

Abstract
Objective
To describe the neuropathologic findings in three LRRK2 G2019S carriers with Parkinson’s disease (PD).
Methods
We cross referenced a list of 956 PD individuals that had been previously genotyped in clinical studies at Columbia University, with 282 subjects with a parkinsonian syndrome who came to autopsy in our brain bank since 1991. We found three autopsies of G2019S mutation carriers. Pathological analyses of the samples were blind to the genetic findings. We retrospectively reviewed the clinical records of the three patients.
Results
All three had a clinical and pathological diagnosis of PD. Cognitive impairment was a late feature in two out of three patients. Cortical involvement varied significantly: one had diffuse Lewy Body (LB) pathology, tau inclusions and amyloid pathology consistent with advanced Alzheimer’s disease; one had diffuse cortical LB and one had only brainstem predominant LB pathology.
Conclusions
Cognitive impairment may be a long term complication in G2019S mutation carriers. However, the extent of cortical involvement is variable. Larger longitudinal follow up of LRRK2 G2019S mutation carriers is required to assess for risk factors for cortical involvement and dementia.
Keywords: Parkinson’s disease, Lewy Bodies, LRRK2 gene mutation, Dementia
Introduction
LRRK2 G2019S mutations are among the most frequently identified genetic causes for Parkinson’s disease (PD) (Healy, Falchi et al. 2008). The International LRRK2 Consortium study estimated G2019S mutation frequency at 1% of sporadic and 4 % of familial PD patients (Healy, Falchi et al. 2008). The highest frequency was found in North African Arabs (36% in familial, 39% in sporadic) (Lesage, Durr et al. 2006) and Ashkenazi Jews (28% and 10% respectively) (Ozelius, Senthil et al. 2006). Clinically, G2019S mutation carriers who develop PD have a very similar disease to non-carriers, including the development of motor symptoms (e.g., rigidity and bradykinesia) and later cognitive difficulties (Healy, Falchi et al. 2008) (Belarbi, Hecham et al. 2010). Drug induced dyskinesia happens frequently, but may happen later than in non-carriers (Healy, Falchi et al. 2008). The neuropathology of mutation carriers is heterogeneous. Twenty-seven autopsy reports of LRRK2 G2019S mutation carriers have been published in the English literature. Twenty-four were obtained from patients with a clinical history of PD. Of these, twenty showed Lewy bodies (LBs) with variable neurofibrillary tangle burden (NFTs). Two had only tau inclusions and two had no distinctive inclusions (Wider, Dickson et al. 2010). However, most studies, including ours, only screened autopsies of patients with a clinical history of PD. Only five of the autopsy studies reporting these G2019S mutation carriers, screened patients with diagnoses other than PD (Gilks, Abou-Sleiman et al. 2005; Rajput, Dickson et al. 2006; Ross, Toft et al. 2006; Dachsel, Ross et al. 2007; Gaig, Ezquerra et al. 2008). To further explore the pathological characteristics, we describe the autopsy findings of three LRRK2 G2019S mutation carriers.
Methods
We have genotyped 956 PD patients for the LRRK2 G2019S mutation in clinical studies at Columbia University (Marder, Tang et al. 2010). Columbia University Institutional Review Board approved the protocols and consent procedures. Written informed consent was obtained from all participants in the study. Inclusion criteria for these studies required U.K. brain bank criteria for PD (Hughes, Daniel et al. 1992; Daniel and Lees 1993). We cross referenced the participants’ list with 282 patients with parkinsonism who had an autopsy in our center since 1991, including 98 PD, 72 Dementia with LB (DLB), 33 Progressive Supranuclear Palsy (PSP), 31 Multiple System Atrophy (MSA), 18 Corticobasal ganglionic degeneration (CBGD), 12 Alzheimer’s disease (AD), five Alzheimer’s disease Lewy body variant (ADLBV), seven Frontotemporal lobar degeneration (FTLD) and six Creutzfeldt-Jakob disease (CJD) patients. We found three G2019S mutation carriers in our brain bank; two out of 98 PD and one out of five ADLBV patients. Pathological analyses of the samples were blind to the genetic findings. We retrospectively reviewed the clinical records of these three patients.
Results
Patient 1 presented at age 44 with right hand rest tremor, micrographia and right foot dragging. PD was diagnosed. The patient had an excellent response to levodopa/carbidopa. Fifteen years after onset, dyskinesia developed on 750mg/day of levodopa. Seventeen years after onset, adrenal medullary transplantation improved anxiety and imbalance for 2 years. Then, its effect dissipated. Twenty six years after onset, falling resulted in physical dependence. Motor complications were improved by subthalamic nucleus deep brain stimulation, 32 years after onset. The patient died 34 years after onset at age 78. Cognitive decline was never documented. No other family member had been diagnosed with PD or other neurodegenerative disease.
Brain autopsy revealed severe neuronal loss in the dorsal nucleus of vagus (dnV), locus coeruleus (LC) and substantia nigra pars compacta (SNpc). LBs were present in the above locations. One LB was found in the substantia innominata (SI). Very rare NFTs were found in the Sommer sector of the hippocampal formation. Neuropathologic stage of PD (NSPD) according to Braak staging was 4/6.(Braak, Del Tredici et al. 2003) (Figure: panel A, D, G, J, M, P).
Figure.
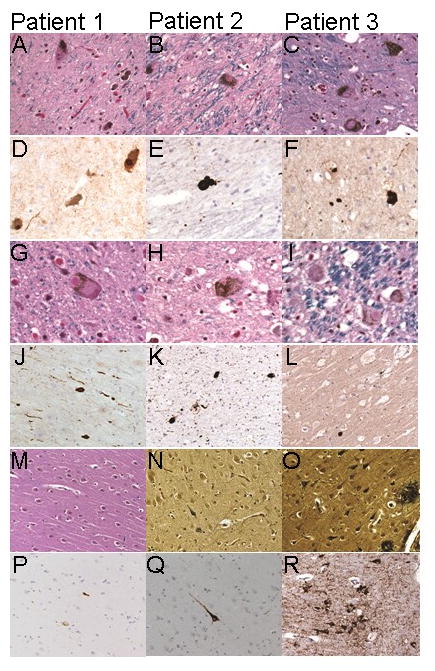
Pathological findings in three LRRK2 G2019S mutation carriers with PD.
Panel A–C [Luxol Haematoxylin Eosin (LHE) stain] shows Lewy Bodies (LB)s in the substantia nigra pars compacta (SNpc) (magnification 400x).
Panel D–F (alpha-synuclein stain) depicts alpha-synuclein reactive LBs and Lewy neurites (LNs) in the SNpc (magnification 400x).
Panel G–I (LHE stain) reveals LBs in the locus coeruleus (LC) (magnification 630x).
Panel J, K (substantia Innominata) and L (motor cortex) shows LBs and LNs reactive to alpha-synuclein stain (magnification 200x).
Picture M (LHE stain) shows a single NFT in the hippocampal formation cornus Ammonis 1 (CA1). Panel N and O (Bielschowsky) reveals NFTs and ghost tangles in CA1.
Panel P–R (AT8 stain) demonstrates tau-immunoreactive NFTs (magnification 200x).
Patient 2 presented at age 39 with right hand clenching, micrographia and foot dragging. The patient was diagnosed with PD and had an excellent response to levodopa. Dyskinesia developed five years after onset on 700mg/day of levodopa. The first fall occurred nine years after onset. Seventeen years after onset, a left pallidotomy improved the motor complications; its effect dissipated seven years later. Dementia was clinically diagnosed 24 years after onset of symptoms. The patient died 28 years after symptom onset. While there was no family history of PD, two siblings developed Crohn’s disease. Brain pathology showed severe neuronal loss in the LC and SNpc and moderate in the SI. LBs were found in the dnV, SNpc, hypothalamus, SI, amygdala and rarely in the entorhinal cortex, cingulate gyrus (3 per 100x microscopic field) and prefrontal cortex (2 throughout the slide). Rare NFTs were present in the Sommer sector, with few neuritic plaques in the parahippocampal gyrus. NSPD was 4/6. (Figure: panel B, E, H, K, N, Q).
Patient 3 presented at age 53 with right hand rest tremor, micrographia and depression. PD was diagnosed with very good response to levodopa/carbidopa. Ten years later, hyposmia was noted by the patient. Twelve years after onset, dyskinesia developed on 1000mg/day of levodopa. The first fall occurred 19 years into her illness. Twenty-one years after symptoms onset, dementia was diagnosed based on clinical impression. Marked worsening of imbalance, dementia and depression preceded death at age 84. The duration of symptoms was 31 years. Family history of PD and AD was positive only in third degree relatives. One sibling died of “colitis” as a young adult.
Autopsy showed severe neuronal loss in the LC, SNpc and the hippocampal Sommer sector. Few LBs were noted in the above locations, dnV, hypothalamus and the thalamus. Abundant LBs (up to 20 per 100x microscopic filed) were found in the SI, amygdala, entorhinal, cingulate, temporal, prefrontal and motor cortices. NFTs and neuritic plaques were few in the thalamus and striatum, but many in the amygdala, hippocampal formation and in all cortical lobes. NSPD was 6/6. The pathological diagnosis was ADLBV (Lippa, Smith et al. 1994) (Figure: panel C, F, I, L, O, R).
Discussion
The neuropathology associated with LRRK2 mutations is very heterogeneous and may include LB, tau inclusions, atrophy and normal pathology. The type of the LRRK2 mutation may explain part of this heterogeneity, where most autopsies of the G2019S and the I2020T mutations’ autopsies show LB pathology, while most of the R1441C mutations do not. In fact, there is a pathological variability even within the same family as shown in the initial report of R1441C carriers (Zimprich, Biskup et al. 2004).
While most of the G2019S autopsies show LB pathology, the literature (including our report) may be biased towards LB pathology, as many autopsy studies genotyped patients with clinical or pathological diagnosis of PD (Giasson, Covy et al. 2006; Gaig, Marti et al. 2007; Silveira-Moriyama, Guedes et al. 2008; Gomez and Ferrer 2010). In particular, all nine autopsy studies, reporting on 27 G2019S carriers, screened 891 patients with LB disorders (Gilks, Abou-Sleiman et al. 2005; Giasson, Covy et al. 2006; Rajput, Dickson et al. 2006; Ross, Toft et al. 2006; Gaig, Marti et al. 2007; Gaig, Ezquerra et al. 2008; Silveira-Moriyama, Guedes et al. 2008; Gomez and Ferrer 2010); 363 with PSP (Rajput, Dickson et al. 2006; Ross, Toft et al. 2006; Gaig, Ezquerra et al. 2008); 62 MSA (Rajput, Dickson et al. 2006; Ross, Toft et al. 2006; Gaig, Ezquerra et al. 2008); seven CBGD (Rajput, Dickson et al. 2006; Gaig, Ezquerra et al. 2008); 654 AD (Ross, Toft et al. 2006); 59 FTLD(Rajput, Dickson et al. 2006; Dachsel, Ross et al. 2007; Gaig, Ezquerra et al. 2008); 102 HD(Gilks, Abou-Sleiman et al. 2005); 16 ET (Rajput, Dickson et al. 2006), and 444 controls (Rajput, Dickson et al. 2006; Ross, Toft et al. 2006; Silveira-Moriyama, Guedes et al. 2008). However, publication bias may also exist, in that autopsies with uncommon findings are more likely to be reported.
None of the three patients reported here had a first degree family history of PD. This may be partially explained by the incomplete penetrance of G2019S mutations (Ozelius, Foroud et al. 2007). Two patients had a first degree family history of colitis. One patient had two siblings with Crohn’s disease. LRRK2 has been associated with Crohn’s disease in genome-wide association studies (Umeno, Asano et al. 2011); however, the genotypes of these siblings with Crohn’s in our cases are unknown.
All three cases reported here had brainstem LB pathology. However, these cases highlight the clinical and pathological heterogeneity of cortical involvement in LRRK2 G2019S mutation carriers. Clinically, cognitive performance in PD is diverse, but up to 83% of PD survivors after 20 years develop dementia (Hely, Reid et al. 2008). In our cases, dementia was present in two patients (disease duration was long in all three, ranging between 28 and 34 years). This sample is very small but not significantly dissimilar from idiopathic PD. On pathology, one case had little cortical involvement, one had LB involvement of the cortex and one had ADLBV. In the latter, it is unknown whether there is a link between the G2019S genotype and the AD pathology. Cortical involvement has not been consistently reported in the literature. Of the 20 G2019S autopsies reported in the English literature with Lewy bodies, seven were reported to have extensive cortical involvement (NSPD stage 5–6), 12 had brainstem-type PD (NSPD stages 3–4) and one did not include details on cortical involvement. Therefore, even in patients with the same genetic lesion, there is widely variable cortical involvement. Based on these cases, we hypothesize that other genetic and environmental risk factors predispose one to cortical involvement in G2019S mutation carriers. Larger series and longitudinal follow-up of LRRK2 G2019S mutation carriers are required to assess risk factors for cortical degeneration and dementia and to explore the pathological heterogeneity in autopsies.
Acknowledgments
Study funding: This study was supported by NIH grants: R01NS36630 (Dr. Marder), R01NS060113 (Dr. Clark), P50AG008702 (Dr. Honig), UL1 RR024156 (Dr. Alcalay) and the Parkinson’s Disease Foundation (Drs. Marder, Fahn and Clark).
Footnotes
Disclosures:
Dr. Poulopoulos, Dr. Cortes, Dr. Vonsattel, Dr. Cote and Ms. Moskowitz report no disclosures.
Dr. Fahn reports receiving support from consulting and advisory board membership with honoraria from Oxford Biomedica (Sept 2009), Proctor-Goodwin (Nov 2009), GE Healthcare (Nov 2009), RJG Foundation (March 2010), IMPAX Pharmaceuticals (May 2010), Lundbeck (June 2010). He is receiving research support from the Parkinson’s Disease Foundation (no salary support). He received a grant from the Smart Family Foundation (no salary support). He received a grant from the US Department of Defense’s Telemedicine and Advanced Technology Research Center (TATRC) for the World Parkinson Congress 2010, and a grant from the National Institutes of Health for the World Parkinson Congress 2010. Dr. Fahn received lecture honoraria from Columbia University (July 2009), Sun Pharmaceuticals India (Sept 2009), World Association of Sleep Medicine (Nov 2009), American Academy of Neurology (April 2010), Columbia University (July 2010). Dr. Fahn reports serving as an editor with author honoraria from “Current Neurology and Neurosurgery Report” (annual); Elsevier for co-author of book
Principles and Practice of Movement Disorders.
Dr. Waters received speaking honorarium from Boehringer and Teva.
Dr. Honig is funded by NIH grant P50AG008702.
Dr. Clark is funded by NIH grant R21NS050487.
Dr. Marder served on the editorial board of Neurology; is funded by NIH [NS36630 (PI), 1UL1 RR024156-01(Director PCIR), PO412196- G (Co-I), PO412196-G (Co-I)], and R01NS36630, and from the Parkinson Disease Foundation, Huntington’s Disease Society of America, the Parkinson Study Group and the Michael J Fox foundation.
Dr. Alcalay is funded by NIH (K12 part of UL1 RR024156) and the Brookdale Foundation, the Parkinson’s Disease Foundation and the Michael J Fox foundation and the Smart Foundation.
References
- Belarbi S, Hecham N, et al. LRRK2 G2019S mutation in Parkinson’s disease: a neuropsychological and neuropsychiatric study in a large Algerian cohort. Parkinsonism Relat Disord. 2010;16(10):676–679. doi: 10.1016/j.parkreldis.2010.09.003. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24(2):197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Dachsel JC, Ross OA, et al. Lrrk2 G2019S substitution in frontotemporal lobar degeneration with ubiquitin-immunoreactive neuronal inclusions. Acta Neuropathol. 2007;113(5):601–606. doi: 10.1007/s00401-006-0178-1. [DOI] [PubMed] [Google Scholar]
- Daniel SE, Lees AJ. Parkinson’s Disease Society Brain Bank, London: overview and research. Journal of neural transmission Supplementum. 1993;39:165–172. [PubMed] [Google Scholar]
- Gaig C, Ezquerra M, et al. Screening for the LRRK2 G2019S and codon-1441 mutations in a pathological series of parkinsonian syndromes and frontotemporal lobar degeneration. J Neurol Sci. 2008;270(1–2):94–98. doi: 10.1016/j.jns.2008.02.010. [DOI] [PubMed] [Google Scholar]
- Gaig C, Marti MJ, et al. G2019S LRRK2 mutation causing Parkinson’s disease without Lewy bodies. J Neurol Neurosurg Psychiatry. 2007;78(6):626–628. doi: 10.1136/jnnp.2006.107904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson BI, Covy JP, et al. Biochemical and pathological characterization of Lrrk2. Ann Neurol. 2006;59(2):315–322. doi: 10.1002/ana.20791. [DOI] [PubMed] [Google Scholar]
- Gilks WP, Abou-Sleiman PM, et al. A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet. 2005;365(9457):415–416. doi: 10.1016/S0140-6736(05)17830-1. [DOI] [PubMed] [Google Scholar]
- Gomez A, Ferrer I. Involvement of the cerebral cortex in Parkinson disease linked with G2019S LRRK2 mutation without cognitive impairment. Acta Neuropathol. 2010;120(2):155–167. doi: 10.1007/s00401-010-0669-y. [DOI] [PubMed] [Google Scholar]
- Healy DG, Falchi M, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 2008;7(7):583–590. doi: 10.1016/S1474-4422(08)70117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hely MA, Reid WG, et al. The Sydney multicenter study of Parkinson’s disease: the inevitability of dementia at 20 years. Mov Disord. 2008;23(6):837–844. doi: 10.1002/mds.21956. [DOI] [PubMed] [Google Scholar]
- Hughes AJ, Daniel SE, et al. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. 1992;55(3):181–184. doi: 10.1136/jnnp.55.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage S, Durr A, et al. LRRK2 G2019S as a cause of Parkinson’s disease in North African Arabs. N Engl J Med. 2006;354(4):422–423. doi: 10.1056/NEJMc055540. [DOI] [PubMed] [Google Scholar]
- Lippa CF, Smith TW, et al. Alzheimer’s disease and Lewy body disease: a comparative clinicopathological study. Annals of neurology. 1994;35(1):81–88. doi: 10.1002/ana.410350113. [DOI] [PubMed] [Google Scholar]
- Marder KS, Tang MX, et al. Predictors of parkin mutations in early-onset Parkinson disease: the consortium on risk for early-onset Parkinson disease study. Archives of neurology. 2010;67(6):731–738. doi: 10.1001/archneurol.2010.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozelius LJ, Foroud T, et al. G2019S mutation in the leucine-rich repeat kinase 2 gene is not associated with multiple system atrophy. Mov Disord. 2007;22(4):546–549. doi: 10.1002/mds.21343. [DOI] [PubMed] [Google Scholar]
- Ozelius LJ, Senthil G, et al. LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. N Engl J Med. 2006;354(4):424–425. doi: 10.1056/NEJMc055509. [DOI] [PubMed] [Google Scholar]
- Rajput A, Dickson DW, et al. Parkinsonism, Lrrk2 G2019S, and tau neuropathology. Neurology. 2006;67(8):1506–1508. doi: 10.1212/01.wnl.0000240220.33950.0c. [DOI] [PubMed] [Google Scholar]
- Ross OA, Toft M, et al. Lrrk2 and Lewy body disease. Ann Neurol. 2006;59(2):388–393. doi: 10.1002/ana.20731. [DOI] [PubMed] [Google Scholar]
- Silveira-Moriyama L, Guedes LC, et al. Hyposmia in G2019S LRRK2-related parkinsonism: clinical and pathologic data. Neurology. 2008;71(13):1021–1026. doi: 10.1212/01.wnl.0000326575.20829.45. [DOI] [PubMed] [Google Scholar]
- Umeno J, Asano K, et al. Meta-analysis of published studies identified eight additional common susceptibility loci for Crohn’s disease and ulcerative colitis. Inflamm Bowel Dis. 2011;17(12):2407–2415. doi: 10.1002/ibd.21651. [DOI] [PubMed] [Google Scholar]
- Wider C, Dickson DW, et al. Leucine-rich repeat kinase 2 gene-associated disease: redefining genotype-phenotype correlation. Neurodegener Dis. 2010;7(1–3):175–179. doi: 10.1159/000289232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44(4):601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]