Akt signaling is required for H. pylori-induced inactivation of FoxO1/3a and its nuclear exclusion. We propose that FoxO1/3a are candidate transcriptional regulators involved in the regulation of H. pylori-mediated PI3K/Akt signaling in gastric epithelial cells.
FoxO1/3a is typically present in the nucleus in a dephosphorylated form and is bound to DNA or other transcription factors to regulate the transcriptional activity of its target genes. In general, stimulus-induced phosphorylation of FoxO1/3a leads to inactivation and cytoplasmic transportation, known as the transcriptionally inactive form, resulting in inhibition of the transcriptional activity of pro-apoptotic genes, apoptosis, and cytokine production (9, 11, 12, 17). This study confirmed that in mock-infected cells unphosphorylated FoxO or low basal phosphorylated FoxO remains in the nucleus, while H. pylori infection is associated with phosphorylation and cytoplasmic translocation of FoxO leading to inactivation of the transcriptional activity of FoxO. Recent reports suggested that post-translational modifications by phosphorylation influenced the cytoplasmic translocation of FoxO and its subcellular localization (18), suggesting that H. pylori-induced post-translational modifications via PI3K/Akt signaling affect FoxO1/3a activity.
We also found that reduced expression of Akt, FoxO1, or FoxO3a associated with use of the respective specific siRNAs reduced H. pylori-induced IL-8 production possibly through nuclear retention, inhibition of phosphorylation, and reduced cytoplasmic translocation of FoxO1/3a. These findings suggest that FoxO family members are novel substrates and effectors that mediate H. pylori-induced PI3K/Akt-dependent signaling to regulate genes responsible for H. pylori-mediated cytokine production. However, 2 recent reports from the same group reported opposite results (ie, that silencing of the FoxO3a expression increased the IL-8 production in human intestinal epithelial H-29 cells induced by tumor necrosis factor 2 or bacterial lipopolysaccharide)(12, 19). In human umbilical vein endothelial cells, however, knockdown of the FoxO1 expression using specific siRNA led to a profound decrease in the IL-8 gene expression (20). In primary aortic endothelial cells, knockdown of the FoxO1 expression using specific siRNA inhibited IL-6 production induced by the complement C5b-9 complex without causing a significant change in IL-8 production. Although the precise mechanism involved in reduced FoxO1 or FoxO3a expression and IL-8 production remained unknown, it is clear that H. pylori-mediated inactivation of FoxO1 and FoxO3a via phosphorylation, cytoplasmic translocation and retention (inactivation), and reduced expression of FoxO1/3a following treatment with specific siRNAs (ie, additional reduction of cytoplasmic FoxO1/3a levels and their phosphorylation) are completely different phenomena. However, it is also important to note that prolonged incubation of gastric epithelial cells with H. pylori (e.g., 21 h) exerted indirect effects on cytokine production such as IL-8 release and therefore further studies will be required to understand the molecular mechanism involved in PI3K/Akt mediated regulation of FoxO and IL-8 production.
We also showed that key H. pylori virulence factors, (ie, cag PAI and OipA) play important roles in the regulation of PI3K, Akt, and FoxO1/3a activation. In contrast to infection with wild-type H. pylori, infection with the oipA mutant resulted in reduced phosphorylation of FoxO1 and FoxO3a serine and threonine residues, suggesting that OipA plays an important role in the PI3K/Akt→FoxO1/3a pathway. In contrast, infection with the cag PAI mutant selectively increased FoxO1 or FoxO3a phosphorylation. Infection with the cag PAI/oipA double mutant resulted in complete inhibition of FoxO1/3a phosphorylation, suggesting that OipA, predominantly through PI3K/Akt signaling, influenced the nuclear retention, phosphorylation, and cytoplasmic translocation of FoxO1/3a. Since both cag PAI and OipA activate PI3K-Akt signaling, it seems unlikely that there are different effectors downstream of Akt activated by cag PAI and Akt activated by OipA. The oipA mutant contains an intact cag PAI type IV secretion system and the CagA oncoprotein, whereas the cag PAI mutant does not have a type IV secretion system or CagA. It is well known that CagA is injected into the host cells by the type IV secretion system and disturbs cellular signaling pathways. Therefore, CagA or other factors introduced into host cells via the type IV secretion system might provide a possible explanation for the unexpected phenomenon. For example, Akt→FoxO1/3a pathways might interact with these factors resulting in a feedback mechanism responsible for the inhibition of cag PAI-mediated FoxO phosphorylation. Our preliminary data showed that CagA formed complexes with paxillin, a downstream signaling molecule of Akt (Tabassam et al., unpublished data), and we suggest that these complexes might play a role in the regulation of Akt→FoxO1/3a pathways. Further studies will be necessary to clarify the roles of cag PAI-related molecules in the activation of Akt→FoxO1/3a pathways.
We showed that the IL-8 induction related to activation and cytoplasmic translocation of FoxO1/3a signaling is mainly regulated by OipA. However, FoxO1/3a signaling was not the major pathway in the IL-8 induction since, in general, the IL-8 production is predominantly regulated by cag PAI in wild-type infections(4). Compared to OipA, inhibition of the cag PAI function resulted in a more evident reduction in PI3K phosphorylation (6). Although OipA plays an important role in the PI3K/Akt→FoxO1/3a→IL-8 production, a FoxO1/3a-independent pathway for the PI3K/Akt→IL-8 production also exists, and cag PAI may be involved in this pathway.
In conclusion, H. pylori infection induced the PI3K/Akt-mediated phosphorylation, inactivation, and cytoplasmic translocation (transcriptionally inactive forms) of FoxO1 and FoxO3a. OipA is one of the major factors involved in post-translational modifications and inhibition of the DNA-binding activity of FoxO1/3a indicating that OipA negatively regulates the transcriptional activity of Fox1/3a. In contrast, cag PAI regulated FoxO1/3a activation and nuclear retention (transcriptionally active forms) and might have opposing effects to OipA in regulating FoxO-regulated signaling. The functional consequences of PI3K/Akt-dependent OipA- and cag PAI-regulated FoxO signaling remain to be determined. Reduced expression of Akt, FoxO1, or FoxO3a partially inhibits H. pylori-induced IL-8 production. We propose that FoxO1/3a may be a novel target in the regulation of H. pylori-induced PI3K/Akt-mediated cell survival signaling and IL-8 production through cytoplasmic translocation and inactivation of the transcriptional activity of FoxO1/3a (regulation of transcriptionally active and inactive forms). Targeting the transcriptionally active and inactive forms of FoxO through FoxO phosphorylation sites might represent a novel therapeutic approach for H. pylori-related pathogenic events.
Acknowledgments
Grant Support:
This report is based on work supported in part by grants from the National Institutes of Health DK62813, a Public Health Service grant (DK56338) which funds the Texas Gulf Coast Digestive Diseases Center and the Office of Research and Development Medical Research Service Department of Veterans Affairs.
Footnotes
Conflicts of interest
The author declares no competing interests.
References
- 1.Yamaoka Y. Mechanisms of disease: Helicobacter pylori virulence factors. Nat Rev Gastroenterol Hepatol. 2010 Nov;7(11):629–641. doi: 10.1038/nrgastro.2010.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kitadai Y, Haruma K, Sumii K, Yamamoto S, Ue T, Yokozaki H, et al. Expression of interleukin-8 correlates with vascularity in human gastric carcinomas. Am J Pathol. 1998 Jan;152(1):93–100. [PMC free article] [PubMed] [Google Scholar]
- 3.Yamaoka Y, Kwon DH, Graham DY. A M(r) 34,000 proinflammatory outer membrane protein (oipA) of Helicobacter pylori. Proc Natl Acad Sci U S A. 2000 Jun 20;97(13):7533–7538. doi: 10.1073/pnas.130079797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yamaoka Y, Kudo T, Lu H, Casola A, Brasier AR, Graham DY. Role of interferon-stimulated responsive element-like element in interleukin-8 promoter in Helicobacter pylori infection. Gastroenterology. 2004 Apr;126(4):1030–1043. doi: 10.1053/j.gastro.2003.12.048. [DOI] [PubMed] [Google Scholar]
- 5.Takeshima E, Tomimori K, Kawakami H, Ishikawa C, Sawada S, Tomita M, et al. NF-kappaB activation by Helicobacter pylori requires Akt-mediated phosphorylation of p65. BMC Microbiol. 2009;9:36. doi: 10.1186/1471-2180-9-36. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 6.Tabassam FH, Graham DY, Yamaoka Y. Helicobacter pylori activate epidermal growth factor receptor- and phosphatidylinositol 3-OH kinase-dependent Akt and glycogen synthase kinase 3beta phosphorylation. Cell Microbiol. 2009 Jan;11(1):70–82. doi: 10.1111/j.1462-5822.2008.01237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagy TA, Frey MR, Yan F, Israel DA, Polk DB, Peek RM., Jr Helicobacter pylori regulates cellular migration and apoptosis by activation of phosphatidylinositol 3-kinase signaling. J Infect Dis. 2009 Mar 1;199(5):641–651. doi: 10.1086/596660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yan F, Cao H, Chaturvedi R, Krishna U, Hobbs SS, Dempsey PJ, et al. Epidermal growth factor receptor activation protects gastric epithelial cells from Helicobacter pylori-induced apoptosis. Gastroenterology. 2009 Apr;136(4):1297–1293. doi: 10.1053/j.gastro.2008.12.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dansen TB, Burgering BM. Unravelling the tumor-suppressive functions of FOXO proteins. Trends Cell Biol. 2008 Sep;18(9):421–429. doi: 10.1016/j.tcb.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 10.Duronio V. The life of a cell: apoptosis regulation by the PI3K/PKB pathway. Biochem J. 2008 Nov 1;415(3):333–344. doi: 10.1042/BJ20081056. [DOI] [PubMed] [Google Scholar]
- 11.Hu MC, Lee DF, Xia W, Golfman LS, Ou-Yang F, Yang JY, et al. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell. 2004 Apr 16;117(2):225–237. doi: 10.1016/s0092-8674(04)00302-2. [DOI] [PubMed] [Google Scholar]
- 12.Snoeks L, Weber CR, Turner JR, Bhattacharyya M, Wasland K, Savkovic SD. Tumor suppressor Foxo3a is involved in the regulation of lipopolysaccharide-induced interleukin-8 in intestinal HT-29 cells. Infect Immun. 2008 Oct;76(10):4677–4685. doi: 10.1128/IAI.00227-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kudo T, Lu H, Wu JY, Graham DY, Casola A, Yamaoka Y. Regulation of RANTES promoter activation in gastric epithelial cells infected with Helicobacter pylori. Infect Immun. 2005 Nov;73(11):7602–7612. doi: 10.1128/IAI.73.11.7602-7612.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Watanabe T, Tada M, Nagai H, Sasaki S, Nakao M. Helicobacter pylori infection induces gastric cancer in Mongolian gerbils. Gastroenterology. 1998 Sep;115(3):642–648. doi: 10.1016/s0016-5085(98)70143-x. [DOI] [PubMed] [Google Scholar]
- 15.Choi IJ, Fujimoto S, Yamauchi K, Graham DY, Yamaoka Y. Helicobacter pylori environmental interactions: effect of acidic conditions on H. pylori-induced gastric mucosal interleukin-8 production. Cell Microbiol. 2007 Oct;9(10):2457–2469. doi: 10.1111/j.1462-5822.2007.00973.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kobayashi I, Semba S, Matsuda Y, Kuroda Y, Yokozaki H. Significance of Akt phosphorylation on tumor growth and vascular endothelial growth factor expression in human gastric carcinoma. Pathobiology. 2006;73(1):8–17. doi: 10.1159/000093087. [DOI] [PubMed] [Google Scholar]
- 17.Aoki M, Jiang H, Vogt PK. Proteasomal degradation of the FoxO1 transcriptional regulator in cells transformed by the P3k and Akt oncoproteins. Proc Natl Acad Sci U S A. 2004 Sep 14;101(37):13613–13617. doi: 10.1073/pnas.0405454101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Calnan DR, Brunet A. The FoxO code. Oncogene. 2008 Apr 7;27(16):2276–2288. doi: 10.1038/onc.2008.21. [DOI] [PubMed] [Google Scholar]
- 19.Snoeks L, Weber CR, Wasland K, Turner JR, Vainder C, Qi W, et al. Tumor suppressor FOXO3 participates in the regulation of intestinal inflammation. Lab Invest. 2009 Sep;89(9):1053–1062. doi: 10.1038/labinvest.2009.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Potente M, Urbich C, Sasaki K, Hofmann WK, Heeschen C, Aicher A, et al. Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. J Clin Invest. 2005 Sep;115(9):2382–2392. doi: 10.1172/JCI23126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fosbrink M, Niculescu F, Rus V, Shin ML, Rus H. C5b-9-induced endothelial cell proliferation and migration are dependent on Akt inactivation of forkhead transcription factor FOXO1. J Biol Chem. 2006 Jul 14;281(28):19009–19018. doi: 10.1074/jbc.M602055200. [DOI] [PubMed] [Google Scholar]