

Abstract
Indoleamine 2,3-dioxygenase (IDO) modifies adaptive immunity, in part by determining the character of inflammatory responses in the tissue microenvironment. Small molecule inhibitors of IDO are being developed to treat cancer, chronic infections and other diseases, so the systemic effects of IDO disruption on inflammatory phenomena may influence the design and conduct of early phase clinical investigations of this new class of therapeutic agents. Here, we report cardiac and gastrointestinal phenotypes observed in IDO deficient mice that warrant consideration in planned assessments of the safety risks involved in clinical development of IDO inhibitors. Calcification of the cardiac endometrium proximal to the right ventricle was a sexually dimorphic strain-specific phenotype with ∼30% penetrance in BALB/c mice lacking IDO. Administration of complete Freund's adjuvant containing Toll-like receptor ligands known to induce IDO caused acute pancreatitis in IDO deficient mice, with implications for the design of planned combination studies of IDO inhibitors with cancer vaccines. In an established model of hyperlipidemia, IDO deficiency caused a dramatic elevation in levels of serum triglycerides. In the large intestine, IDO loss only slightly increased sensitivity to induction of acute colitis, but it markedly elevated tumor incidence, multiplicity and staging during inflammatory colon carcinogenesis. Together, our findings suggest potential cardiac and gastrointestinal risks of IDO inhibitors that should be monitored in patients as this new class of drugs enter early clinical development.
Keywords: IDO, heart calcification, pancreatitis, colitis, colon carcinoma, hyperlipidemia
Introduction
Inflammation is an important contributory factor in the development and progression of many age-associated human diseases including cancer.1,2 Recent work on the immune modulatory enzyme indoleamine 2,3-dioxygenase (IDO) suggests that it may support an essential defining feature of chronic pathologic inflammations associated with cancer, chronic infections, and other disorders.3–5 Based on a number of genetic and pharmacological studies of IDO function, several structurally distinct IDO inhibitors are presently entering clinical trials to evaluate their use in restoring anti-tumor immune functions that are restrained during malignant progression.6,7 Indeed, preclinical studies demonstrate that IDO activation poses a common immune escape barrier erected by tumors to thwart immune attack, the degradation of which can dramatically heighten responses to chemotherapy.8–11 These findings fit more generally with the growing evidence that cancer therapeutic outcomes might be improved by immunochemotherapy regimens capable of destroying cancer cells more effectively than either immunotherapy or chemotherapy alone.12–14 While IDO has been implicated in immune modulation, its genetic ablation in the mouse does not produce any gross immune deficiencies or inflammatory liabilities under laboratory conditions. However, there has been little study of how IDO disruption may affect inflammatory and immune processes that are induced under stress-related conditions that may better mimic a patient's health status. In this study, we report observations made over several years of study of IDO deficient mice that suggest possible sources of risk for which to monitor during clinical development of IDO inhibitors. These results are timely because of the multiple academic and pharmaceutical groups now advancing this new class of immunomodulators into early phase human trials.
Results
Calcification of the cardiac endometrium in IDO deficient mice is a partially penetrant strain-specific phenotype that is sexually dimorphic.
A comparison of tissue pathologies and histologies in C57BL6/J and BALB/c strains of IDO-deficient mice revealed a high incidence of heart calcification on the endometrium proximal to the right ventricle in BALB/c mice (Fig. 1). This phenotype was observed exclusively in female animals as young as 3 mo of age suggesting rapid onset. It was not observed in male animals on either the BALB/c or C57BL6/J genetic backgrounds, indicating sexual dimorphism as well as strain dependency. Examination of numerous animals at various ages indicated that the phenotype was ∼30% penetrant. Male BALB/c mice are reported to exhibit heart calcification but at a far lower penetrance of ∼3% observed.15 These observations suggested that in mice IDO acts as a strain specific modifier of risks of cardiac calcification in a sexually dimorphic manner.
Figure 1.
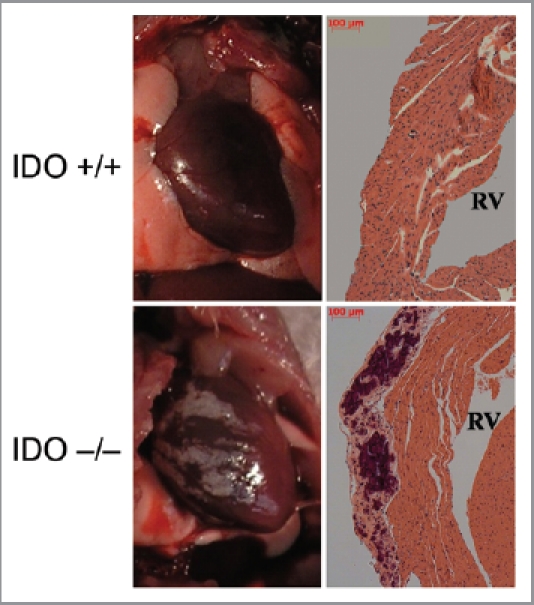
Calcification of the cardiac endometrium in IDO deficient BALB/c mice. Representative illustrations of gross pathology (left panels) and representative histology (right panels) are shown of calcification of the cardiac endometrium proximal to the right ventricle which were observed in female animals with a ∼30% penetrance. RV, right ventricle.
Complete Freund's adjuvant induces acute pancreatitis under conditions of IDO deficiency.
IDO exerts an important function in dendritic cells where it determines whether antigen presentation leads to immune activation or tolerization to the antigen. Consistent with this function IDO activity exerts a restraining influence on vaccine responses.16,17 In exploring the possible effects of IDO on vaccination regimens that produce autoimmune myocarditis in the mouse, we found that when co-administered with a peptide antigen the use of complete Freund's adjuvant (CFA) triggered the onset of acute pancreatitis in IDO-/- C57BL6/J mice, a phenotype that was evident by gross pathology in animals sacrificed 28 d after treatment (Fig. 2A). This phenotype was fully penetrant in female mice that were tested. No differences were observed in the autoimmune myocarditis induced by the peptide antigen, nor were there signs of a similarly acute inflammation in any other of the other major organs examined at necropsy (not shown). In humans, pancreatitis occurs as a rare side-effect of vaccination18 or as a rare response after infection by Mycobacterium tuberculosis, a component of CFA.19 In fact, we noted that CFA also produced signs of a mild pancreatitis in mice that were wild-type (WT), but not the far more florid pathology displayed in IDO-/- mice (Fig. 2B). Histological and immunohistological analyses performed at an endpoint of 28 d included alterations to the pancreatic insulin-producing islets (Fig. 2C). Insulin production was not abolished nor was there any significant alteration in serum glucose levels (data not shown), limiting the extent of this inflammatory condition over the course of 28 d. Together, these observations suggested that IDO deficiency increased risks of acute pancreatitis caused by Mycobacterium tuberculosis or CFA when administered with vaccine adjuvant.
Figure 2.
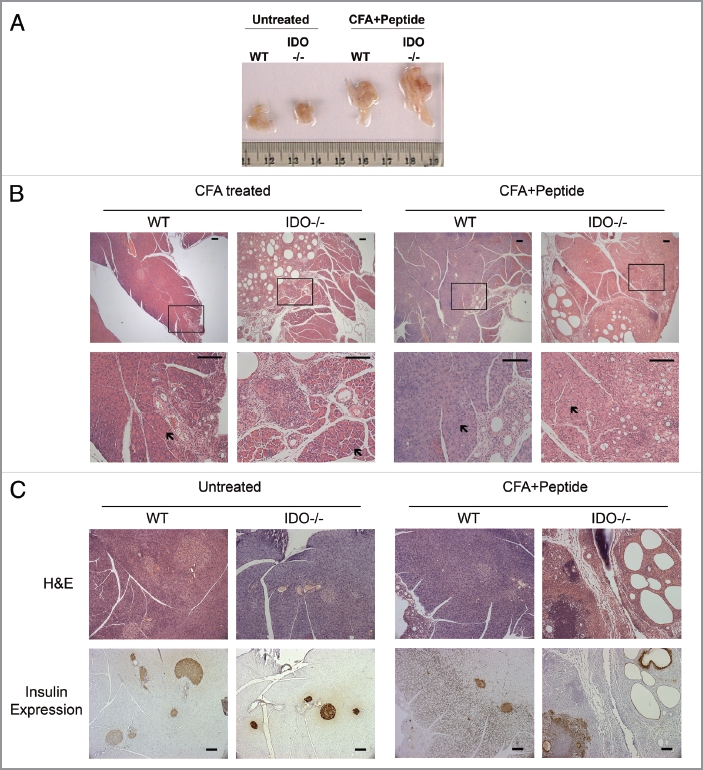
Acute pancreatitis induced by complete Freund's adjuvant in IDO deficient mice. (A) Gross pathology. Freshly dissected pancreata are shown to illustrate the marked increase in organ size produced in mice 28 d after treatment with CFA + peptide (untreated WT n = 5, untreated IDO-/- n = 4, CFA + Peptide treated WT n = 5, CFA + Peptide treated IDO-/- n = 5) (see Material and Methods). (B) Pancreas histology. Representative samples are shown to illustrate pancreatitis that developed after treated with CFA or CFA + Peptide in WT and IDO-/- mice. Lower magnification images are presented in the upper row and boxed areas are magnified in the lower row. Areas indicated by arrows show normal acinar regions of pancreatic tissue. Scale bar = 100 µM. (CFA treated WT n = 5, CFA treated IDO-/- n = 5, CFA + Peptide treated WT n = 5, CFA + Peptide treated IDO-/- n = 5). (C) Insulin expression. Pancreatic tissue sections were stained with H&E for histology or processed for immunohistochemical expression of insulin in islet β-cells. Brown staining demonstrates the presence of insulin containing β-cells in these tissues. Scale bar = 100 µM.
IDO deficiency promotes hyperlipidemia.
Another effect of IDO deficiency was determined in a mouse model of hyperlipidemia, where loss of IDO was sufficient to dramatically elevate the accumulation of lipids, specifically triglycerides, in blood serum (Fig. 3A). These experiments were conducted on theC57BL6/J strain background and while the phenotype was fully penetrant no sexual dimorphism was observed. Hypertriglyceridemia in the context of low HDL levels and insulin resistance contributes to increased risks of cardiovascular disease.20–22 Despite some controversy as an independent risk factor, an increase in average serum triglyceride levels in the U.S. has occurred since the 1980s with an estimated 13% of U.S. individuals having elevated triglycerides.20,23 While the mechanism and consequences of elevated triglycerides in the context of genetic IDO deficiency were not defined in our model, this phenotype illustrates an additional potential concern of IDO inhibition in the presence of an existing condition of hyperlipidemia or hypertriglyceridemia.
Figure 3.
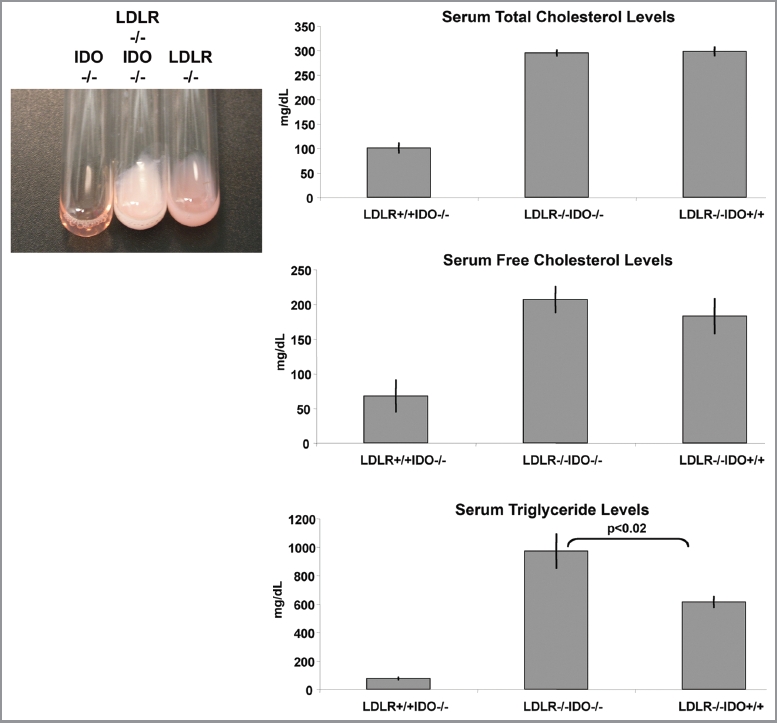
IDO deficiency accentuates hyperlipidemia in a mouse model of atherosclerosis driven by high fat diet. Blood serum samples were analyzed from WT, LDLR-/- or IDO-/-LDLR-/- mice on the C57BL6/J strain background. A representative illustration of hyperlipidemia present in mice fed a high fat diet is shown. Quantification of serum lipids demonstrated a significant increase in serum triglycerides in IDO-/-LDLR-/- mice fed an atherogenic diet (p < 0.02). No significant differences in levels of serum cholesterol were observed. Total mice tested in each group was IDO-/- (n = 10), IDO-/-LDLR-/- (n = 10), LDLR-/- (n = 9).
IDO deficiency increases sensitivity to colon carcinogenesis.
IDO has been reported to modify cancer-associated inflammation in skin and to alter inflammatory responses in the colon.10,24 To evaluate the effects of IDO as a modifier of intestinal inflammation, we used a common model of colitis that is induced by administration of low-molecular weight dextran sodium sulfate (DSS) in drinking water. By causing damage to the epithelium,25 DSS produces a strong inflammatory response due to destruction of mucin content or altered macrophage function associated with increased exposure to luminal antigens.26–28 After 3 d treatment with 3% DSS in drinking water, IDO-deficient mice exhibited slightly more sensitivity to colitis. However, this subtle effect was no longer apparent by 7 d when WT and IDO-/- mice both presented with short, bloody colons and severe colitis characterized by loss of crypts and epithelium and an accumulation of inflammatory cells (Table 1 and Fig. 4A). To assess this effect in a longer term assay, we compared the effects of IDO deletion on inflammatory colon carcinogenesis promoted by DSS. Briefly, tumors were initiated in WT or IDO-/- cohorts of mice by administrating a single i.p. dose of 20 mg/kg of dimethylhydrazine (DMH), a DNA alkylating carcinogen that induces tumors in the descending colon with a histopathology similar to sporadic colon cancer in humans. All mice then received a one week treatment of 2% DSS in drinking water followed by normal drinking water during the remaining course of the experiment. At the 18 week endpoint, we observed that 100% (8/8) of IDO-/- mice presented with colon tumors whereas only 25% (2/8) of WT mice presented with colon tumors (Table 2 and Fig. 4B). Additionally, tumor multiplicity was 4-fold higher in IDO-/- mice (n = 2.2 ± 0.3) compared with WT mice (n = 0.5 ± 0.4). IDO-/- mice also developed greater numbers of multiple carcinoma than WT mice (100 vs. 63%). In contrast, histological staging of tumors from IDO deficient mice indicated that they were not significantly more invasive in character than tumors from WT mice (Table S1). These results were obtained from experiments conducted on the C57BL6/J strain background and no sexual dimorphism was observed. The negative modifier effect of IDO was dose-dependent, insofar as administration of higher doses of 30 mg/kg DMH and 3% DSS in an otherwise identical carcinogenesis protocol nearly abolished the difference in tumor incidence, multiplicity and staging in WT and IDO-/- mice (Fig. S1 and Tables S2 and S3). In support of the notion that IDO acts as a negative modifier in this context, the expression level of IDO enzyme was elevated in colon tumors dissected from WT mice (data not shown). Overall, these results argued that IDO deficiency acted as a positive modifier of inflammatory insults in the colon to accentuate their impact on the formation and progression of chemically induced colon carcinomas.
Table 1.
Slight sensitizing effect of IDO deletion on acute induction of inflammatory colitis. WT and IDO-/- mice (n = 10) on the C57BL6/J strain background were provided 3% DSS in drinking water to induce colitis. Mice were euthanized 3 or 7 d after treatment was initiated and colons were dissected and processed for histological evaluation
| Genotype | Days DSS | Normal colon | Minimal colitis | Mild colitis | Severe colitis |
| WT | 3 | 4/10 (40%) | 0/10 (0%) | 6/10 (60%) | 0/10 (0%) |
| IDO-/- | 3 | 2/10 (20%) | 2/10 (20%) | 6/10 (60%) | 0/10 (0%) |
| WT | 7 | 0/10 (0%) | 0/10 (0%) | 0/10 (0%) | 10/10 (100%) |
| IDO-/- | 7 | 0/10 (0%) | 0/10 (0%) | 0/10 (0%) | 10/10 (100%) |
Figure 4.
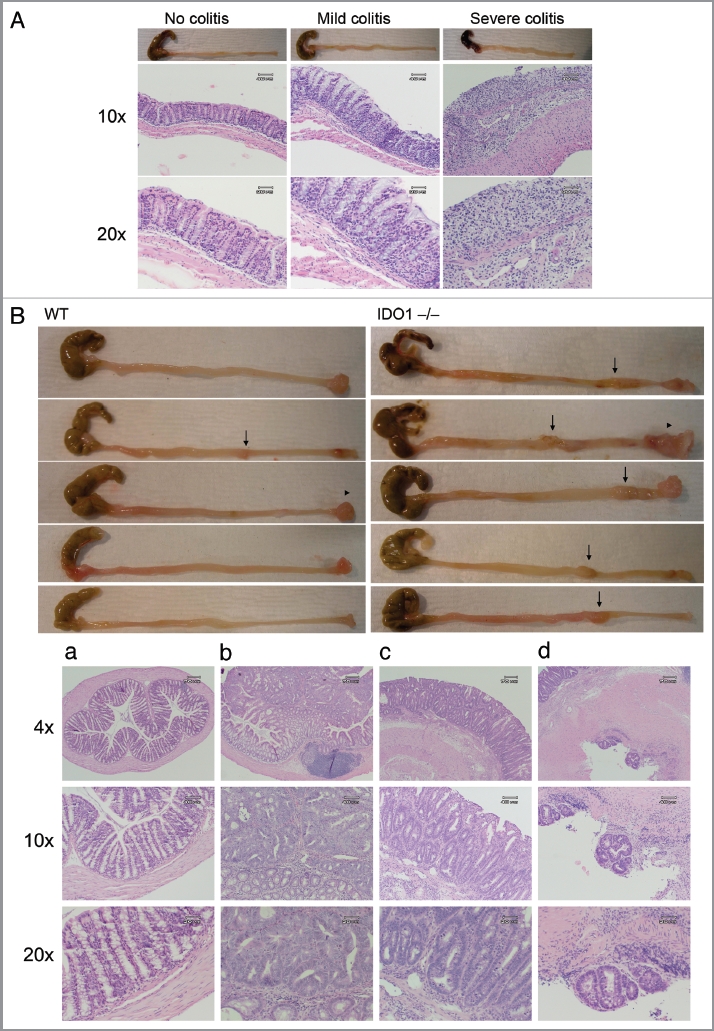
IDO deficiency accentuates the effects of inflammatory insults that promote colon carcinogenenesis. (A) Colitis induction. Colons from WT and IDO-/- mice treated 3 d with 3% DSS in drinking water were fixed in paraffin and processed for H&E staining. Representative examples scored for no colitis, mild colitis and severe colitis are shown at original magnifications of 10× (middle) or 20× (bottom). (B) Colon carcinogenesis. WT and IDO-/- mice were treated once i.p. with 20 mg/kg DMH and then 7 d 2% DSS in drinking water followed by normal drinking water to an endpoint of 18 wks. Colons were harvested from euthanized mice, fixed in paraffin and processed for H&E staining. Representative images of gross pathology in WT or IDO-/- mice (top panels) illustrate observations of macroscopic tumors (arrows) or prolapsed rectums (arrowheads). Representative images of histopathology in IDO-/- mice (bottom panels) illustrate observations of (a) no tumor, (b) mucosal invasion, (c) submucosal invasion and (d) muscular invasion. Original magnifications are 4× (top), 10× (middle), 20× (bottom).
Table 2.
IDO loss increases tumor incidence and multiplicity during inflammatory colon carcinogenesis
| Genotype | Tumor incidence | Tumors per mouse | Tumor multiplicity | Absence of carcinoma | Early carcinoma | Carcinoma | Multiple carcinoma |
| WT | 2/8 (25%) | 0.5 ± 0.4 | 1/2 (50%) | 1/8 (13%) | 0/8 (0%) | 2/8 (25%) | 5/8 (63%) |
| IDO-/- | 6/6 (100%) | 2.2 ± 0.3 | 5/6 (83%) | 0/6 (0%) | 0/6 (0%) | 0/6 (0%) | 6/6 (100%) |
Colon tumors were initiated in WT and IDO-/- mice on the C57BL6/J strain background with a single i.p. injection of 20 mg/kg DMH followed by 7 d treatment with 2% DSS in drinking water. All animals then received regular drinking water through the course of the experiment. Mice were euthanized at an endpoint of 18 weeks and colons were dissected for processing and histological evaluation.
Discussion
In documenting several cardiac and gastrointestinal liabilities exhibited in IDO-deficient mice, the observations presented here offer a source of possible safety concerns for oncologists to consider monitoring in patients who are recruited to trials of IDO inhibitors, a new class of small molecule drugs being investigated by a growing number of pharmaceutical, biotechnology and academic groups in early Phase I/II human trials. While our observations emerged from several years using these animals in preclinical studies of cancer, they also relate more widely to likely clinical trials of IDO inhibitors in other diseases characterized by pathologic chronic inflammations supported by IDO activity, including HIV, HCV and influenza infections, allergies, asthma and a variety of other autoimmune diseases like rheumatoid arthritis and lupus.4
Our findings deepen and extend the pathological relevance of IDO to inflammation in the gastrointestinal tract. IDO is a regulator of adaptive T-cell immunity but its functions as a modifier of innate inflammatory processes are still emerging.29 Genetic studies of IDO in the mouse do not support any function in classical acute inflammatory responses. However, IDO-deficient mice exhibit resistance to skin and lung cancers driven by chronic inflammation (ref. 10 and C. Smith, M.Y. Chang, G.C. Prendergast, A.J. Muller and colleagues, unpublished observations), suggesting that IDO is essential to generate cancer-associated chronic inflammatory states that support tumoral immune escape. Indeed, studies in IDO deficient mice have prompted the concept that cancer-associated inflammation and immune escape are genetically synonymous.4 Inflammatory bowel diseases (IBDs) such as Crohn's disease and ulcerative colitis30 are implicated in promoting tumorigenesis of the intestinal epithelium.2,31 Intestinal immunity downregulates the inflammatory response against dietary antigens and commensal bacteria present in the intestine. IDO is highly expressed in normal colon and its expression is upregulated by inflammation,32,33 perhaps as a feedback control to prevent colitis.34 In contrast to skin and lung, our findings argued that IDO functions as a negative modifier of inflammatory colon carcinogenesis, in contrast to the positive modifying effects of IDO observed in skin and lung carcinogenesis as noted above. A negative modifier effect in the colon is consistent with the evidence that IDO restrains inflammatory colitis.24 Moreover, it is notable that this phenotype is correlated inversely with the increased susceptibility to gastrointestinal inflammation and colon carcinogenesis displayed by mice lacking Bin1, a tumor suppressor that is a key IDO regulator.35 What is the basis for the different tumor suppressive effects of IDO in colon? One explanation may be based on the ability of IDO to promote formation of T regulatory cells (Tregs) in cancer.36 While Tregs are generally supportive in most settings of cancer, Tregs actually limit rather than promote tumorigenesis in the colon.37,38 Thus, the different contributions of IDO as a modifier of susceptibility to inflammatory cancers may parallel the different contributions made by Tregs in cancer, possibly explaining why IDO is suppressive like Tregs to colon cancer. Whether or not this interpretation is borne out by future work, in considering the impact of our findings on IDO inhibitor development it seems reasonable to raise concerns of increased risks of progression for polyps or occult dormant tumors that may exist in patients receiving treatment with an IDO inhibitor.
We also noted that IDO deficiency increased hyperlipidemia in the context of an established mouse model of atherosclerosis development and progression, namely, diet-induced atherosclerosis in LDLR-deficient mice. While the basis for this phenotype was undefined, our observations raise concerns of an increased risk of morbidly elevated serum lipid profiles in patients who receive treatment with an IDO inhibitor.
IDO deficiency also produced a dramatic sensitivity to pancreatitis triggered by administration of Freund's adjuvant, containing Mycobacterium tuberculosis. This classical vaccine adjuvant contains tuberculin antigens that stimulate Toll receptor (TLR) signaling and immune responsiveness. IDO expression is strongly upregulated by TLR signals in a variety of tissues possibly as a negative feedback control on TLR-mediated immune stimulation.11,24,39–41 Thus, a more potent TLR stimulation elicited by Freund's adjuvant in the absence of IDO may lead to an unrestrained inflammatory response in the pancreas. The reason for the exquisite sensitivity of this organ in particular is obscure, but may relate to the development of a rare clinical condition, tuberculosis of the pancreas.19 Clinical testing of IDO inhibitors is expected to include a wide variety of combinations with approved and experimental agents, including cancer vaccines. Thus, in terms of their clinical relevance, our observations suggest caution in the use of Freund's adjuvant, other adjuvants or components of adjuvants containing TLR ligands in patients receiving treatment with an IDO inhibitor, due to the risks of inducing an acute pancreatitis that could require emergency attention.
Heart calcification was another liability to IDO deficiency that we observed, albeit only in the BALB/c mouse strain where it is known to have a very low incidence, and only at partial but substantial penetrance in females. While survival was impacted in affected animals through several months of age, consistent with earlier studies of this phenotype in BALB/c mice,42 we did not characterize specific effects on heart function or associated morbidities. Thus, it is clear that IDO deficiency is insufficient on its own to elicit heart calcification, and also insufficient to limit survival. However, in considering clinical development of an IDO inhibitor, the emergence of heart calcification proximal to the right ventricle in even a small number of patients receiving treatment would clearly raise safety concerns that might be insurmountable. In conclusion, an extensive histopathological analysis of IDO deficient mice reveals a number of potential concerns to clinical development of IDO inhibitors, perhaps offering a source of preclinical information that could assist in the design and conduct of suitable Phase I/II protocols to assess the relative safety of this new class of immunomodulatory agents as therapeutic adjuvants or neoadjuvants being developed by a growing number of research groups.
Material and Methods
Mouse strains and diets.
C57BL6/J and BALB/c strains of IDO-/- mice43 were kindly provided by A. Mellor (Medical College of Georgia). LDLR-/- mice on the C57BL6/J strain as a genetic model of hyperlipidemia were obtained from Jackson Laboratories (002207). LDLR-/-IDO-/- double knockout animals were created by backcrossing F1 offspring produced by interbreeding single knockout animals on the same congenic C57BL6/J strain. PCR genotyping by standard methods was performed as described43 or Jackson Laboratory website for the LDLR allele. In the hyperlipidemia studies LDLR-/-, IDO-/- and LDLR-/-IDO-/- mice were fed a “Western” atherogenic diet (Harlan/Teklad, 88137; 21% anhydrous milkfat-butterfat, 34% sucrose and a total of 0.2% cholesterol) for a total of 3 mo beginning at 4 weeks of age. All experimental procedures were approved by the Lankenau Institute Animal Care and Use Committee.
Vaccination.
Two-month-old mice were injected s.c. with complete Freund's adjuvant (CFA) with or without 100 µg cardiac myosin heavy chain peptide that produced experimental autoimmune myocarditis (EAM), an inflammation of the heart.44,45 Immunizations were repeated by injecting with or without the peptide in CFA at day 7. All mice were sacrificed 28 d after first injection.
Chemically induced colitis and colon carcinogenesis.
To induce colitis, mice on the C57BL6/J background strain at 7 wk of age were administered 3% dextran sodium sulfate (DSS, MP Biomedicals, 160110, MW 36–50kDa) in drinking water. At 3 and 7 d after DSS treatment animals were sacrificed and colons were inspected for macroscopic and microscopic pathological lesions. For colon carcinogenesis, mice on the same background strain at 7 wk of age were administered a single intraperitoneal dose of the genotoxic colon carcinogen 1,2-dimethylhydrazine (DMH) at 20 or 30 mg/kg body weight as indicated in the Results. One week later, animals were administered 2 or 3% DSS in drinking water as indicated for 7 d followed by regular water for 16–18 weeks46 at which time they were euthanized and examined for pathologic colon lesions.
Histopathological and expression analysis.
Mice were euthanized as indicated and tissues isolated at necropsy were fixed in 10% neutral buffered formalin or 4% paraformaldehyde, sectioned, and stained for histopathological analysis with hemotoxylin and eosin using standard methods. IDO expression was documented in tissues where indicated by immunoprecipitationprotein gel analysis as described elsewhere.47 Briefly, protein gel blots were processed by standard methods using anti-mouse IDO (mIDO-48 clone, BioLegend, 122401) and HRP-conjugated goat anti-rat secondary antibody (1:1000 dilution) (Southern Biotechnology Associates, 3010-05), using a commercial luminescence kit for detection (ECL-Western, Amersham).
Serum lipid analysis.
Total and free serum cholesterol and triglyceride levels were measured from atherogenic diet fed LDLR-/-, IDO-/- and LDLR-/-IDO-/- mice using colorimetric assays (Wako Chemicals USA, Inc.). Data were calculated as average +/2SEM and were examined using t-tests to test for differences in mean values.
Supplementary Material
Disclosure of Potential Conflicts of Interest
J.B.D., A.J.M and G.C.P. communicate conflicts of interest with regard to their roles as shareholders, grantees and advisors to NewLink Genetics Corporation, which is developing IDO clinical trials.
References
- 1.Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7:211–217. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 3.Muller AJ, Mandik-Nayak L, Prendergast GC. Beyond immunosuppression: reconsidering indoleamine 2,3-dioxygenase as a pathogenic element of chronic inflammation. Immunotherapy. 2010;2:293–297. doi: 10.2217/imt.10.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prendergast GC, Metz R, Muller AJ. Towards a genetic definition of cancer-associated inflammation: role of the IDO pathway. Am J Pathol. 2010;176:2082–2087. doi: 10.2353/ajpath.2010.091173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. 2007;117:1147–1154. doi: 10.1172/JCI31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muller AJ, Scherle PA. Targeting the mechanisms of tumoral immune tolerance with small-molecule inhibitors. Nat Rev Cancer. 2006;6:613–625. doi: 10.1038/nrc1929. [DOI] [PubMed] [Google Scholar]
- 7.Prendergast GC. Immune escape as a fundamental trait of cancer: focus on IDO. Oncogene. 2008;27:3889–3900. doi: 10.1038/onc.2008.35. [DOI] [PubMed] [Google Scholar]
- 8.Muller AJ, DuHadaway JB, Sutanto-Ward E, Donover PS, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunomodulatory target of the tumor suppressor gene Bin1, potentiates cancer chemotherapy. Nat Med. 2005;11:312–319. doi: 10.1038/nm1196. [DOI] [PubMed] [Google Scholar]
- 9.Hou DY, Muller AJ, Sharma MD, DuHadaway J, Banerjee T, Johnson M, et al. Inhibition of indoleamine 2,3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res. 2007;67:792–801. doi: 10.1158/0008-5472.CAN-06-2925. [DOI] [PubMed] [Google Scholar]
- 10.Muller AJ, Sharma MD, Chandler PR, Duhadaway JB, Everhart ME, Johnson BA, 3rd, et al. Chronic inflammation that facilitates tumor progression creates local immune suppression by inducing indoleamine 2,3 dioxygenase. Proc Natl Acad Sci USA. 2008;105:17073–17078. doi: 10.1073/pnas.0806173105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muller AJ, DuHadaway JB, Chang MY, Ramalingam A, Sutanto-Ward E, Boulden J, et al. Non-hematopoietic expression of IDO is integrally required for inflammatory tumor promotion. Cancer Immunol Immunother. 2010;59:1655–1663. doi: 10.1007/s00262-010-0891-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muller AJ, Prendergast GC. Marrying immunotherapy with chemotherapy: why say IDO? Cancer Res. 2005;65:8065–8068. doi: 10.1158/0008-5472.CAN-05-2213. [DOI] [PubMed] [Google Scholar]
- 13.Prendergast GC, Jaffee EM. Cancer immunologists and cancer biologists: why we didn't talk then but need to now. Cancer Res. 2007;67:3500–3504. doi: 10.1158/0008-5472.CAN-06-4626. [DOI] [PubMed] [Google Scholar]
- 14.Prendergast GC, Jaffee EM, editors. Cancer immunotherapy: Immune suppression and tumor growth. New York: Academic Press; 2007. [Google Scholar]
- 15.Matsumori A, Kawai C. Coxsackie virus B3 perimyocarditis in BALB/c mice: experimental model of chronic perimyocarditis in the right ventricle. J Pathol. 1980;131:97–106. doi: 10.1002/path.1711310202. [DOI] [PubMed] [Google Scholar]
- 16.Eleftheriadis T, Liakopoulos V, Antoniadi G, Stefanidis I, Galaktidou G. Indoleamine 2,3-dioxygenase is increased in hemodialysis patients and affects immune response to hepatitis B vaccination. Vaccine. 2011;29:2242–2247. doi: 10.1016/j.vaccine.2011.01.051. [DOI] [PubMed] [Google Scholar]
- 17.Wobser M, Voigt H, Houben R, Eggert AO, Freiwald M, Kaemmerer U, et al. Dendritic cell based antitumor vaccination: impact of functional indoleamine 2,3-dioxygenase expression. Cancer Immunol Immunother. 2007;56:1017–1024. doi: 10.1007/s00262-006-0256-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haviv YS, Sharkia M, Galun E, Safadi R. Pancreatitis following hepatitis A vaccination. Eur J Med Res. 2000;5:229–230. [PubMed] [Google Scholar]
- 19.Franco-Paredes C, Leonard M, Jurado R, Blumberg HM, Smith RM. Tuberculosis of the pancreas: report of two cases and review of the literature. Am J Med Sci. 2002;323:54–58. doi: 10.1097/00000441-200201000-00010. [DOI] [PubMed] [Google Scholar]
- 20.Kannel WB, Vasan RS. Triglycerides as vascular risk factors: new epidemiologic insights. Curr Opin Cardiol. 2009;24:345–350. doi: 10.1097/HCO.0b013e32832c1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lavie CJ, Milani RV, O'Keefe JH. Dyslipidemia intervention in metabolic syndrome: emphasis on improving lipids and clinical event reduction. Am J Med Sci. 2011;341:388–393. doi: 10.1097/MAJ.0b013e31821483fa. [DOI] [PubMed] [Google Scholar]
- 22.Watts GF, Karpe F. Triglycerides and atherogenic dyslipidaemia: extending treatment beyond statins in the high-risk cardiovascular patient. Heart. 2011;97:350–356. doi: 10.1136/hrt.2010.204990. [DOI] [PubMed] [Google Scholar]
- 23.Ghandehari H, Kamal-Bahl S, Wong ND. Prevalence and extent of dyslipidemia and recommended lipid levels in US adults with and without cardiovascular comorbidities: the National Health and Nutrition Examination Survey 2003–2004. Am Heart J. 2008;156:112–119. doi: 10.1016/j.ahj.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 24.Ciorba MA, Bettonville EE, McDonald KG, Metz R, Prendergast GC, Newberry RD, et al. Induction of IDO-1 by immunostimulatory DNA limits severity of experimental colitis. J Immunol. 2010;184:3907–3916. doi: 10.4049/jimmunol.0900291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roediger WE, Moore J, Babidge W. Colonic sulfide in pathogenesis and treatment of ulcerative colitis. Dig Dis Sci. 1997;42:1571–1579. doi: 10.1023/A:1018851723920. [DOI] [PubMed] [Google Scholar]
- 26.Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98:694–702. doi: 10.1016/0016-5085(90)90290-h. [DOI] [PubMed] [Google Scholar]
- 27.Ni J, Chen SF, Hollander D. Immunological abnormality in C3H/HeJ mice with heritable inflammatory bowel disease. Cell Immunol. 1996;169:7–15. doi: 10.1006/cimm.1996.0084. [DOI] [PubMed] [Google Scholar]
- 28.Kitajima S, Takuma S, Morimoto M. Changes in colonic mucosal permeability in mouse colitis induced with dextran sulfate sodium. Exp Anim. 1999;48:137–143. doi: 10.1538/expanim.48.137. [DOI] [PubMed] [Google Scholar]
- 29.Katz JB, Muller AJ, Metz R, Prendergast GC. Indoleamine 2,3-dioxygenase in T-cell tolerance and tumoral immune escape. Immunol Rev. 2008;222:206–221. doi: 10.1111/j.1600-065X.2008.00610.x. [DOI] [PubMed] [Google Scholar]
- 30.Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347:417–429. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- 31.Clevers H. At the crossroads of inflammation and cancer. Cell. 2004;118:671–674. doi: 10.1016/j.cell.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 32.Takikawa O, Yoshida R, Kido R, Hayaishi O. Tryptophan degradation in mice initiated by indoleamine 2,3-dioxygenase. J Biol Chem. 1986;261:3648–3653. [PubMed] [Google Scholar]
- 33.Munn DH. Indoleamine 2,3-dioxygenase, tumor-induced tolerance and counter-regulation. Curr Opin Immunol. 2006;18:220–225. doi: 10.1016/j.coi.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 34.Gurtner GJ, Newberry RD, Schloemann SR, McDonald KG, Stenson WF. Inhibition of indoleamine 2,3-dioxygenase augments trinitrobenzene sulfonic acid colitis in mice. Gastroenterology. 2003;125:1762–1773. doi: 10.1053/j.gastro.2003.08.031. [DOI] [PubMed] [Google Scholar]
- 35.Chang MY, Boulden J, Katz JB, Wang L, Meyer TJ, Soler AP, et al. Bin1 ablation increases susceptibility to cancer during aging, particularly lung cancer. Cancer Res. 2007;67:7605–7612. doi: 10.1158/0008-5472.CAN-07-1100. [DOI] [PubMed] [Google Scholar]
- 36.Mellor AL, Munn DH. Physiologic control of the functional status of Foxp3+ regulatory T cells. J Immunol. 2011;186:4535–4540. doi: 10.4049/jimmunol.1002937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Erdman SE, Poutahidis T, Tomczak M, Rogers AB, Cormier K, Plank B, et al. CD4+ CD25+ regulatory T lymphocytes inhibit microbially induced colon cancer in Rag2-deficient mice. Am J Pathol. 2003;162:691–702. doi: 10.1016/S0002-9440(10)63863-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Erdman SE, Rao VP, Poutahidis T, Ihrig MM, Ge Z, Feng Y, et al. CD4(+)CD25(+) regulatory lymphocytes require interleukin 10 to interrupt colon carcinogenesis in mice. Cancer Res. 2003;63:6042–6050. [PubMed] [Google Scholar]
- 39.Braun D, Longman RS, Albert ML. A two-step induction of indoleamine 2,3 dioxygenase (IDO) activity during dendritic-cell maturation. Blood. 2005;106:2375–2381. doi: 10.1182/blood-2005-03-0979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Luca A, Bozza S, Zelante T, Zagarella S, D'Angelo C, Perruccio K, et al. Non-hematopoietic cells contribute to protective tolerance to Aspergillus fumigatus via a TRIF pathway converging on IDO. Cell Mol Immunol. 2010;7:459–470. doi: 10.1038/cmi.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wingender G, Garbi N, Schumak B, Jungerkes F, Endl E, von Bubnoff D, et al. Systemic application of CpG-rich DNA suppresses adaptive T cell immunity via induction of IDO. Eur J Immunol. 2006;36:12–20. doi: 10.1002/eji.200535602. [DOI] [PubMed] [Google Scholar]
- 42.Matsumori A, Kawai C, Sawada S, Yamamoto K. Experimental coxsackievirus B3 perimyocarditis in the right ventricle in BALB/c mice: a one-year follow-up study. Jpn Circ J. 1980;44:842–847. doi: 10.1253/jcj.44.842. [DOI] [PubMed] [Google Scholar]
- 43.Baban B, Chandler P, McCool D, Marshall B, Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase expression is restricted to fetal trophoblast giant cells during murine gestation and is maternal genome specific. J Reprod Immunol. 2004;61:67–77. doi: 10.1016/j.jri.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 44.Marsland BJ, Nembrini C, Grun K, Reissmann R, Kurrer M, Leipner C, et al. TLR ligands act directly upon T cells to restore proliferation in the absence of protein kinase C-theta signaling and promote autoimmune myocarditis. J Immunol. 2007;178:3466–3473. doi: 10.4049/jimmunol.178.6.3466. [DOI] [PubMed] [Google Scholar]
- 45.Marty RR, Dirnhofer S, Mauermann N, Schweikert S, Akira S, Hunziker L, et al. MyD88 signaling controls autoimmune myocarditis induction. Circulation. 2006;113:258–265. doi: 10.1161/CIRCULATIONAHA.105.564294. [DOI] [PubMed] [Google Scholar]
- 46.Kohno H, Suzuki R, Sugie S, Tanaka T. Beta-Catenin mutations in a mouse model of inflammation-related colon carcinogenesis induced by 1,2-dimethylhydrazine and dextran sodium sulfate. Cancer Sci. 2005;96:69–76. doi: 10.1111/j.1349-7006.2005.00020.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith C, Chang MY, DuHadaway J, Boulden J, Sutanto-Ward E, Soler AP, Ostrand-Rosenberg S, et al. Immune escape mediator IDO is a crucial driver of lung cancer and metastasis that supports IL-6 production and and Gr1+Cd11b+ myeloid suppressor cell function. In press. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.