

Abstract
In order to identify novel candidate tumor suppressor genes (TSGs) implicated in renal cell carcinoma (RCC), we performed genome-wide methylation profiling of RCC using the HumanMethylation27 BeadChips to assess methylation at >14,000 genes. Two hundred and twenty hypermethylated probes representing 205 loci/genes were identified in genomic CpG islands. A subset of TSGs investigated in detail exhibited frequent tumor methylation, promoter methylation associated transcriptional silencing and reactivation after demethylation in RCC cell lines and downregulation of expression in tumor tissue (e.g., SLC34A2 specifically methylated in 63% of RCC, OVOL1 in 40%, DLEC1 in 20%, TMPRSS2 in 26%, SST in 31% and BMP4 in 35%). As OVOL1, a putative regulator of c-Myc transcription, and SST (somatostatin) had not previously been linked to cancer and RCC, respectively, we (1) investigated their potential relevance to tumor growth by RNAi knockdown and found significantly increased anchorage-independent growth and (2) demonstrated that OVOL1 knockdown increased c-Myc mRNA levels.
Key words: renal cell carcinoma (RCC), methylation, epigenetics
Introduction
Kidney cancers account for approximately 2% of all cancers and more than 200,000 new cases of kidney cancer are diagnosed in the world each year.1 The most common form of kidney cancer in adults is renal cell carcinoma (RCC) of which the majority (>75%) is classified as clear cell (conventional).2 Though familial RCC accounts for only about 3% of all cases, the investigation of rare familial cases of kidney cancer has provided important insights into the pathogenesis of sporadic RCC. In particular, the finding that germline mutation of the VHL gene caused a syndromic form of inherited RCC and that somatic inactivation of VHL occurred in most sporadic clear cell RCC.3–7 VHL inactivation leads to stabilization of hypoxia-inducible transcription factors HIF-1 and HIF-2 and activation of a wide repertoire of hypoxic response genes (reviewed in ref. 8 and 9). HIF-mediated RCC growth may be antagonized by multi-tyrosine kinase inhibitors, which are now widely used in the treatment of metastatic kidney cancer.10 Hence, elucidation of the genetic mechanisms of tumorigenesis in RCC has provided a basis for novel therapeutic interventions. Though additional genes such as MET, FLCN, FH and SDHB have been shown to cause inherited RCC, their contribution to the pathogenesis of sporadic RCC is limited11 (www.sanger.ac.uk/genetics/CGP/cosmic). Candidate gene and exome sequencing studies of RCC have recently identified a small number of genes (e.g., PBRM1, SETD1, KDM6A/UTX) that are somatically mutated in sporadic RCC but, while PBRM1 is mutated in about a third of RCC, the others are mutated in less than 5% of tumors.12,13
Promoter region methylation and transcriptional silencing is now recognized as a major mechanism of tumor suppressor gene (TSG) inactivation in a wide range of human cancers (reviewed in ref. 14) and epigenetic inactivation of VHL in RCC was one of the first examples of this phenomenon.5,6 Though VHL inactivation in RCC is more commonly caused by mutation than methylation,4,6,7 the demonstration that the RASSF1A TSG was frequently inactivated by promoter methylation, but rarely mutated, in RCC and other cancers,15–17 suggested that strategies to identify epigenetically inactivated TSGs could represent an important approach to elucidating the molecular pathogenesis of RCC. Consistent with this hypothesis others and we have identified TSGs that are frequently inactivated in RCC (e.g., HAI-2/SPINT2, BNC1, SFRP1).18
The identification of epigenetically inactivated RCC TSGs has been facilitated by advances in genomic technologies. Thus, gene expression microarrays enabled the use of a functional genomic approach (changes in gene expression are detected following global demethylation of cancer cell line genomes) to identify candidate methylated genes. Though successful, this strategy was limited by the large number of candidate genes that were not found to be methylated (and for which changes in expression were presumably secondary to gene regulatory changes induced by reactivation of TSGs that did exhibit promoter methylation) or were found to be methylated in normal tissue.19–22 Further technological advances provided strategies such as MeDIP, which can be utilized to assay methylation status in cancer tissues, and resulted in the identification of additional epigenetically inactivated RCC TSGs.23 A further recent development has been the use of the Illumina BeadArray technology to directly assay methylated CpG status in normal and cancer tissues. In order to identify candidate novel renal TSGs and potential methylated biomarkers, we performed the CpG methylation status analysis of RCC for ∼27,500 CpGs and >14,000 genes using the Illumina Infinium HumanMethylation27 BeadChip Array.
Results
Illumina Infinium HumanMethylation27 array data was obtained from all 38 sporadic renal cell carcinomas (27 male and 11 female) and 9 age-matched normal kidney controls (6 male and 3 female).
An experimental replicate demonstrated high repeatability between HumanMethylation27 arrays with a correlation co-efficient of R2 = 0.9955 (Fig. S1A). Comparison of overall DNA methylation between normal kidney samples demonstrated little variance, with high correlation co-efficient values (R2) for male normal kidney (n = 6) ranging from R2 = 0.9462 to 0.9880 and for female normal kidney (n = 3) ranging from R2 = 0.9746 to 0.9829 (Examples Fig. S1B and S1C). Comparison of overall DNA methylation between normal kidney samples irrelevant of gender ranging from R2 = 0.9136 to 0.9880. Comparisons between the average β-values of the normal kidneys and the kidney tumors (in a gender specific manner) demonstrated a range of variance with several tumors having high degrees of aberrant methylation (Examples Fig. S1D–F). Both aberrant hypermethylation and hypomethylation was observed. This demonstrates that aberrant methylation is an important feature of some, but not all tumors and is not a generalized by product of tumorigenesis.
In all 9 normal kidney samples, 8,336 probes were methylated (β values ≥ 0.25), and in at least 1 of the 9 normal samples 10,845 probes were methylated. 171 probes (168 genes) were methylated in all three normal kidney samples from patients aged >60 y but not in two normal kidney samples from patients aged <45 y and 46 probes (43 genes) were methylated in both normal kidney samples from patients aged <45 y but were unmethylated in normal kidney samples from patients aged >60 y (Fig. S2).
Identification of hypermethylated CpG loci.
Selection was performed on the genes/probes by two differing methods: A “methylation only” method, from which the most highly methylated genes/probes or genes with multiple positive probes were selected, or a “combined analysis” method from which genes/probes were selected on the basis of methylation data and functional annotation (see Methods section) (Fig. S3). Probes that represented imprinted or X chromosome encoded genes (to avoid confusion related to gender and/or chromosomal loss/gain) or for which one or more normal samples demonstrated a β value >0.25 were removed from both analysis strategies.
For the “methylation only” selection, 43 probes demonstrated β-values ≥0.5 representing 42 genes and 48 probes demonstrated β-difference values ≥0.4 representing 44 genes in 7 or more tumor samples. This produced a combined high stringency list containing 60 probes representing 55 genes (31 genes selected by both criteria) (Table 1, Figs. S4 and S5). The three most frequently methylated genes (FOXL1-36.8%, SLC34A2-34.2% and TM6SF1-34.2%) and the five genes for which 2 probes satisfied the “methylation only” selection criteria (COL1A2, OVOL1, SOCS2, TNFRSF10C and ZNF154) were selected for further methylation analysis. The concurrence of hypermethylation between the two probes for each of these five genes was good, with the more heavily methylated probe being positive in 92% (33/36) of the tumors when the less methylated probe was positive (Fig. S6). The distance between probes did not produce any obvious differences (Fig. S6). Additionally, two other genes were chosen as cancer related candidate genes, DLEC1, a tumor suppressor gene known to be methylated in kidney cancer, and, TMPRSS2, which is known to form a gene fusion in prostate cancer. To investigate whether the selection had enriched for genes likely to be methylated, the selection was compared with the PubMeth data (www.pubmeth.org). This demonstrated that 16.4% (9/55) of genes were methylated in at least one other cancer type and that one gene (GSTP1) was known to be methylated in kidney (Table S1).
Table 1.
Methylation only hypermethylated gene probes
| Sporadic Rccs | ||||||
| Selected Probes | Symbol | Target Id | Meth + (B ≥ 0.5 Or Diff ≥ 0.4) | Normal Kidneys | Chr | Product |
| 1 | FOXL1 | cg06995715 | 14/38 (36.8%) | 0/9 | 16 | forkhead box L1 |
| 2 | TM6SF1 | cg14696396 | 13/38 (34.2%) | 0/9 | 15 | transmembrane 6 superfamily member 1 |
| 3 | SLC34A2 | cg19616230 | 13/38 (34.2%) | 0/94 | 4 | solute carrier family 34 (sodium phosphate); member 2 |
| 4 | SOCS2 | cg04797323 | 12/38 (32.6%) | 0/9 | 12 | suppressor of cytokine signaling-2 |
| 5 | TNFRSF10C | cg27090216 | 12/38 (32.6%) | 0/9 | 8 | tumor necrosis factor receptor superfamily; member 10c precursor |
| 6 | OVOL1 | cg20909686 | 12/38 (32.6%) | 0/9 | 11 | OVO-like 1 binding protein |
| 7 | TRIM58 | cg07533148 | 12/38 (32.6%) | 0/9 | 1 | tripartite motif-containing 58 |
| 8 | TLX3 | cg25720804 | 12/38 (32.6%) | 0/9 | 5 | T-cell leukemia; homeobox 3 |
| 9 | UTF1 | cg09053680 | 12/38 (32.6%) | 0/9 | 10 | undifferentiated embryonic cell transcription factor 1 |
| 10 | POU4F2 | cg24199834 | 11/38 (28.9%) | 0/9 | 4 | POU domain; class 4; transcription factor 2 |
| 11 | NOTCH3 | cg06650786 | 11/38 (28.9%) | 0/9 | 19 | Notch homolog 3 |
| 12 | ZNF177 | cg09643544 | 11/38 (28.9%) | 0/9 | 19 | zinc finger protein 177 |
| 13 | C1orf104 | cg22234962 | 10/38 (26.3%) | 0/9 | 1 | hypothetical protein LOC284618 |
| 14 | ICAM4 | cg21494776 | 10/38 (26.3%) | 0/9 | 19 | intercellular adhesion molecule 4 isoform 1 precursor |
| 15 | ZNF454 | cg03355526 | 10/38 (26.3%) | 0/9 | 5 | zinc finger protein 454 |
| 16 | AEBP1 | cg02126753 | 10/38 (26.3%) | 0/9 | 7 | adipocyte enhancer binding protein 1 precursor |
| 17 | SCARF2 | cg14785479 | 10/38 (26.3%) | 0/9 | 22 | scavenger receptor class F; member 2 isoform 2 |
| 18 | PENK | cg04598121 | 10/38 (26.3%) | 0/9 | 8 | proenkephalin |
| 19 | HCG9 | cg04623837 | 9/38 (23.7%) | 0/9 | 6 | hypothetical protein LOC10255 |
| 20 | PRAC | cg12374721 | 9/38 (23.7%) | 0/9 | 17 | small nuclear protein PRAC |
| 21 | GPC2 | cg18691434 | 9/38 (23.7%) | 0/9 | 7 | glypican 2 |
| 22 | COL1A2 | cg25300386 | 9/38 (23.7%) | 0/9 | 7 | α2 type I collagen |
| 23 | PRPH | cg09595479 | 9/38 (23.7%) | 0/9 | 12 | peripherin |
| 24 | ZNF154 | cg08668790 | 9/38 (23.7%) | 0/9 | 19 | zinc finger protein 154 (pHZ-92) |
| 25 | UNQ9433 | cg17162024 | 9/38 (23.7%) | 0/9 | 8 | hypothetical protein LOC389658 |
| 26 | IRX2 | cg15433631 | 9/38 (23.7%) | 0/9 | 5 | iroquois homeobox protein 2 |
| 27 | TMPRSS2 | cg24901042 | 8/38 (21.1%) | 0/9 | 21 | transmembrane protease; serine 2 |
| 28 | CDKN2B | cg10210238 | 8/38 (21.1%) | 0/9 | 9 | cyclin-dependent kinase inhibitor 2B isoform 2 |
| 29 | RAMP1 | cg03270167 | 8/38 (21.1%) | 0/9 | 2 | receptor activity-modifying protein 1 precursor |
| 30 | GSTP1 | cg04920951 | 8/38 (21.1%) | 0/9 | 11 | glutathione transferase |
| 31 | SLC15A3 | cg21992250 | 8/38 (21.1%) | 0/9 | 11 | solute carrier family 15; member 3 |
| 32 | FBN2 | cg25084878 | 8/38 (21.1%) | 0/9 | 5 | fibrillin 2 precursor |
| 33 | DLEC1 | cg23881725 | 8/38 (21.1%) | 0/9 | 3 | deleted in lung and esophageal cancer 1 isoform DLEC1-L1 |
| 34 | ZNF540 | cg03975694 | 8/38 (21.1%) | 0/9 | 19 | zinc finger protein 540 |
| 35 | IRX4 | cg03963198 | 8/38 (21.1%) | 0/9 | 5 | iroquois homeobox protein 4 |
| 36 | FLT4 | cg00489401 | 8/38 (21.1%) | 0/9 | 5 | fms-related tyrosine kinase 4 isoform 1 |
| 37 | ZNF154 | cg21790626 | 8/38 (21.1%) | 0/9 | 19 | zinc finger protein 154 (pHZ-92) |
| 38 | FBXO39 | cg20723355 | 8/38 (21.1%) | 0/9 | 17 | F-box protein 39 |
| 39 | RIMS4 | cg19332710 | 8/38 (21.1%) | 0/9 | 20 | regulating synaptic membrane exocytosis 4 |
| 40 | BCAN | cg21475402 | 7/38 (18.4%) | 0/9 | 1 | brevican isoform 2 |
| 41 | CA3 | cg18674980 | 7/38 (18.4%) | 0/9 | 8 | carbonic anhydrase III |
| 42 | SOCS2 | cg23412850 | 7/38 (18.4%) | 0/9 | 12 | suppressor of cytokine signaling-2 |
| 43 | OXR1 | cg17176732 | 7/38 (18.4%) | 0/9 | 8 | oxidation resistance 1 |
| 44 | CBX4 | cg04398978 | 7/38 (18.4%) | 0/9 | 17 | chromobox homolog 4 |
| 45 | ABCA3 | cg00949442 | 7/38 (18.4%) | 0/9 | 16 | ATP-binding cassette; sub-family A member 3 |
| 46 | DGKE | cg01344452 | 7/38 (18.4%) | 0/9 | 17 | diacylglycerol kinase epsilon |
| 47 | TNFRSF10C | cg14015044 | 7/38 (18.4%) | 0/9 | 8 | tumor necrosis factor receptor superfamily; member 10c precursor |
| 48 | SPAG6 | cg25802093 | 7/38 (18.4%) | 0/9 | 10 | sperm associated antigen 6 isoform 1 |
| 49 | COL1A2 | cg18511007 | 7/38 (18.4%) | 0/9 | 7 | α2 type I collagen |
| 50 | HTR7 | cg06291867 | 7/38 (18.4%) | 0/9 | 10 | 5-hydroxytryptamine receptor 7 isoform d |
| 51 | OVOL1 | cg13496736 | 7/38 (18.4%) | 0/9 | 11 | OVO-like 1 binding protein |
| 52 | HIST1H1A | cg10146929 | 7/38 (18.4%) | 0/9 | 6 | H1 histone family; member 1 |
| 53 | HLF | cg04219321 | 7/38 (18.4%) | 0/9 | 17 | hepatic leukemia factor |
| 54 | CCDC37 | cg00891278 | 7/38 (18.4%) | 0/9 | 3 | hypothetical protein LOC348807 |
| 55 | UQCRH | cg21576698 | 7/38 (18.4%) | 0/9 | 1 | ubiquinol-cytochrome c reductase hinge protein |
| 56 | TCFL5 | cg10729531 | 7/38 (18.4%) | 0/9 | 20 | transcription factor-like 5 protein |
| 57 | SALL3 | cg15191648 | 7/38 (18.4%) | 0/9 | 18 | sal-like 3 |
| 58 | CACNA1G | cg18454685 | 7/38 (18.4%) | 0/9 | 17 | voltage-dependent calcium channel α1G subunit isoform 1 |
| 59 | GRM6 | cg14859460 | 7/38 (18.4%) | 0/9 | 5 | glutamate receptor; metabotropic 6 precursor |
| 60 | CHD5 | cg08080029 | 7/38 (18.4%) | 0/9 | 1 | chromodomain helicase DNA binding protein 5 |
Methylation of three of 18 “methylation only” selected genes (with methylation frequencies >25%) was associated with significantly poorer survival by Kaplan-Meier analysis [C1ORF104 (log rank analysis χ2 = 5.41 p = 0.02), ICAM4 (χ2 = 3.91 p = 0.048), TM6SF1 (χ2 = 4.1 p = 0.044)]. However, an analysis of an independent, publicly available methylome data set from the Cancer Genome Atlas for survival and methylation of C1ORF104, ICAM4 and TM6SF1 did not provide statistically significant differences though there was a trend for poorer survival in patients with C1ORF104 or TM6SF1 tumor methylation (χ2 = 4.1 p = 0.044).
For the “combined analysis” selection strategy, 179 probes demonstrated β-values ≥0.4 representing 166 genes and 178 probes demonstrated β-difference values ≥0.3 representing 164 genes in 7 or more tumor samples producing a total of 220 probes representing 205 genes (125 genes were selected by both criteria) (Table S2 and Fig. S5). Functional analysis of this selection using the DAVID bioinformatics resource (david.niaid.nih.gov) demonstrated an enrichment of genes involved functions related to carcinogenesis: cell-cell signaling (17 genes), regulation of cell proliferation (17 genes), regulation of cell death (16 genes), cell-cell adhesions (15 genes), angiogenesis/blood vessel development (9 genes), EGF genes (5 genes), tumor suppressor (5 genes) and cadherins (3 genes) (Table S3-Genes present in the “methylation only” selection are underlined). Ingenuity® Systems Pathway Analysis (www.ingenuity.com) highlighted enrichment of the TGFβ signaling pathway (5 genes) and the Somatostatin anti-apoptosis pathway (3 genes-SST, GNB4 and GUCY2D) (Table S3 and Fig. 4A). Three genes, BMP4, SST and CDKN2B, were identified by both enrichment analyses. BMP4 and SST were selected for further methylation analysis as the third gene, CDKN2B, was previously known to be methylated in kidney cancer.18
Figure 4.

Methylation Analysis of SST. (A) The anti-proliferative role of Somatostatin Receptor 2 pathway demonstrated to be enriched for by the combined analysis selected gene set using Ingenuity® Systems Pathway Analysis Software (www.ingenuity.com). The selected genes are marked on the pathway. (B) Demonstrates the methylation status of example renal cell carcinoma cell lines (Un = undigested, Di = digested) in comparison the mRNA expression of the relevant gene before and after 5-Aza-2′-deoxycytidine de-methylating treatment (-ve = no treatment, 5-aza = de-methylating treatment). (C) Examples of tumor/associated normal (T/N) CoBRA pairs (numbered 1–20) showing tumor specific/enriched methylation in tumors and no methylation in normal kidney samples and certain tumors (Un = undigested, Di = digested). (D) Demonstrates the loss or depletion of methylated SST gene mRNA in tumor/associated normal (T/N) mRNA pairs (numbered 1–15). Kidney tumor (K) numbers were added to compare CoBRA and mRNA T/N pairs.
Confirmation of probe methylation by bisulphite sequencing and CoBRA.
Before further analysis was performed, three hypermethylated genes (DLEC1, OVOL1 and SOCS2) were selected for bisulphite sequencing to confirm the predicted β-values and demonstrate how representative the probes values were for CpG island methylation level. DLEC1 was previously reported to be methylated in RCC26 and OVOL1 and SOCS2 both had two selected Infinium probes in their respective CpG islands. Examples of tumors with either high or low β-values were selected for each gene and the bisulphite sequencing demonstrated that β-values >0.5 generally indicated highly methylation at the probe position and wider CpG island methylation, while low β-values showed little or no methylation (Fig. 1). Furthermore, CoBRA analysis was performed on selected tumors and normal kidney samples for DLEC1, OVOL1 and SOCS2. For these genes, promoter methylation was detected by CoBRA for high β-values and no methylation was present in the normal kidney samples (examples in Fig. S7A). A gene in the methylation only selection, FBN2, had been recently shown by our group to be methylated in kidney cancer by MeDIP assessment.22 Comparison of the CoBRA results using the primers from that paper and the primers based around the Infinium probe demonstrated good correlation between the two primer sets and the Infinium probe β-value (examples in Fig. S7B). No methylation was seen in the normal kidney samples for either primer set. In combination, these results were consistent with the validity of the β-values, provided evidence for accepting the assumption that the single probed CpG was representative of the CpG island and validated the use of CoBRA for further investigation. Evaluation of the other genes identified by the MeDIP study demonstrated varied levels of methylation. This variability between the two investigational approaches is likely to reflect differences between the positioning of the probes in the Infinium array and those used for the MeDIP studies (though variability between tumor sample sets cannot be excluded) (Fig. S8).
Figure 1.

Confirmation of probe β-value methylation by bisulphite sequencing. The regions surrounding a positive probe was amplified and direct sequenced for bisulphite-modified tumor DNA. For each gene, three tumors had 10 clone sequences analyzed and methylation indexes (M.I.s) were calculated for both the target CpG of the relevant Infinium probe or for the total number of CpGs analyzed in the sequence and displayed next to the clone sequencing. Black dots represent methylation present and BstUI digestion sites have been marked on the sequence. The positions of the Infinium probe or probes and the CpG island are mapped to the first exon of the respective genes.
Cluster analysis of the “methylation only” hypermethylated probes.
Euclidean hierarchical clustering was performed on the 60 selected “methylation only” probes for the 38 sporadic RCC tumor samples. This clustered the samples into 5 specific groups: a highly methylated/CpG Island Methylator Phenotype (CIMP) group, 2 medium methylated groups separated by the probes that were methylated, a low methylation group and a group containing a methylation signature similar to the normal kidney samples (Fig. 2). Using the average “methylation only” probe β-values, the groups were all shown to have statistically different levels of methylation, apart from the two medium methylation groups and the normal-like methylation group and the actual normal kidney samples (Fig. S9 and Table S5). There was no statistical association between VHL mutation status (present in 55.2% of tumors-Table S5) and level of methylation or cluster group.
Figure 2.

Euclidean hierarchical clustering for the selected “methylation only” probes. Euclidean hierarchical clustering for the 60 selected “methylation only” probes was performed using Cluster 3.0 (bonsai.hgc.jp/∼mdehoon/software/cluster) and visualized using Java TreeView (jtreeview.sourceforge.net). Tumor samples were labeled for the presence of VHL mutation or VHL methylation (assessed using the Infinium HumanMethylation27 array probes).
Methylation analysis of selected renal cell carcinoma hypermethylation genes.
Twelve selected genes (FOXL1, SLC34A2, TM6SF1, COL1A2, OVOL1, SOCS2, TNFRSF10C, ZNF154, DLEC1, TMPRSS2, SST and BMP4) were initially analyzed by CoBRA to confirm the absence and presence of methylation in normal and malignant samples respectively. In addition, RT-PCR was performed to document re-expression (or increased expression) in RCC cell lines after 5-Aza-2′-deoxycytidine treatment. When relevant, further analysis was performed by CoBRA to demonstrate whether specific or increased methylation was present in 19 tumor/associated normal DNA pairs and by RT-PCR to demonstrate loss or depletion of gene mRNA in 15 tumor/associated normal mRNA pairs.
From the 12 selected genes, 8 (SLC34A2, TM6SF1, COL1A2, OVOL1, DLEC1, TMPRSS2, SST and BMP4) demonstrated methylation in kidney cancer cell lines, re-expression in kidney cancer cell lines after 5-Aza-2′-deoxycytidine treatment and no methylation in the normal kidney samples (Table 2 and Figs. 3A and 4B). FOXL1, TNFRSF10C and ZNF154 all demonstrated methylation in normal kidney samples, while SOCS2 demonstrated no re-expression in methylated kidney cancer cell lines following global demethylation.
Table 2.
Methylation analysis of selected renal cell carcinoma hypermethylation genes
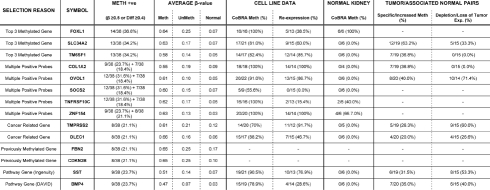 |
Figure 3.

Analysis of the Hypermethylated “methylation only” Genes. (A) Demonstrates the methylation status of example renal cell carcinoma cell lines (Un = undigested, Di = digested) in comparison the mRNA expression of the relevant gene before and after 5-Aza-2′-deoxycytidine de-methylating treatment (-ve = no treatment, 5-aza = de-methylating treatment). (B) Examples of tumor/associated normal (T/N) CoBRA pairs (numbered 1–20) showing tumor specific/enriched methylation in tumors and no methylation in normal kidney samples and certain tumors (Un = undigested, Di = digested). (C) Demonstrates the loss or depletion of methylated gene mRNA in tumor/associated normal (T/N) mRNA pairs (numbered 1–15). Kidney tumor (K) numbers were added to compare CoBRA and mRNA T/N pairs.
From the remaining 8 genes, 5 demonstrated tumor specific methylation (TM6SF1-36.8%, OVOL1-40.0%, DLEC1-20.0%, TMPRSS2-26.3% and SST-31.5%) and 3 genes demonstrated tumor specific/increased methylation with a low level methylation in several associated normal samples (SLC34A2-63.2%, COL1A2-36.8% and BMP4-35.0%) (Table 2 and Figs. 3B and 4C). Furthermore, 6 genes demonstrated loss or depletion of mRNA expression in tumor tissue compared with the associated normal tissue (SLC34A2-33.3%, OVOL1-71.4%, DLEC1-26.6%, TMPRSS2-60.0%, SST-53.3% and BMP4-40.0%) (Table 2 and Figs. 3C, 4D and S10).
All selected genes were also investigated for known mutations within the catalog of somatic mutations in cancer (COSMIC) database (www.sanger.ac.uk/genetics/CGP/cosmic). Only one reported variant in kidney cancer was identified, a silent mutation in SLC34A2, but a significant number of variants of COL1A2 and DLEC1 occur in a variety of cancers (Table S4).
Anchorage-independent growth assays and real-time RT-PCR analysis.
To investigate the functional impact of loss of OVOL1 and SST, anchorage-independent soft agar growth assays were performed after siRNA knockdown of the relevant genes in 293 embryonic kidney cells. SST was chosen due to its known function in an anti-apoptosis pathway (Fig. 4A) and OVOL1 was chosen due to its candidate function as a repressor of c-Myc (Fig. 5B). DLEC1 was also analyzed as a known gene methylated in kidney for which re-expression in RCC cell lines have been shown to inhibit clonogenicity, but knockdown has yet to be shown to enhance anchorage-independent growth.27 A statistically significant enhancement of anchorage-independent growth was observed for all three genes (OVOL1 p = 0.0003, DLEC1 p = 0.0004 and SST p = 0.0145) with OVOL1 demonstrating the greatest effect (Fig. 5A).
Figure 5.

Anchorage-independent growth assays and Real-time RT-PCR analysis. (A) Anchorage-independent soft agar assays were performed to compare siRNA knockdown of OVOL1, SST and DLEC1 to non-specific scrambled siRNA knockdown in 293 embryonic kidney cells. The graph represents the average from triplicate experiments presented as fold change relative to scrabbled siRNA with error bars for standard deviation and p-values calculated using the student's t-test. (B) The known interactive pathways and functions of the OVOL1 gene transcription factor product obtained from GeneGo Analysis (www.genego.com). Red lines indicate suppression and green lines indicate upregulation. (C) Real time RT-PCR analysis of MYC expression either with non-specific scrabbled siRNA knockdown or without siRNA knockdown of the OVOL1 gene. The graph represents the average from triplicate experiments presented as fold change relative to scrabbled siRNA with error bars for standard deviation.
RT-PCRs demonstrated an effective knockdown for all three genes at the mRNA level (Data not shown). OVOL1 negatively regulates the expression of c-Myc and c-Myb (Fig. 5B) by directly binding to its promoter.28 As OVOL1 knockdown resulted in the greatest anchorage-independent growth advantage we wished to determine if this correlated with an increase in the expression of c-MYC. Real-Time RT-PCR showed that the OVOL1 Silencer® Select siRNA knockdown total mRNA demonstrated a 1.78 fold increase in MYC expression compared with the Silencer® Negative Control #1 siRNA total mRNA (Fig. 5C).
Discussion
Previously others and we have used the Illumina Goldengate methylation BeadArray assay (which interrogates the methylation status of 1505 specific CpG sites in 807 genes) to identify methylated TSGs in RCC.25 However, to our knowledge, this is the first application of the much more comprehensive Illumina Infinium HumanMethylation27 BeadChip Array (∼27,500 CpGs and >14,000 genes) in RCC. Furthermore recent reports of the use of this platform in other cancer types such as colorectal,29 breast,24 ovary,30 and head and neck31 cancers are consistent with our findings that the methodology is accurate and that it enables high-throughput methylation analysis. Nevertheless, our findings illustrate that, though the direct assay of CpG methylation facilitates the identification of epigenetically inactivated TSGs, the strategy is not entirely specific, as some genes with apparent tumor-specific methylation are not confirmed on further analysis. In part this relates to the limitations of the assay used. Not all CpGs are analyzed and there is no information on the functional effect of the detected methylation on gene transcription. In order to ameliorate these concerns we used a twin strategy to prioritize candidate genes that was intended to give highest priority to genes with evidence of the most intense tumor specific methylation and, among those with lesser degrees of methylation, take into account their functional relevance to cancer. In total we identified 205 candidate methylated TSGs by our high-throughput analysis. Further testing of a subset of these suggests that in the majority of these cases the finding of frequent tumor-specific methylation is correct and that CpG promoter methylation would be associated with transcriptional silencing. Given that in a recent literature review we found reports of only 43 genes that were methylated in >20% of RCC, the current study significantly expands the catalog of methylated RCC genes that can be investigated for application as potential biomarkers for the detection, diagnosis, prognostication and therapy of RCC.18 It was noticeable that the number of genes methylated in individual tumors was extremely variable with some tumors showing large numbers of methylated genes and others with β values similar to that of normal kidney. Previously we found evidence of a “CpG island methylator phenotype” (CIMP+) in a subset of RCC from patients with VHL disease and sporadic RCC without VHL mutations.25 Thus, our latest findings are consistent with this and also with those of Dulaimi et al.32 who reported that a subset of RCC (∼3%) were methylated for at least five out of the ten genes studied. In sporadic colorectal cancer the CIMP+ tumors are preferentially associated with the presence of a BRAF mutation and microsatellite instability (from MLH1 promoter methylation).33 However, we did not find any evidence of an association between VHL mutations and methylation phenotype, which is in agreement with our previous observation that CIMP+ tumors were not specific to either VHL disease linked RCC or VHL-wild type sporadic RCC.25 Epigenetic therapies (e.g., decitabine, the clinical form of the demethylating agent 5-aza-2′-deoxycytidine) have been investigated in several clinical trials for neoplasia and promising responses have been reported in hematological malignancies.34 Hence, it would be interesting to see whether the extent of tumor promoter region methylation in individual RCC might predict response to epigenetic therapies.
Methylation profiling might also inform targeted therapeutic strategies by identifying RCC in which specific signaling pathways were aberrant. We investigated whether epigenetic inactivation of DLEC1, OVOL1 and SST was likely to influence growth of renal cancer cells and observed that knockdown of each of these genes was associated with enhanced anchorage-independent cell growth. DLEC1 encodes a cytoplasmic protein without any significant homology to known proteins and the mechanism of tumor suppression has not been delineated.35 To our knowledge, somatostatin (SST) has not previously been reported to be inactivated in RCC. However, frequent epigenetic inactivation of the promoter region methylation has been described in colorectal, esophageal, cervical and gastric cancers.36–39 Somatostatin analogs (e.g., octreotide and lanreotide) have been reported to have anti-proliferative effects and clinical benefit in neuroendocrine tumors40 and it is interesting to speculate that somatostatin analogs might also prove to be beneficial in non-neuroendocrine tumors with SST silencing.
OVOL1, a human homolog of the Drosophila ovo gene, has not previously been implicated in a human cancer. Ovol1 knockout mice are small and display hypogenitalism, abnormal hair formation and renal developmental abnormalities.41 OVOL1 encodes a zinc-finger protein transcription factor and is a downstream target of the TGFβ/BMP7-Smad4 signaling pathway.42 In addition, it has been reported that cutaneous suprabasal cells from Ovol1-deficient mice demonstrate increased c-myc activity and that Ovol1 represses c-myc transcription by directly binding to its promoter.28 Consistent with these observations, we demonstrated that in human kidney cells siRNA knockdown of OVOL1 increased c-Myc transcript levels. Furthermore, of the three genes tested, siRNA knockdown of OVOL1 was associated with the most significant effect on cell growth. Multiple lines of evidence indicate that c-Myc has a critical role in the pathogenesis of RCC and c-Myc expression may be dysregulated by a variety of mechanisms. Thus copy number analysis of RCC demonstrates c-Myc as the most likely driver for chromosome 8q amplification in RCC43 and that c-Myc pathway is frequently activated in RCC.44 However, c-Myc pathway activation might result from a variety of mechanisms, including genomic amplification, HIF-2 overexpression secondary to VHL inactivation (though HIF-1 expression downregulates c-Myc),45–47 and epigenetic silencing of OVOL1. These findings highlight how potential c-Myc based therapeutic approaches might be targeted in RCC.
In summary, the simultaneous methylation profiling of >27,000 CpGs and >14,000 genes has demonstrated both the utility of this approach to identify novel insights into the pathogenesis of RCC and also some of the limitations of this approach. Nevertheless, recent technological advances have resulted in higher-density methylation assays becoming available and it is likely that these, in combination with exome/genome sequencing and gene expression analyses, will facilitate the identification of further genes implicated in the pathogenesis of RCC such that the challenge will no longer be to elucidate the molecular pathology of RCC but to understand how comprehensive knowledge of RCC genomics can be best translated into new diagnostic and therapeutic advances.
Materials and Methods
Patient DNA samples and cell line DNA/total RNA.
DNA was extracted from 38 sporadic renal cell carcinomas as described previously in reference 22. Samples were selected to contain the most amount of tumor but were not micro-dissected. Corresponding normal kidney DNA was extracted for 19 patients and matched RCC and normal tissue mRNA was extracted for 15 cases. For control studies, normal kidney DNA was extracted from 9 control kidney samples extracted during non-cancer related surgeries upon the kidney. All participants gave informed written consent for research studies. The study was conducted according to the principles expressed in the Declaration of Helsinki and was approved by the relevant Institutional Review Board/Ethics committees.
RNA (with and without 5-Aza-2′-deoxycytidine treatment) and DNA were extracted from 15 RCC cell lines (769-P, 786-O, A498, ACHN, Caki-1, Caki-2, CAL54, RCC4, RCC11, RCC12, RCC48, SKRC18, SKRC39, SKRC45, SKRC47) and DNA from a further 7 (RCC6, KTCL26, KTCL140, RCC1, SKRC54, UMRC2, UMRC3). The Human Embryonic Kidney (HEK293) cell line was used for Anchorage-independent growth assay and was grown under standard tissue culture conditions (as described previously in ref. 22).
Infinium array.
Tumor DNA (n = 39; a single tumor sample was run twice as a test for reproducibility) and the normal kidney DNA (n = 9) from non-cancerous kidneys were assayed using the Illumina Infinium HumanMethylation27 BeadChips (Illumina, San Diego, CA USA). The BeadChip contains probes for 27,578 CpG sites covering over 14,000 human RefSeq genes. Bisulphite modification of DNA, chip-processing and data acquisition was performed following the manufacturer's manual by the Wellcome Trust Centre for Human Genetics Genomics Lab, Oxford, UK. Initial array results were run through the BeadStudio software to generate β-values ranging for 0 (designating no methylation) to 1 (designating complete methylation) (Illumina, San Diego, CA).
Candidate gene selection.
Selection was performed using two approaches: first, a “methylation only” method in which the most highly methylated genes/probes or genes with multiple positive probes were selected. Second, a “combined analysis” in which methylation and functional information were combined. In addition to using the background normalized average β-values, β-difference values were calculated for each probe for each tumor sample by subtracting the mean normal β-value for that probe.
For both selection strategies, first all the probes for which any of the 9 normal samples demonstrated a β-value ≥0.25 were removed. For the “methylation only” selection, background normalized average β-values were considered hypermethylated if the β-value was ≥0.5 or the β-difference value (between sample β value and mean normal β value) was ≥0.4 in 7 or more tumor samples.24 For the “combined analysis” selection process, background normalized average β-values were considered hypermethylated if the β-value was ≥0.4 or the β-difference value was ≥0.3 in 7 or more tumor samples and then the selected genes were analyzed using the Database for Annotation, Visualization and Integrated Discovery (DAVID) (david.abcc.ncifcrf.gov) and the Ingenuity® Systems Pathway Analysis Software (www.ingenuity.com) to identify those genes implicated in cancer-related pathways.
Promoter methylation analysis of selected candidate genes.
Previously we have validated the Illumina BeadArray GoldenGate CpG methylation assay in familial RCC samples.25 In this study further validation was undertaken using combined bisulphite restriction analysis (CoBRA) to assess the methylation status of selected genes in cell line DNA, normal kidney DNA, tumor DNA and associated normal kidney DNA where appropriate. All DNA was modified using an EpiTect kit (Qiagen, Heidelberg, Germany) according to manufacturers' instructions. CoBRA PCR primers were designed around the probed CpG (or CpGs) for each gene and all were initially designed to be semi-nested and altered to fully-nested if required. Both rounds of PCR were touchdowns with five cycles lowered 1°C per cycle down to a gene specific final annealing temperature for a further 35 cycles, 5 µl of primary product was added to the secondary PCR. PCR products were digested with BstUI (Fermentas UK, York, UK) overnight at 37°C prior to visualization on a 2% agarose gel.
For particular genes, several tumor samples with varied β-values were cloned and sequenced to validate the array and confirm CoBRA results. PCR products were cloned into the pGEM-T easy vector (Promega, Madison, WI USA) according to manufacturers' instructions. Up to 15 colonies were picked per sample and sequenced following single-colony PCR using T7 and SP6 primers sites present within the pGEM-T easy vector. Methylation indexes were calculated as a percentage of the number of methylated CpGs out of the total CpGs sequenced.
mRNA expression analysis of selected candidate genes.
Expression analysis was performed for selected genes by comparing either total RNA from 15 renal cell carcinoma cell lines both with and without 5-Aza-2′-deoxycytidine treatment (as described in ref. 22) or total RNA from kidney tumor and its associated normal kidney.
All cell lines were maintained in DMEM (Sigma-Aldrich, St. Louis, MO USA) supplemented with 2 mM glutamine and 10% FCS at 37°C, 5% CO2. The cell lines were demethylated by daily treatments of fresh media with 5 µM 5-Aza-2′-deoxycytidine (Sigma-Aldrich, Dorset, UK) over 5 d. The frozen tumor and associated normal samples were ground under liquid nitrogen into a fine powder. Total RNA was isolated from both using RNA-Bee reagent following the manufacturer's instructions (AMS Biotechnology, Oxford, UK), followed by purification using RNeasy Mini-columns (Qiagen, Crawle, UK). One microgram total RNA was converted to cDNA using Superscript III (Invitrogen, Carlsbad, CA USA) and random hexamer primers (Fermentas UK, York, UK). The kidney tumor cDNAs were converted from 1.5 µg of total RNA.
RT-PCR primers were designed for each gene such that the primers were always positioned in different exons and had a 56°C annealing temperature. RT-PCRs were performed using a touchdown PCR program with five cycles lowered 1°C per cycle down from 61°C to 57°C followed by a further 35 cycles with a final annealing temperature of 56°C. Primer details are available on request.
Anchorage-independent growth assays and real-time RT-PCR analysis.
Silencer® Select siRNAs against OVOL1 (s9938), SST (s13494) and DLEC1 (s19295) or Silencer® Negative Control #1 siRNA (Ambion, Austin, TX) was transfected into HEK293 cells using Interferin reagent (Polyplus, Illkirch, France) following the manufacturer's instructions. After 24 h incubation, cells were seeded into 2 ml DMEM in 10% FCS and 3% agar. Cells were maintained by addition of 200 µl of DMEM in 10% FCS weekly. After 3 weeks of growth, a final count of colonies was performed. Cells not seeded into agar were incubated for a further 24 h before efficiency of knockdown was assessed by RT-PCR using previous conditions. Additionally, the total RNA from the OVOL1 knockdown was assessed by real-time RT-PCR for increased expression of the mRNA for the oncogenic MYC gene. One microliter of the OVOL1 Silencer® Select siRNA knockdown cDNA or the Silencer® Negative Control #1 siRNA cDNA was used for PCR amplification in an ABI 7000 real-time PCR system (Applied Biosystems) as recommended by the manufacturer. The MYC (Hs00905030_m1) Taqman® Gene Expression Assay (Applied Biosystems) primers and fluorogenic probe were used and normalized using the ACTB (Hs99999903_m1) Taqman® Gene Expression Assay (Applied Biosystems) as an internal control. Samples were run in triplicate and CT values obtained were compared by the Delta CT method. Results are expressed as an average fold-change compared the Silencer® Negative Control #1 siRNA cDNA.
Cluster analysis and statistical analysis.
Cluster analysis was performed using Cluster 3.0 (bonsai.hgc.jp/∼mdehoon/software/cluster) and visualized using Java TreeView (jtreeview.sourceforge.net). Statistical analysis was performed as indicated using MedCalc (www.medcalc.org) with a significance level of 5%.
Acknowledgments
We thank Cancer Research UK for financial support.
Supplementary Material
References
- 1.Ferlay J, Autier P, Boniol M, Heanue M, Colombet M, Boyle P. Estimates of the cancer incidence and mortality in Europe in 2006. Ann Oncol. 2007;18:581–592. doi: 10.1093/annonc/mdl498. [DOI] [PubMed] [Google Scholar]
- 2.Mancini V, Battaglia M, Ditonno P, Palazzo S, Lastilla G, Montironi R, et al. Current insights in renal cell cancer pathology. Urol Oncol. 2008;26:225–238. doi: 10.1016/j.urolonc.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 3.Latif F, Tory K, Gnarra J, Yao M, Duh FM, Orcutt ML, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260:1317–1320. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- 4.Foster K, Prowse A, van den Berg A, Fleming S, Hulsbeek MM, Crossey PA, et al. Somatic mutations of the von Hippel-Lindau disease tumour suppressor gene in non-familial clear cell renal carcinoma. Hum Mol Genet. 1994;3:2169–2173. doi: 10.1093/hmg/3.12.2169. [DOI] [PubMed] [Google Scholar]
- 5.Herman JG, Latif F, Weng Y, Lerman MI, Zbar B, Liu S, et al. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci USA. 1994;91:9700–9704. doi: 10.1073/pnas.91.21.9700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clifford SC, Prowse AH, Affara NA, Buys CH, Maher ER. Inactivation of the von Hippel-Lindau (VHL) tumour suppressor gene and allelic losses at chromosome arm 3p in primary renal cell carcinoma: evidence for a VHL-independent pathway in clear cell renal tumourigenesis. Genes Chromosomes Cancer. 1998;22:200–209. doi: 10.1002/(SICI)1098-2264(199807)22:3<200::AID-GCC5>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 7.Banks RE, Tirukonda P, Taylor C, Hornigold N, Astuti D, Cohen D, et al. Genetic and epigenetic analysis of von Hippel-Lindau (VHL) gene alterations and relationship with clinical variables in sporadic renal cancer. Cancer Res. 2006;66:2000–2011. doi: 10.1158/0008-5472.CAN-05-3074. [DOI] [PubMed] [Google Scholar]
- 8.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 9.Kaelin WG., Jr The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer. 2008;8:865–873. doi: 10.1038/nrc2502. [DOI] [PubMed] [Google Scholar]
- 10.Chowdhury S, Larkin JM, Gore ME. Recent advances in the treatment of renal cell carcinoma and the role of targeted therapies. Eur J Cancer. 2008;44:2152–2161. doi: 10.1016/j.ejca.2008.06.028. [DOI] [PubMed] [Google Scholar]
- 11.Morris MR, Maina E, Morgan NV, Gentle D, Astuti D, Moch H, et al. Molecular genetic analysis of FIH-1, FH and SDHB candidate tumour suppressor genes in renal cell carcinoma. J Clin Pathol. 2004;57:706–711. doi: 10.1136/jcp.2003.011767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463:360–363. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Varela I, Tarpey P, Raine K, Huang D, Ong CK, Stephens P, et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011;469:539–542. doi: 10.1038/nature09639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsai HC, Baylin SB. Cancer epigenetics: linking basic biology to clinical medicine. Cell Res. 2011;21:502–517. doi: 10.1038/cr.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morrissey C, Martinez A, Zatyka M, Agathanggelou A, Honorio S, Astuti D, et al. Epigenetic inactivation of the RASSF1A 3p21.3 tumor suppressor gene in both clear cell and papillary renal cell carcinoma. Cancer Res. 2001;61:7277–7281. [PubMed] [Google Scholar]
- 16.Dallol A, Forgacs E, Martinez A, Sekido Y, Walker R, Kishida T, et al. Tumour specific promoter region methylation of the human homologue of the Drosophila Roundabout gene DUTT1 (ROBO1) in human cancers. Oncogene. 2002;21:3020–3028. doi: 10.1038/sj.onc.1205421. [DOI] [PubMed] [Google Scholar]
- 17.Morris MR, Hesson LB, Wagner KJ, Morgan NV, Astuti D, Lees RD, et al. Multigene methylation analysis of Wilms' tumour and adult renal cell carcinoma. Oncogene. 2003;22:6794–6801. doi: 10.1038/sj.onc.1206914. [DOI] [PubMed] [Google Scholar]
- 18.Morris MR, Maher ER. Epigenetics of renal cell carcinoma: the path towards new diagnostics and therapeutics. Genome Med. 2010;2:59. doi: 10.1186/gm180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morris MR, Gentle D, Abdulrahman M, Maina EN, Gupta K, Banks RE, et al. Tumor suppressor activity and epigenetic inactivation of hepatocyte growth factor activator inhibitor type 2/SPINT2 in papillary and clear cell renal cell carcinoma. Cancer Res. 2005;65:4598–4606. doi: 10.1158/0008-5472.CAN-04-3371. [DOI] [PubMed] [Google Scholar]
- 20.Ibanez d, Caceres I, Dulaimi E, Hoffman AM, Al-Saleem T, Uzzo RG, Cairns P. Identification of novel target genes by an epigenetic reactivation screen of renal cancer. Cancer Res. 2006;66:5021–5028. doi: 10.1158/0008-5472.CAN-05-3365. [DOI] [PubMed] [Google Scholar]
- 21.Morris MR, Gentle D, Abdulrahman M, Clarke N, Brown M, Kishida T, et al. Functional epigenomics approach to identify methylated candidate tumour suppressor genes in renal cell carcinoma. Br J Cancer. 2008;98:496–501. doi: 10.1038/sj.bjc.6604180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morris MR, Ricketts C, Gentle D, Abdulrahman M, Clarke N, Brown M, et al. Identification of candidate tumour suppressor genes frequently methylated in renal cell carcinoma. Oncogene. 2010;29:2104–2117. doi: 10.1038/onc.2009.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morris MR, Ricketts CJ, Gentle D, McRonald F, Carli N, Khalili H, et al. Genome-wide methylation analysis identifies epigenetically inactivated candidate tumour suppressor genes in renal cell carcinoma. Oncogene. 2011;30:1390–1401. doi: 10.1038/onc.2010.525. [DOI] [PubMed] [Google Scholar]
- 24.Hill VK, Ricketts C, Bieche I, Vacher S, Gentle D, Lewis C, et al. Genome-wide DNA methylation profiling of CpG islands in breast cancer identifies novel genes associated with tumorigenicity. Cancer Res. 2011;71:2988–2999. doi: 10.1158/0008-5472.CAN-10-4026. [DOI] [PubMed] [Google Scholar]
- 25.McRonald FE, Morris MR, Gentle D, Winchester L, Baban D, Ragoussis J, et al. CpG methylation profiling in VHL related and VHL unrelated renal cell carcinoma. Mol Cancer. 2009;8:31. doi: 10.1186/1476-4598-8-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Daigo Y, Nishiwaki T, Kawasoe T, Tamari M, Tsuchiya E, Nakamura Y. Molecular cloning of a candidate tumor suppressor gene, DLC1, from chromosome 3p21.3. Cancer Res. 1999;59:1966–1972. [PubMed] [Google Scholar]
- 27.Zhang Q, Ying J, Li J, Fan Y, Poon FF, Ng KM, et al. Aberrant promoter methylation of DLEC1, a critical 3p22 tumor suppressor for renal cell carcinoma, is associated with more advanced tumor stage. J Urol. 2010;184:731–737. doi: 10.1016/j.juro.2010.03.108. [DOI] [PubMed] [Google Scholar]
- 28.Nair M, Teng A, Bilanchone V, Agrawal A, Li B, Dai X. Ovol1 regulates the growth arrest of embryonic epidermal progenitor cells and represses c-myc transcription. J Cell Biol. 2006;173:253–264. doi: 10.1083/jcb.200508196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oster B, Thorsen K, Lamy P, Wojdacz TK, Hansen LL, Birkenkamp-Demtröder K, et al. Identification and validation of highly frequent CpG island hypermethylation in colorectal adenomas and carcinomas. Int J Cancer. 2011;129:2855–2866. doi: 10.1002/ijc.25951. [DOI] [PubMed] [Google Scholar]
- 30.Bauerschlag DO, Ammerpohl O, Bräutigam K, Schem C, Lin Q, Weigel MT, et al. Progression-free survival in ovarian cancer is reflected in epigenetic DNA methylation profiles. Oncology. 2011;80:12–20. doi: 10.1159/000327746. [DOI] [PubMed] [Google Scholar]
- 31.Poage GM, Houseman EA, Christensen BC, Butler RA, Avissar-Whiting M, McClean MD, et al. Global hypomethylation identifies Loci targeted for hypermethylation in head and neck cancer. Clin Cancer Res. 2011;17:3579–3589. doi: 10.1158/1078-0432.CCR-11-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dulaimi E, Ibanez de Caceres I, Uzzo RG, Al-Saleem T, Greenberg RE, Polascik TJ, et al. Promoter hypermethylation profile of kidney cancer. Clin Cancer Res. 2004;10:3972–3979. doi: 10.1158/1078-0432.CCR-04-0175. [DOI] [PubMed] [Google Scholar]
- 33.Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38:787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 34.Issa JP, Kantarjian HM. Targeting DNA methylation. Clin Cancer Res. 2009;15:3938–3946. doi: 10.1158/1078-0432.CCR-08-2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qiu GH, Salto-Tellez M, Ross JA, Yeo W, Cui Y, Wheelhouse N, et al. The tumor suppressor gene DLEC1 is frequently silenced by DNA methylation in hepatocellular carcinoma and induces G1 arrest in cell cycle. J Hepatol. 2008;48:433–441. doi: 10.1016/j.jhep.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 36.Mori Y, Cai K, Cheng Y, Wang S, Paun B, Hamilton JP, et al. A genome-wide search identifies epigenetic silencing of somatostatin, tachykinin-1, and 5 other genes in colon cancer. Gastroenterology. 2006;131:797–808. doi: 10.1053/j.gastro.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 37.Jin Z, Mori Y, Hamilton JP, Olaru A, Sato F, Yang J, et al. Hypermethylation of the somatostatin promoter is a common, early event in human esophageal carcinogenesis. Cancer. 2008;112:43–49. doi: 10.1002/cncr.23135. [DOI] [PubMed] [Google Scholar]
- 38.Ongenaert M, Wisman GB, Volders HH, Koning AJ, Zee AG, van Criekinge W, et al. Discovery of DNA methylation markers in cervical cancer using relaxation ranking. BMC Med Genomics. 2008;1:57. doi: 10.1186/1755-8794-1-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jackson K, Soutto M, Peng D, Hu T, Marshal D, El-Rifai W. Epigenetic silencing of somatostatin in gastric cancer. Dig Dis Sci. 2011;56:125–130. doi: 10.1007/s10620-010-1422-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Auernhammer CJ, Göke B. Therapeutic strategies for advanced neuroendocrine carcinomas of jejunum/ileum and pancreatic origin. Gut. 2011;60:1009–1021. doi: 10.1136/gut.2009.204453. [DOI] [PubMed] [Google Scholar]
- 41.Dai X, Schonbaum C, Degenstein L, Bai W, Mahowald A, Fuchs E. The ovo gene required for cuticle formation and oogenesis in flies is involved in hair formation and spermatogenesis in mice. Genes Dev. 1998;12:3452–3463. doi: 10.1101/gad.12.21.3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kobayashi Y, Absher DM, Gulzar ZG, Young SR, McKenney JK, Peehl DM, et al. DNA methylation profiling reveals novel biomarkers and important roles for DNA methyltransferases in prostate cancer. Genome Res. 2011;21:1017–1027. doi: 10.1101/gr.119487.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beroukhim R, Brunet JP, Di Napoli A, Mertz KD, Seeley A, Pires MM, et al. Patterns of gene expression and copy-number alterations in von-hippel lindau disease-associated and sporadic clear cell carcinoma of the kidney. Cancer Res. 2009;69:4674–4681. doi: 10.1158/0008-5472.CAN-09-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang SW, Chang WH, Su YC, Chen YC, Lai YH, Wu PT, et al. MYC pathway is activated in clear cell renal cell carcinoma and essential for proliferation of clear cell renal cell carcinoma cells. Cancer Lett. 2009;273:35–43. doi: 10.1016/j.canlet.2008.07.038. [DOI] [PubMed] [Google Scholar]
- 45.Gordan JD, Bertout JA, Hu CJ, Diehl JA, Simon MC. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell. 2007;11:335–347. doi: 10.1016/j.ccr.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, et al. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell. 2007;11:407–420. doi: 10.1016/j.ccr.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 47.Gordan JD, Lal P, Dondeti VR, Letrero R, Parekh KN, Oquendo CE, et al. HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell. 2008;14:435–446. doi: 10.1016/j.ccr.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.