

Abstract
Background
IBD5, IL23R, and ATG16L1 genetic variations are established Crohn’s disease (CD) risks alleles. We evaluated these in a population-based case-control study within a cohort to determine their penetrance, population attributable risk, independence, and relationship to other established CD risk factors, including NOD2.
Methods
DNA from 213 CD, 118 ulcerative colitis, and 315 healthy control subjects from the population-based University of Manitoba IBD Research Registry were genotyped for IBD5 and IL23R single-nucleotide polymorphisms (SNPs), and for the Thr300Ala ATG16L1 SNP. Univariate and multivariate analyses were performed for these and nongenetic risk factors. We introduce multidimensionality reduction (MDR) to explore gene– gene interactions.
Results
ATG16L1, IBD5, and IL23R SNPs were significantly associated with CD. Multivariate analysis showed independent CD association for carriers of ATG16L1 (odds ratio [OR] = 1.8, 95% confidence interval [CI] 1.09–3.24), IBD5-IGR2230 (OR = 2.16, 95% CI 1.30–3.59), and IL23R-rs10889677 (OR = 2.13, 95% CI 1.39–3.28) while retaining association for NOD2 mutation carriers (OR = 4.45, 95% CI 2.68–7.38), IBD family history (OR = 2.75, 95% CI 1.42–5.31), tobacco (OR = 2.06, 95% CI 1.35–3.14), and Jewish ethnicity (OR = 20.1, 95% CI 2.16–186.8). IL23R minor variants for Arg381Gln and Intron 6 rs7517848 showed independent, CD protection and 3′ untranslated variant rs108896778 showed risk. MDR analysis suggested an interaction between IBD5, ATG16L1, and IL23R risk alleles. Penetrance values for ATG16L1 and IBD5 were 0.27% for heterozygotes, and 0.35% and 0.44%, respectively, for homozygotes. IL23R rs108896778 penetrance was 0.37%.
Conclusions
A population-based analysis of CD risk factors is useful for characterizing the epidemiology of multiple CD genetic and nongenetic risk factors. Gene– gene interactions are likely, but require further evaluation in large population-based cohorts.
Keywords: Crohn’s disease, IL23R, ATG16L1, IBD5, genetics
Crohn’s disease (CD) and ulcerative colitis (UC), the 2 major forms of inflammatory bowel disease (IBD), develop from both genetic and environmental etiologies. Genetic etiologies were hypothesized by consistently identified familial clustering, increased risk in Ashkenazi Jews, and greater monozygotic versus dizygotic twin concordance rates.1 However, molecular genetic risk factors have now been established. The most well-replicated include 3 common functional mutations in the NOD2 gene (G908R, R702W, and Cins1007fs), the IBD5 risk haplotype on chromosome 5q31 (including functional polymorphisms on SLC22A4 and SLC22A5), and recently discovered risk polymorphisms in interleukin 23 receptor (IL23R) and the autophagy gene ATG16L1.2–10 It is noteworthy that these risk polymorphisms have been consistently observed in persons of European ancestry but have not been found to be risk factors in the Japanese, Korean, and Chinese populations.11–14 Consistent environmental risk factors include residence in Western industrialized countries, especially more northern latitudes, and increased CD but decreased UC risk from smoking.15
To understand the effect of the various established IBD risk factors on a population, to determine gene penetrance for genetic counseling purposes, and to determine interactions, it is best to assess these risk factors in a population-based study. Understanding gene penetrances and gene interaction for genetic counseling purposes is becoming increasingly more critical, with multiple companies now offering the public at large the opportunity to purchase CD gene testing so individuals can know their disease risk. We have previously reported an assessment of known risk factors of IBD family history, Jewish ethnicity, tobacco, geography, and the 3 common NOD2/CARD15 mutations in a study cohort drawn from the population-based University of Manitoba IBD Research Registry (UMIBDRR).15 In this population-based case-control study within a cohort, the increased odds ratio (OR) of developing CD from a single mutant NOD2 gene was 3.7 and 2 mutant genes was 40.0. For the first time, we reported that NOD2 mutation risks were independent of risks for CD family history (OR = 6.2), Jewish ethnicity (OR = 18.5), and smoking (OR = 3.0 for current and 1.7 for ex-smokers, respectively). Population-based NOD2 penetrance was 4.9% for 2 mutant chromosomes, 0.54% for a single mutant chromosome and 0.184% for persons wildtype for NOD2. NOD2 was also a risk for stricturing and internal fistulizing CD complications.
We have now expanded our assessment of CD genetic risk factors in our population-based IBD registry to include the recent 3 additional CD genetic risk factors that have been consistently replicated: IBD5 risk haplotype including functional variants on SLC22A4 and SLC22A5 genes, IL23R gene variants including the protective missense polymorphism Arg381Gln, and the established associated coding variant on ATG16L1, Thr300Ala. We now report the population-based risk and penetrance of these more recently established molecular genetic risk factors in the context of NOD2 status, IBD family history, tobacco, and Jewish ethnicity. We have also explored their potential gene– gene interactions, assessed the relative strength of different risk alleles for IBD5 and IL23R, and have examined the effect of multiple risk alleles from different genes on CD risk in a defined nonreferral-based population.
MATERIALS AND METHODS
Study Population
The study population included persons recruited to enter an IBD risk factor study in 2002. The cases were age 18 to 50, with diagnoses of CD or UC that had been registrants of the UMIBDRR, a registry that was developed at the time of establishment of the University of Manitoba IBD Epidemiology Database.16 This database was created by accessing subjects with IBD through the Manitoba Health administrative databases that register all health system contacts of all Manitoba residents. Manitoba Health is the single health insurance provider for the province. Controls (HC) were individuals recruited from a 10:1 mailing to a random sample of age, gender, and geographically matched persons drawn also from the administrative data of Manitoba Health. Details of the development of this risk factor cohort used for this study, CD and UC diagnoses confirmation methods, and phenotype classification are as reported previously.15 Data on ethnicity, tobacco use, family history in a first-degree relative, and residency were obtained from subjects through administered questionnaires. Informed consent was obtained from all participants for a multiple risk factor study including genetic risks.
Genotyping
Genotyping was performed on DNA purified from study subjects consenting to phlebotomy using a PURE-GENE DNA Isolation Kit according to the manufacturer’s protocol (Gentra Systems, Minneapolis, MN). Genotyping was performed using the TaqMan biallelic discrimination system (Applied Biosystems, Foster City, CA) with alleles determined on an Applied Biosystems 7900HT Fast Real-Time polymerase chain reaction (PCR) system analyzer. (Primers and probes used in this study are available upon request.) All single-nucleotide polymorphism (SNP) assays were verified by comparison with 24 CD samples that had been genotyped by DNA sequencing. Genotypes were performed blinded to IBD or HC status.
Statistical Method
Hardy–Weinberg equilibrium was tested among controls for each SNP using Pearson’s χ2 test, to determine whether the proportion of each genotype obtained was in agreement with expected values as calculated from allele frequencies. Genotype frequency differences between cases and controls were evaluated using standard contingency χ2 tests. Two-sample t-tests were used for continuous variables of which the normal distribution assumption was appropriate. Nominal data were analyzed using the χ2 test or Fisher’s exact test. A 2-tailed P-value <0.05 was considered significant. Logistic regression was used to assess the association between genotypes and CD and UC risks. The OR and 95% confidence intervals (CIs) were calculated under a dominant and codominant (homozygote and heterozygote genotypes which carry the variant allele versus homozygote wildtype, the reference group) models.
For haplotype analysis, D′ and r2 were used to estimate the magnitude of pairwise linkage disequilibrium for IL23R and IBD5 using the PROC ALLELE procedure implemented in SAS 9.1/Genetics package program (SAS Institute, Cary, NC).17,18 We applied a global score test to assess the difference in overall haplotype frequency profiles between cases and controls.19 Permutated P-values for the global tests were calculated from empirical distribution created from a minimum of 10,000 permutated datasets. The association between each haplotype and risk of CD was estimated by regression substitution, a function implemented in the module haplo.glm in the program HaploStat (http://www.mayo.edu/hsr/Sfunc.html) for the R programming language.20 Additive effect of haplotypic association with the risk of CD in the generalized linear model implemented in the HaploStat was assumed to obtain the odds ratios.
The penetrance values for genotypes and population attributable risk (PAR) were estimated as per Hampe et al21 and per Schlesseman,22 respectively. When a genotype was not represented in the control group, the OR was estimated with the method recommended by Hampe et al,21 using the corresponding Manitoba CD prevalence of 270 cases per 100,000 in 200023 and assuming Hardy–Weinberg equilibrium for the alleles in the general population.
The tests for gene– gene and gene– environmental interactions using logistic regression analysis were conducted by the Wald test for the β coefficient of the cross-product term in a model that contained the main effects of 2 different genetic loci in addition to the interaction term. The statistical significance of the interaction in logistic regression analyses was evaluated by comparing nested models with and without the cross-product terms using a likelihood ratio test and was accomplished in SAS v. 9.1. To further explore the potential multiloci gene– gene interactions, we used the method of multidimensionality reduction (MDR), which is a data complexity reduction strategy of detecting and modeling potential gene– gene interactions and can be used to select models of high-order combinations of genes.24,25 With limited power to detect high-order gene– gene interactions using traditional logistic regression analysis in a moderate-sized study, MDR can be used to explore potential gene– gene interactions and as a tool of hypothesis generation for underlying complex biological mechanisms. The data are divided into a training set and an independent testing set for 10-fold cross-validation. A set of n genetic markers and their possible multifactor combinations (n = 1, 2, 3, and 4) are searched. The ratio of the number of cases to the number of controls is calculated within each multifactor combinations and a single MDR model that has the fewest misclassifications is selected. For multiple testing correction, a permutation-based (1,000 permutation times) empirical P-value is calculated for each selected best model.
RESULTS
Baseline Characteristics
The study population consisted of 213 CD, 118 UC and 315 HC that completed informed consent, interview, had CD and UC disease diagnoses confirmed, had DNA samples available, and were self-identified as white race. In addition to limiting the study to persons of white race, the population study subjects differed from our prior report on CARD15/NOD2 study of the Manitoba IBD Risk Factor Cohort15 in that we did not include 18 white study subjects (11 CD and 7 UC, and 0 HC) for which DNA samples were no longer available or adequate for genotyping, and we added an additional 27 white study subjects from our Risk Factor Database (6 with confirmed CD, 13 with confirmed UC, and 8 HC) that had consented to phlebotomy and had new DNA samples become available since our prior report.
The main clinical characteristics of IBD patients and HC included in this study are summarized in Table 1. As noted previously, although Manitoba Health sent 10 letters in the same postal region for each male and each female IBD risk factor registrant, a significantly greater percentage of females joined in the study as HC.15 Sixty-four percent of study subjects were urban dwellers with no differences in residency among CD, UC, and HC, as expected by matched geographic recruitment of HC. The proportion of Jewish CD and UC cases was significantly greater than that of HC. IBD cases exhibited younger mean age at time of study entry than HC. Tobacco usage and family history of IBD in first-degree relatives were more frequent for the CD patients compared with HC, but showed no differences between UC patients and HC.
TABLE 1.
Study Subjects Characteristics Among Self-Reported White Population
| HC (n=315) | CD (n=213) | CD vs. HC P-value | UC (n=118) | UC vs. HC P-value | |
|---|---|---|---|---|---|
| Mean age at study entry (y) | 40.0 | 37.2 | <0.001 | 37.8 | 0.001 |
| Gender | 0.005 | <.001 | |||
| Male | 86 (27%) | 83 (39%) | 59 (50%) | ||
| Female | 229 (73%) | 130 (61%) | 59 (50%) | ||
| Jewish (%) | 1 (0.3%) | 14 (6.5%) | <0.001 | 10 (8.5%) | <0.001 |
| Residency: urban (%) | 188 (60%) | 135 (63%) | 0.21 | 76 (66%) | 0.15 |
| Tobacco use ever (%) | 137 (46%) | 116 (55%) | 0.01 | 61 (52%) | 0.12 |
| Family history of IBD in first-degree relative (%) | 24 (8%) | 39 (18.6%) | <0.001 | 13 (11.3%) | 0.17 |
Single SNP Marker Association Analysis of IBD5, IL23R, and ATG16L1
We genotyped 5 candidate SNP variants for the IBD5 locus including the reported functional variants in the SLC22A4 and SLC22A5 transporter genes present on the risk haplotype, 5 SNP variants for IL23R, and the established ATG16L1 missense risk polymorphism Thr300Ala (coding nucleotide 898A>G). All of the investigated SNPs were in Hardy–Weinberg equilibrium in controls except IL23R SNP rs1495965 (P = 0.03). The percentage of samples that could be genotyped was greater than 98%. Genotype distributions are shown in Tables 2 and 3.
TABLE 2.
Association of SNPs in Genes With Crohn’s Disease (213 CD vs. 315 Controls)
| 0 Copy of Variant
|
1 Copy of Variant
|
2 Copies of Variant
|
Carrier of Variant
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| Case/Control | OR (ref) | Case/control | OR (95% CI) P | Case/control | OR (95% CI) P | OR (95% CI) P | |||
| ATG16L1 | Thr300Ala | rs2241880a 898 A → G | 28/76 | 1.00 | 103/150 | 1.86 (1.13–3.07) 0.02 | 77/88 | 2.38 (1.40–4.04) 0.001 | 2.05 (1.28–3.30) 0.003 |
| IBD5 | SLC22A4(OCTN1) L503F | rs1050152 1672 C → T | 49/109 | 1.00 | 107/159 | 1.50 (0.99–2.27) 0.06 | 51/45 | 2.52 (1.49–4.26) 0.001 | 1.72 (1.16–2.56) 0.007 |
| IBD5 | SLC22A5(OCTN2) promoter | rs2631367 –207 G → C |
37/97 | 1.00 | 109/160 | 1.79 (1.14–2.80) 0.01 | 61/56 | 2.86(1.69–4.86) <0.001 | 2.06 (1.34–3.17) 0.001 |
| IBD5 | SLC22A5(IGR2230) Intron 2 | rs17622208 G → A |
38/96 | 1.00 | 108/162 | 1.68 (1.08–2.64) 0.02 | 61/55 | 2.80 (1.66–4.73) <0.001 | 1.97 (1.28–3.01) 0.002 |
| IBD5 | IGR2198 | rs11739135 C → G |
51/116 | 1.00 | 109/153 | 1.62 (1.07–2.44) 0.02 | 47/44 | 2.42 (1.43–4.11) 0.001 | 1.80 (1.22–2.66) 0.003 |
| IBD5 | IGR2096 | rs12521868 G → T |
51/112 | 1.00 | 109/154 | 1.55 (1.03–2.35) 0.04 | 47/47 | 2.20 (1.30–3.70) 0.003 | 1.70 (1.15–2.52) 0.008 |
| IL23R | Intron 6 | rs7517847 T → G |
84/91 | 1.00 | 91/157 | 0.63 (0.42–0.93) 0.02 | 36/66 | 0.59 (0.36–0.98) 0.04 | 0.62 (0.43–0.89) 0.01 |
| IL23R | Intron 7 | rs2201841 T → C |
63/152 | 1.00 | 120/130 | 2.23 (1.52–3.27) <0.001 | 28/32 | 2.11 (1.17–3.79) 0.01 | 2.20 (1.52–3.19) <0.001 |
| IL23R | Exon 9 Arg381Gln | rs11209026a 1142 G → A | 202/285 | 1.00 | 9/28 | 0.45 (0.21–0.98) 0.05 | 0/1 | — | 0.44 (0.20–0.94) 0.04 |
| IL23R | 3′ UTR | rs10889677 C → A |
62/159 | 1.00 | 121/125 | 2.48 (1.69–3.65) <0.001 | 28/30 | 2.39 (1.32–4.32) 0.004 | 2.47 (1.70–3.57) <0.001 |
| IL23R | intergenic | rs1495965 A → G |
47/90 | 1.00 | 117/167 | 1.34 (0.88–2.05) 0.18 | 47/57 | 1.58 (0.94–2.66) 0.09 | 1.40 (0.93–2.10) 0.10 |
Nonsynonymous SNPs.
TABLE 3.
Association of SNPs in Genes With UC (Only White population, 118 UC vs. 315 Controls)
| 0 Copy of Variant
|
1 Copy of Variant
|
2 Copies of Variant
|
Carrier of Variant
|
|||||
|---|---|---|---|---|---|---|---|---|
| Case/Control | OR (ref) | Case/Control | OR (95% CI) P | Case/Control | OR (95% CI) P | OR (95% CI) P | ||
| ATG16L1 | rs2241880a | 27/76 | 1.00 | 58/150 | 1.09 (0.64–1.86) 0.77 | 28/88 | 0.90 (0.49–1.65) 0.72 | 1.02 (0.61–.168) 0.95 |
| IBD5_OCTN1 | rs1050152a | 41/109 | 1.00 | 53/159 | 1.30 (0.70–2.42) 0.41 | 22/45 | 0.89 (0.55–1.42) 0.62 | 0.98 (0.63–1.53) 0.92 |
| IBD5_OCTN2 | rs2631367 | 34/97 | 1.00 | 59/160 | 1.05 (0.64–1.72) 0.84 | 23/56 | 1.17 (0.63–2.18) 0.62 | 1.08 (0.68–1.73) 0.74 |
| IBD5_IGR2230 | rs17622208 | 33/96 | 1.00 | 60/162 | 1.08 (0.66–1.77) 0.77 | 23/55 | 1.22 (0.65–2.28) 0.54 | 1.11 (0.70–1.78) 0.66 |
| IBD5_IGR2198 | rs11739135 | 42/116 | 1.00 | 54/153 | 0.97 (0.61–1.56) 0.92 | 20/44 | 1.26 (0.66–2.37) 0.48 | 1.04 (0.67–1.62) 0.87 |
| IBD5_IGR2096 | rs12521868 | 42/112 | 1.00 | 51/154 | 0.88 (0.55–1.42) 0.61 | 23/47 | 1.30 (0.71–2.41) 0.39 | 0.98 (0.63–1.53) 0.94 |
| IL23R | rs7517847 | 46/91 | 1.00 | 45/157 | 0.57 (0.35–0.92) 0.02 | 24/66 | 0.72 (0.40–1.29) 0.27 | 0.61 (0.39–0.96) 0.03 |
| IL23R | rs2201841 | 50/152 | 1.00 | 52/130 | 1.22 (0.77–1.91) 0.40 | 13/32 | 1.24 (0.60–2.54) 0.57 | 1.22 (0.79–1.88) 0.37 |
| IL23R | rs11209026a | 109/285 | 1.00 | 6/28 | 0.56 (0.23–1.39) 0.21 | 0/1 | — | 0.54 (0.22–1.34) 0.18 |
| IL23R | rs10889677 | 51/159 | 1.00 | 50/125 | 1.25 (0.79–1.97) 0.34 | 14/30 | 1.45 (0.72–2.95) 0.30 | 1.29 (0.84–1.98) 0.25 |
| IL23R | rs1495965 | 36/90 | 1.00 | 55/167 | 0.82 (0.50–1.35) 0.44 | 24/57 | 1.05 (0.57–1.95) 0.87 | 0.88 (0.55–1.40) 0.60 |
Nonsynonymous SNPs.
All SNPs, with the exception of rs1495965 of IL23R, showed significant association with CD risk for heterozygotes, for carriers, and, when present, for homozygotes. For IL23R, SNP rs10889677 showed the strongest association with CD risk (OR = 2.47, 95% CI 1.70–3.57). Variant minor alleles for SNPs rs11209026 and rs7517848 of IL23R were inversely associated with CD risk (OR = 0.44, 95% CI 0.20 – 0.94; and OR = 0.62, 95% CI 0.43– 0.89, respectively). For IBD5, the most significant associations were observed for homozygotes of rs2631367 (OCTN2-207) (OR = 2.86, 95% CI 1.69 – 4.86) and OCTN2 intron 2 variant IGR2230 (OR = 2.80, 95% CI 1.66 – 4.73). ATG16L1 Thr300Ala showed strong association with CD risk (homozygote OR = 2.38, 95% CI 1.40 – 4.04, P = 0.001). For both ATG16L1 and IBD5, there was a tendency for a gene dosage effect. For IL23R risk variants the homozygote and heterozygote risks were very similar. No significant associations for SNPs between UC (n = 118) and controls (n = 315) were observed with the exception of heterozygotes for the rs7517848 minor allele of IL23R which, similar to that observed for the CD phenotype, was also protective of UC (OR = 0.57, 95% CI 0.35–0.92, P < 0.02).
Genotype and Phenotype Correlation
We evaluated the correlation between genotype and phenotypes of ileal disease site and intestinal complications (internal fistulizing or stricturing disease complications) (Table 4). Carriers of the rs7517848 minor allele of IL23R, particularly homozygotes (2 copy carriers), were at risk for ileal disease and hence protected against development of colonic-only CD (20% any ileal site versus 7% colon-only for homozygotes [OR = 4.25, 95% CI 1.18–15.3, P = 0.03] and 65% any ileal site versus 49% colon-only for any carriers [OR = 1.91, 95% CI 0.97–3.77, P = 0.06]) (Table 4a). None of the SNPs tested showed differences in genotype or carrier frequencies for comparisons between CD patients with a history of complications of internal fistulas and/or strictures and CD patients with only inflammation (Table 4b).
TABLE 4A.
Association of SNPs in Genes With CD Behavior (Fistula and/or Stricture n=127 vs. Inflammatory Only n=77)
| 0 Copy of Variant
|
1 Copy of Variant
|
2 Copies of Variant
|
Carrier of Variant
|
|||||
|---|---|---|---|---|---|---|---|---|
| Case/Control | OR (ref) | Case/Control | OR (95% CI) P | Case/Control | OR (95% CI) P | OR (95% CI) P | ||
| ATG16L1 | rs2241880a | 14/12 | 1.00 | 60/39 | 1.32 (0.55–3.15) 0.53 | 51/23 | 1.90 (0.76–4.74) 0.17 | 1.53 (0.67–3.52) 0.31 |
| IBD5_OCTN1 | rs1050152a | 32/16 | 1.00 | 61/40 | 0.76 (0.37–1.57) 0.46 | 31/18 | 0.86 (0.37–1.98) 0.73 | 0.79 (0.40–1.57) 0.51 |
| IBD5_OCTN2 | rs2631367 | 20/16 | 1.00 | 68/36 | 1.51 (0.70–3.27) 0.29 | 36/22 | 1.31 (0.56–3.05) 0.53 | 1.43 (0.69–2.98) 0.33 |
| IBD5_IGR2230 | rs17622208 | 21/16 | 1.00 | 67/36 | 1.25 (0.54–2.89) 0.61 | 36/22 | 1.42 (0.66–3.05) 0.37 | 1.35 (0.65–2.80) 0.41 |
| IBD5_IGR2198 | rs11739135 | 33/17 | 1.00 | 63/41 | 0.79 (0.39–1.60) 0.52 | 28/16 | 0.90 (0.39–2.11) 0.81 | 0.82 (0.42–1.62) 0.57 |
| IBD5_IGR2096 | rs12521868 | 34/16 | 1.00 | 61/43 | 0.67 (0.33–1.36) 0.27 | 29/15 | 0.91 (0.38–2.15) 0.83 | 0.73 (0.37–1.44) 0.37 |
| IL23R | rs7517847 | 50/26 | 1.00 | 57/34 | 0.87 (0.46–1.65) 0.67 | 19/16 | 0.62 (0.27–1.40) 0.25 | 0.79 (0.44–1.43) 0.44 |
| IL23R | rs2201841 | 32/27 | 1.00 | 78/39 | 1.69 (0.89–3.20) 0.11 | 16/10 | 1.35 (0.53–3.46) 0.53 | 1.62 (0.87–3.00) 0.13 |
| IL23R | rs11209026a | 119/74 | 1.00 | 7/2 | 2.18 (0.44–10.8) 0.34 | 0/0 | — | 2.18 (0.44–10.8) 0.34 |
| IL23R | rs10889677 | 32/26 | 1.00 | 77/41 | 1.53 (0.80–2.90) 0.20 | 17/9 | 1.53 (0.59–4.01) 0.38 | 1.53 (0.82–2.84) 0.18 |
| IL23R | rs1495965 | 22/21 | 1.00 | 76/38 | 1.91 (0.94–3.90) 0.08 | 28/17 | 1.57 (0.67–3.67) 0.30 | 1.80 (0.91–3.57) 0.09 |
Nonsynonymous SNPs.
TABLE 4B.
Association of SNPs in Genes With CD Location (Ileum Involved n=163 vs. Ileum Noninvolved n=43)
| 0 Copy of Variant
|
1 Copy of Variant
|
2 Copies of Variant
|
Carrier of Variant
|
|||||
|---|---|---|---|---|---|---|---|---|
| Case/Control | OR (ref) | Case/Control | OR (95% CI) P | Case/Control | OR (95% CI) P | OR (95% CI) P | ||
| ATG16L1 | rs2241880a | 20/4 | 1.00 | 80/21 | 0.76 (0.24–2.47) 0.65 | 58/18 | 0.64 (0.19–2.13) 0.47 | 0.71 (0.23–2.19) 0.55 |
| IBD5_OCTN1 | rs1050152a | 40/8 | 1.00 | 78/25 | 0.62 (0.26–1.51) 0.30 | 39/10 | 0.78 (0.28–2.18) 0.64 | 0.67 (0.29–1.56) 0.35 |
| IBD5_OCTN2 | rs2631367 | 31/5 | 1.00 | 79/26 | 0.49 (0.17–1.39) 0.18 | 47/12 | 0.63 (0.20–1.97) 0.43 | 0.53 (0.19–1.47) 0.23 |
| IBD5_IGR2230 | rs17622208 | 31/6 | 1.00 | 79/25 | 0.61 (0.23–1.63) 0.33 | 47/12 | 0.76 (0.26–2.23) 0.62 | 0.66 (0.26–1.70) 0.39 |
| IBD5_IGR2198 | rs11739135 | 42/8 | 1.00 | 79/26 | 0.58 (0.24–1.39) 0.22 | 36/9 | 0.76 (0.27–2.18) 0.61 | 0.63 (0.27–1.46) 0.28 |
| IBD5_IGR2096 | rs12521868 | 43/7 | 1.00 | 78/27 | 0.47 (0.19–1.16) 0.11 | 36/9 | 0.65 (0.22–1.92) 0.44 | 0.52 (0.21–1.25) 0.14 |
| IL23R | rs7517847 | 57/22 | 1.00 | 71/18 | 1.52 (0.75–3.11) 0.25 | 33/3 | 4.25 (1.18–15.3) 0.03 | 3.44 (1.00–11.8) 0.05 |
| IL23R | rs2201841 | 52/10 | 1.00 | 90/26 | 0.67 (0.30–1.49) 0.32 | 19/7 | 0.52 (0.17–1.57) 0.25 | 0.64 (0.29–1.39) 0.26 |
| IL23R | rs11209026a | 153/42 | 1.00 | 8/1 | 2.20 (0.27–18.1) 0.46 | 0/0 | — | 2.20 (0.27–18.1) 0.46 |
| IL23R | rs10889677 | 51/10 | 1.00 | 90/27 | 0.65 (0.29–1.46) 0.30 | 20/6 | 0.65 (0.21–2.04) 0.46 | 0.65 (0.30–1.43) 0.29 |
| IL23R | rs1495965 | 38/7 | 1.00 | 85/29 | 0.54 (0.22–1.34) 0.18 | 38/7 | 1.00 (0.32–3.13) 1.00 | 0.63 (0.26–1.53) 0.31 |
Nonsynonymous SNPs.
Haplotype Analysis
Haplotype analysis of IBD5 and IL23R variants are shown in Table 5. Strong linkage disequilibrium was observed between the variants of IBD5, but less in IL23R variants. For IBD5 variants, maximum pairwise D′ values were greater than 0.965 and r2 values larger than 0.790, and for IL23R variants, maximum pairwise D′ values were greater than 0.02 and r2 values greater than 0.0003. When the most common IBD5 haplotype consisting of the 5 major alleles CGGCG (SLC22A4-SLC22A5-IGR2230-IGR2198-IGR2096) is considered as reference (global test P = 0.001), there was as expected a significant risk for CD for the all-minor-allele haplotype TCAGT (OR = 1.62, 95% CI 1.23–2.14; P < 0.0006). Two other haplotypes showed nominal evidence for association with CD: a haplotype with only the IGR2230 and OCTN2 minor alleles (haplotype CCACG OR = 2.12, 95% CI 1.07–4.20; P < 0.03) and a rare haplotype with IGR2230, OCTN2 and IGR2198 (haplotype TCAGG OR = 3.09, 95% CI 0.98–9.74; P = 0.05). However, these 2 less common haplotypes do not retain significant association after correcting for multiple comparisons.
TABLE 5.
Associations of Haplotypes in IBD5, IL23R Genes and CD Risk
| Locus/Gene | Haplotype | Proportion in Cases | Proportion in Controls | OR (95% CI) | P-value |
|---|---|---|---|---|---|
| IBD5 (SLC22A4, SLC22A5, IGR2230, IGR2198, IGR2096) | |||||
| CGGCG | 0.437 | 0.554 | Reference | 0.001 (global) | |
| TCAGT | 0.466 | 0.372 | 1.62 (1.23–2.14) | 0.0006 | |
| CCACG | 0.048 | 0.030 | 2.12 (1.07–4.20) | 0.03 | |
| TCAGG | 0.019 | 0.008 | 3.09 (0.98–9.74) | 0.05 | |
| IL23R (rs1495965, rs10889677, rs11209026, rs2201841, rs7517847) | |||||
| ACGTT | 0.198 | 0.240 | Reference | 0.0001 (global) | |
| GAGCT | 0.243 | 0.163 | 1.59 (0.98–2.56) | 0.06 | |
| GCGTG | 0.035 | 0.121 | 0.33 (0.15–0.74) | 0.007 | |
| ACGTG | 0.197 | 0.204 | 0.95 (0.60–1.49) | 0.82 | |
| GCGTT | 0.099 | 0.072 | 1.40 (0.76–2.59) | 0.28 | |
| GAGCG | 0.086 | 0.076 | 1.46 (0.84–2.55) | 0.18 | |
| AAGCT | 0.044 | 0.028 | 1.58 (0.66–3.83) | 0.31 | |
| ACATG | 0.011 | 0.019 | 0.55 (0.14–2.15) | 0.39 | |
| ACATT | 0.004 | 0.026 | 0.18 (0.02–1.42) | 0.10 | |
For 5-marker haplotype of IL23R for rs1495965-rs10889677-rs11209026-rs2201841-rs7517847, the GCGTG haplotype revealed significant protection for developing CD relative to the reference major SNP haplotype ACGTT (OR = 0.33, 95% CI 0.60–1.49, P < 0.007; global P = 0.0001). This haplotype has borderline significance for protection against CD after stringent Bonferroni correction (P = 0.056). The haplotype protection shows even greater significance when comparing its presence in CD versus HC relative to all other haplotypes as reference (OR = 0.09, 95% CI 0.02–0.33; P = 0.0003). No significant associations were found for any haplotypes between UC and controls (data not shown).
Both the IL23R associated haplotype (GCGTG) and the reference haplotype (ACGTT) have identical alleles for rs10889677, rs11209026, and rs2201841, suggesting that there may be independent risk of CD arising from rs1495965 and/or rs7517847. Because the rs7517847 G allele alone is protective of CD but the rs1495965 G allele alone (and in all other haplotypes) is a risk for CD (Table 2), we hypothesized that the protective association of the GCGTG haplotype (compared to the most common reference haplotype ACGTT) on the background of the rs11209026 common G allele may be driven by an independent protective effect of rs7517847 minor allele. We therefore evaluated rs7517847 conditioned on rs11209026 (Arg381Gln) and vice versa. We observed that the minor alleles for both SNPs showed an inverse association with CD independent of each other: the OR for rs7517847 adjusting for rs11209026 was 0.62 (95% CI 0.43–0.90, P = 0.01) and for rs11209026 adjusting for rs7517847 was 0.45 (95% CI 0.21–0.98, P = 0.04). However, compared to persons with wildtype genotypes at both SNPs, we did not observe a stronger inverse association while carrying variant protective alleles at both SNPs in comparison to those carrying variant allele in only 1 of the 2 SNPs. Therefore, although these 2 SNPs show association with CD risk and both SNPs should be considered in assessing CD risk for IL23R, their independence does not appear to be complete.
Gene–Gene Interaction Analysis
We performed the MDR analysis to explore the potential gene– gene interaction for CD risk between variants genotyped, with or without including variants in NOD2.15 The MDR exhaustively searched multifactor combinations (1-, 2-, 3-, and 4-marker combinations) of SNPs and picked the best model, which has the lowest prediction error, to explain the observed CD risk in the data. Among the 11 SNPs in ATG16L1, IBD5, and IL23R, after cross-validation and permutation tests, the 1-marker model result suggested that IL23R_rs10889677 has the lowest prediction error (Table 6a). This prediction error 40% is statistically significant, with an empirical P-value 0.007 based on 1000 permutations. Subjects carrying “AA” or “AC” genotype have a high risk for CD and were classified as affected individuals. This result is compatible with the single-marker association result, which suggested carrying variant allele in IL23R_rs10889677 has the strongest risk for CD among carriers (Table 2).
TABLE 6A.
Multidimensionality Reduction (MDR) (IL23R as Dominant, IBD5, and ATG16L1 as Genotypic, NOD2 Not Considered)
| No. of Markers Combinations | Best Model | Cross-Validation Consistency | Prediction Error | Significance, P-value (Based on 1000 Permutations) |
|---|---|---|---|---|
| 1 | IL23R_rs10889677a | 10 | 0.40 | 0.007 |
| 2 | IBD5_IGR2230, IL23R_rs10889677b | 5 | 0.44 | 0.205 |
| 3 | IBD5_IGR2198, ATG16L1, IL23R_rs10889677c | 7 | 0.38 | 0.001 |
| 4 | IBD5_IGR2198, ATG16L1, IL23R_rs2201841, IL23R_rs7517847d | 7 | 0.39 | 0.003 |
IL23R_rs10889677 in dominant mode (AA/AC vs. CC).
IBD5_IGR2230 (rs17622208) in genotypic mode (AA, GA, GG); IL23R_rs108 (rs10889677) in dominant mode (AA/AC vs. CC).
IBD5_IGR2198 (rs11739135) in genotypic mode (GG, GC, CC); ATG16L1 (rs2241880) in genotypic mode (GG, AG, AA); IL23R_rs10889677 in dominant mode (AA/AC vs. CC).
IBD5_IGR2198 (rs11739135) in genotypic mode (GG, GC, CC); ATG16L1 (rs2241880) in genotypic mode (GG, AG, AA); IL23R_rs2201841 in dominant mode (CC/CT vs. TT); IL23R_rs7517847 in dominant mode (GG/TG vs. TT).
When SNPs were considered 2 at a time, the best selected model suggested interaction between IL23R_rs10889677 and IBD5_IGR2230 (rs17622208) (Table 6a). However, this prediction error 44% is not statistically significant, with an empirical P-value 0.205 based on 1000 permutations. Interestingly, when interaction between these 2 markers was also suggested using a logistic regression approach, it was noted that IBD5_IGR2230 risk was only significant in the presence of IL23R_rs10889677 risk allele (Table 7). However, the test for interaction did not reach statistical significance (Pinteraction = 0.17), while testing in a traditional logistic regression model by adding interaction terms except the main effects variables. When SNPs were considered 3 at a time, the best selected model which suggested interaction between IL23R_rs10889677, IBD5_IGR2198 (rs11739135), and ATG16L1 (rs2241880) showed the lowest prediction error among all combinations of 3 SNPs. This prediction error 38% is statistically significant, with an empirical P-value 0.001 based on 1000 permutations. When SNPs were considered 4 at a time, the best selected model which suggested interaction between IL23R_rs2201841, IL23R_rs7517847, IBD5_IGR2198 (rs11739135), and ATG16L1 (rs2241880) had the lowest prediction error among all combinations of 4 SNPs. This prediction error 39% is statistically significant, with an empirical P-value 0.003 based on 1000 permutations (Table 6a).
TABLE 7.
Association Between IL23R rs10889677 Carrier and CD Risk, Stratified by Carrier Status of IBD5_IGR2230
| Noncarrier of IGR2230 (n=134)
|
Carrier of IGR2230 (n=383)
|
|||
|---|---|---|---|---|
| OR (95% CI) | P-value | OR (95% CI) | P-value | |
| Carrier of rs10889677 | 1.47 (0.69–3.16) | 0.32 | 2.73 (1.77–4.20) | <0.001 |
Pinteraction = 0.17.
We also performed MDR analysis including the presence of NOD2 mutations (in jointly additive mode) as well as ATG16L1, IBD5, and IL23R (adding NOD2 mutation wildtypes, heterozygotes, and homozygotes: data from Brant et al15). After cross-validation and permutation tests, the selected multifactor models: 1-marker model (NOD2, empirical P-value 0.019); 2-marker model (NOD2-IBD5, empirical P-value 0.001); 3-marker model (NOD2-IBD5-ATG16L1, empirical P-value 0.003); and 4-marker model (NOD2-IBD5-ATG16L1-IL23R, empirical P-value 0.006) (Table 6b). This result is illustrated in Figure 1.
TABLE 6B.
Multidimensionality Reduction (MDR) (IL23R as Dominant, IBDs and ATG16 as Genotypic, NOD2 Considered as Addictive)
| No. of Markers Combinations | Best Model | Cross-Validation Consistency | Prediction Error | Significance, P-value (Based on 1000 Permutations) |
|---|---|---|---|---|
| 1 | NOD2a | 9 | 0.40 | 0.019 |
| 2 | NOD2, IBD5_OCTN2b | 6 | 0.37 | 0.001 |
| 3 | NOD2, IBD5_OCTN1, ATG16L1c | 4 | 0.38 | 0.003 |
| 4 | NOD2, IBD5_OCTN1, ATG16L1, IL23R_rs10889677d | 5 | 0.39 | 0.006 |
NOD2 in jointly additive mode (o variant, 1 variant, 2 variants).
IBD5_OCTN2 (rs2631367) in genotypic mode (AA, GA, GG); NOD2 in jointly additive mode (o variant, 1 variant, 2 variants).
IBD5_OCTN1 (rs1050152) in genotypic mode (TT, CT, CC); ATG16L1 (rs2241880) in genotypic mode (GG, AG, AA); NOD2 in jointly additive mode (0 variant, 1 variant, 2 variants).
IBD5_OCTN1 (rs1050152) in genotypic mode (TT, CT, CC); ATG16L1 (rs2241880) in genotypic mode (GG, AG, AA); NOD2 in jointly additive mode (0 variant, 1 variant, 2 variants); IL23R_rs10889677 in dominant mode (AA/AC vs. CC).
FIGURE 1.
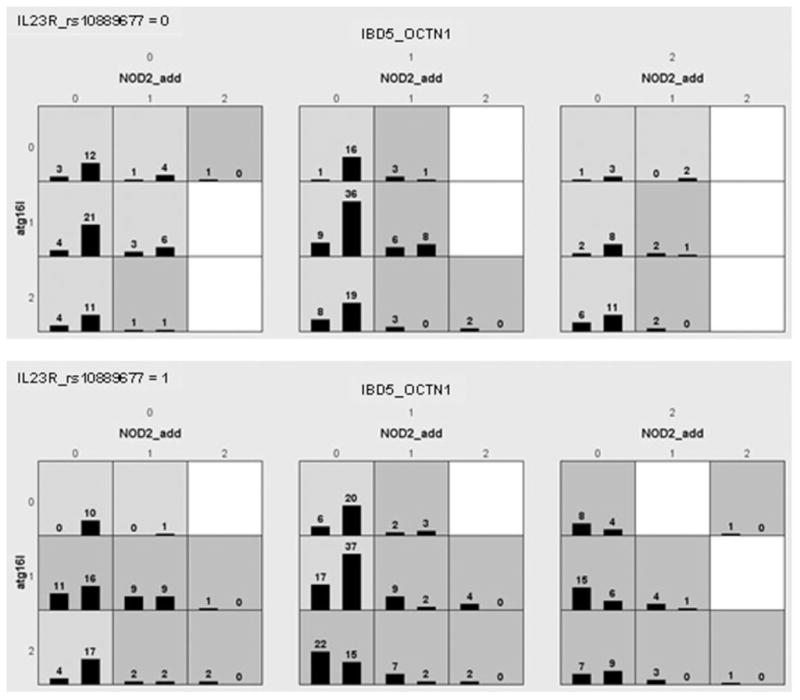
Histograms of cases and controls illustrating evidence observed in multidimensional reduction (MDR) analysis of interaction among NOD2 mutations, and specific risk variants for IL23R, IBD5, and ATG16L1. For each small square the left bar represents number of cases and right bar number of controls. As seen, cases are more represented in the bars in the bottom panel showing the presence of IL23R_rs10889677 3′ untranslated risk allele as compared to absence of this allele in the top panel, especially in the 2 right large squares representing heterozygotes and homozygotes (respectively) for OCTN1 (SLC22A4) rs1050152 risk variants, and in the bottom 2 rows of each large square, representing heterozygotes and homozygotes (respectively) for the ATG16L1 Ala300 risk variant. The dark-shaded squares are higher risk for CD and light-shaded squares are lower risk for CD. Absence of NOD2 mutations in the left column of each large square is associated with lower CD risk except for combined homozygotes for IBD_OCTN1 and carriers for IL23R_rs10889677 risk allele (bottom, right, large square).
Estimation of Effects Using Logistic Regression
We evaluated all potential CD risk factors assessed in our population-based cohort, controlling for sex, attained age, and geography (the 3 bases of control recruitment) using traditional logistic regression both with and without inclusion of risk polymorphisms for ATG16L1, IBD5 (IGR2230), IL23R (rs10889677) and the presence of any of the major NOD2 mutations (data from Brant et al15). As seen in Table 8, risk alleles for each of these 4 genes significantly contribute to overall CD risk with point estimated OR, for carrying a risk allele, of 1.9 for ATG16L1 carriers, 2.2 for IBD5_IGR2230 carriers, 2.2 for IL23R_rs10889677 carriers, and 4.4 for NOD2 carriers. No statistically significant gene–gene or gene–environment interactions were observed in our data. Interestingly, risks for Jewish ethnicity, tobacco, and IBD family history remained significant (and were each slightly increased) with addition of the 4 molecular genetic risk factors (Table 8).
TABLE 8.
Logistic Regression Modeling Without and With Risk Genes
| Without Risk Genes in Model
|
With Risk Genes in Model
|
|||
|---|---|---|---|---|
| OR 95% CI | P-value | OR 95% CI | P-value | |
| Age (<30 y/o vs. >=30 y/o) | 3.88 (1.96–7.69) | <0.001 | 3.57 (1.71–7.46) | 0.001 |
| Gender (female vs. male) | 0.56 (0.37–0.85) | 0.006 | 0.58 (0.37–0.91) | 0.02 |
| Residency (urban vs. rural) | 1.01 (0.68–1.49) | 0.98 | 1.05 (0.68–1.62) | 0.82 |
| Positive family history of IBD | 2.41 (1.31–4.42) | 0.005 | 2.75 (1.42–5.31) | 0.003 |
| Tobacco use | 1.84 (1.25–2.71) | 0.002 | 2.06 (1.35–3.14) | 0.001 |
| Jewish ethnicity | 17.1 (2.12–138.3) | 0.008 | 20.1 (2.16–186.8) | 0.008 |
| Carrier of NOD2 mutation | — | 4.45 (2.68–7.38) | <0.001 | |
| Carrier of ATG16L1 | — | 1.88 (1.09–3.24) | 0.02 | |
| Carrier of rs10889677 | — | 2.13 (1.39–3.28) | 0.001 | |
| Carrier of IBD5_IGR2230 | — | 2.16 (1.30–3.59) | 0.003 | |
Penetrance and Population Attribution Risk
We next evaluated the penetrance and population attributable risks for developing CD for the 3 new genes studied (IL23R risk polymorphisms of rs10889677, independent protective polymorphisms Arg381Gln [rs11209026] and rs7517847, and IBD5_IGR2230 and ATG16L1 Thr300Ala polymorphisms) in our population-based case-control study within a cohort utilizing penetrance figures derived from the same population epidemiologic database as the cases and controls were derived (Table 9). For reference we utilized the Manitoba CD prevalence of 270/100,000 from the year 2000.23 Note that for SNPs where the minor allele is protective for CD (i.e., IL23R SNPs rs7517848 and Arg381Gln), population penetrance is decreased in comparison to the wildtype “risk” allele (Table 9).
TABLE 9.
Penetrance and Population Attributable Risks of Selected Variants in Candidate Genes
| IL23R_rs10889677a | IL23R_rs7517847b | IL23R_rs11209026c | IBD5_IGR2230 | ATG16L1 (Thr300Ala) | |
|---|---|---|---|---|---|
| Pen −/− | 0.0015 | 0.0037 | 0.0028 | 0.0016 | 0.0015 |
| Pen −/+ | 0.0038 | 0.0023 | 0.0013 | 0.0027 | 0.0027 |
| Pen +/+ | 0.0037 | 0.0022 | — | 0.0044 | 0.0035 |
| PAR −/+ | 38.1% | −22.7% | −5.1% | 26.3% | 29.1% |
| PAR +/+ | 12.3% | −9.4% | — | 24.0% | 27.7% |
| PAR carrier | 43.1% | −36.9% | −5.1% | 40.2% | 44.3% |
PAR for rs10889677 (carrier): 43% of the CD in the underlying population of this study derived from could be preventable if the whole population does not carry variant A allele. In other words, 43% of CD in the whole population was attributed to carrying variant A allele in the whole population.
Carrying variant G allele has reduced risk of CD, the interpretation of PAR for IL23R_rs7517847 would be: PAR for rs7517847 (carrier): a 37% increase of CD in the underlying population of this study derived from could be observed if the whole population does not carry variant G allele.
Carrying variant A allele has reduced risk of CD, the interpretation of PAR for IL23R_rs11209026 would be: PAR for rs11209026 (carrier): a 5% increase of CD in the underlying population of this study derived from could be observed if the whole population does not carry protective A allele.
DISCUSSION
Evaluation of IBD5, ATG16L1, and IL23R as IBD Genetic Risk Factors
After NOD2, the loci/candidate genes IBD5, IL23R, and ATG16L1 have been observed to have the most consistent evidence for CD risk. Prior to recent genomewide association studies, IBD5 has been the only other established CD risk haplotype with SLC22A4 (OCTN1) and SLC22A5 (OCTN2) functional variants as the leading candidate genes. Furthermore, of multiple genes identified by recent genome-wide association studies, IL23R and ATG16L1 have the strongest risk for CD, with ORs generally greater than 2.0 (or less than 0.5), with the other genes having effects generally OR less than 1.5.8,10,26,27 Therefore, after NOD2, these 3 loci are most relevant to study in a population-based cohort to better understand their individual and compound effects, and effects relative to other IBD risk factors. Although the size of this study was suboptimal, perhaps because of the nature of the population-based cohort, we were able to observe significant association for nearly all of the previously identified associated SNPs found within each of these loci. The magnitude of risk was similar to that observed in other cohorts. As expected for common risk alleles with modest effects (OR between 1.5 and 2.5) and for a disease that is present in 0.1%–0.3% of a given population, penetrance values were less than 0.5% for even the higher-risk homozygote carriers for risk alleles of IBD5, IL23R, and ATG16L1, with penetrance 0.13% for the uncommon IL23R Arg381Gln protective variant.
In terms of actual risk for CD, even presence of risk alleles at all four loci will result in only a very low risk for developing CD, too low for clinical use: i.e., for persons that carry risk alleles at ATG16L1, IBD5 and IL23R, and are also heterozygous for NOD2, we estimate population penetrance at less than 2%. However, for persons with a CD family history, because of a greater pretest probability of developing CD, genotyping these four loci may potentially be useful for genetic counseling. Assuming a 5% estimated lifetime risk of developing CD for siblings of CD patients, and assuming familial risk is largely independent of genotype at these loci (as previously observed for NOD2),15 for siblings of CD patients that are carriers of risk alleles at all four loci, we estimate a 27% ± 21% CD risk. In contrast, we estimate only a 1.6% CD risk for siblings of CD patients that carry only one risk allele (heterozygous) at only one of the four loci (or one or two risk alleles at IL23R rs10889677). Therefore, testing siblings of CD patients for these four risk loci together may be useful to identify siblings at greater or lesser risk of developing CD, perhaps for monitoring for early signs of CD or for potential preventative therapy trials. Note that large population-based studies will be needed to better pinpoint the actual risks for genetic counseling purposes.
For IBD5 and ATG16L1 but not for IL23R, homozygotes had greater risk than heterozygotes, suggesting a potential gene dosage effect. This is compatible with multiple studies of IBD5 including the discovery cohort, and suggests that modeling multiple CD risk factors in larger cohorts should take into account IBD5 gene dosage.5,28,29 For ATG16L1 a gene dosage effect has not been observed.30
In contrast to NOD2 and ATG16L1, the specific functional risk alleles for IBD5 and IL23R have not been established. Therefore, we genotyped several potential risk alleles and performed haplotype analyses. Nearly all SNPs except for 1 showed significant association; however, for neither of these loci was genotyping able to localize the risk to a single polymorphism. For IL23R, haplotype analyses and conditioning analyses showed independent CD protection associated with rs7517847 located on intron 6 and the conserved, Arg381Gln nonsynonymous variant. SNP rs7517847 was observed to have risk independent of Arg381Gln in the NIDDK IBD Genetics Consortium discovery cohort and in an Oxford, UK cohort.8,31 Raelson et al32 in a recently reported French Canadian whole genome association study observed 2 protective and 2 risk haplotypes for IL23R, and suggested that a distinct risk haplotype involves epistatic interactions of variants in the 5′ and 3′ regions of the gene. Additionally, the MDR analyses suggested that the untranslated 3′ message variant, rs10889677, shows the strongest effect. Whether any of these IL23R SNPs alter IL23R function remains to be determined. Nonetheless, to understand the mechanism underlying IL23R genetic risk and for optimal genetic counseling purposes, high-density association analyses in very large cohorts and functional studies may be necessary to pinpoint the apparently multiple IL23R functional risk alleles. Consistent with other studies of IBD5, linkage disequilibrium across the SNPs genotyped was too great to localize risk to 1 or more individual SNPs genotyped.
Phenotype Assessment
Of the 3 genes evaluated, only IL23R has been consistently shown to be associated, albeit less strongly, with UC.8,33–35 In our study, for all 3 loci genotyped, only 1 SNP, IL23R-rs7517847, showed significant UC association: protection from UC similar to the CD association analysis. Lack of association for other IL23R SNPs is most likely because of the smaller size of the UC cohort and weaker effect of IL23R on UC.
We did not observe association of IBD5 and ATG16L1 variants with UC; however, with a few exceptions36–38 significant association with these loci with UC has not been observed,39–41 and, in general, IBD5 and ATG16L1 are considered as mainly CD-specific risk loci. Analyses of the different loci and subphenotypes of CD disease showed only 1 association, rs7517848 as a protective allele for colonic CD. Interestingly, in our cohort the minor allele of IL23R-rs7517848 exhibits protection for both UC and colon-only CD, suggesting that IL23R may have a predilection for an effect on colon site for all IBD phenotypes. Nonetheless, the CD site association was only nominal and, though of interest, it does not retain significance when corrected for the multiple alleles tested. CD subphenotype association analyses and IL23R has been negative in several cohorts, including the Oxford study where rs7517847 was evaluated.31 We did not find any evidence of subphenotype association with IBD5 or ATG16L1. ATG16L1 was observed to be associated with ileal CD in 1 British cohort but this has not been replicated in several other cohorts.30,38,42 IBD5 association with subphenotypes have been variable and have especially included perianal site and development of fistulizing/stricturing disease.29,40,43,44 Our analyses did not include perianal site.
Compound Role for Disease Risk Prediction
Although several individual gene polymorphisms have been identified for the disease association and increasing individual risk, only a limited number of studies have assessed the risk of CD by way of combining information from multiple predisposing polymorphisms. Traditional logistic regression modeling showed that all 4 genes/loci significantly contributed to CD risk as well as Jewish ethnicity, tobacco, and IBD family history. It is noteworthy that as in our prior study of NOD2, there remained a significant risk for IBD family history even after incorporating 3 additional risk genes/loci. This may not be surprising, given recent findings (from combined analyses of recent genomewide association studies) that there are numerous additional CD risk genes as well as several familial IBD genetic loci independent of these risk genes (i.e., IBD2, IBD4, IBD6, IBD7, IBD8, and IBD9).45,46 Of course, there also remains the possibility that some of the observed familial risk is due to common, non-genetic risk factors shared within families, in addition to tobacco and geography.
Our logistic regression model did not find significant evidence for gene– gene or gene– environment interaction, perhaps in part because of the limited size of our study, although as noted (Table 7) there was a trend for interaction of IBD5 and IL23R. It is particularly noteworthy that a study of the Oxford, UK cohort also observed that, with the exception of rs11209026, IL23R risk polymorphisms showed significant CD association only in the subgroup of persons positive for IBD5.31
The MDR analyses suggested that there is likely significant interaction between all 4 of these most influential CD genes and loci. This may not be surprising, as one may expect that there may be overlap in the pathways of the CD risk effects of these genes—in particular NOD2 and ATG16L1, both believed to produce CD risk by altering innate immunity against gut bacteria. An examination of Figure 1 illustrates that study subjects having few risk alleles are much more represented in the controls, whereas study subjects with NOD2 mutations and additional risk alleles are more represented among the cases (Fig. 1). The MDR we performed searched all possible combinations of 1-, 2-, 3-, and 4-SNP and selected the best models with the lowest prediction errors. This approach provides a clue for hypothesis generation in exploring the underlying biological mechanism. Evidence for interaction suggests that pathophysiologic mechanisms of 1 genetic locus involves that of the other locus, and hence this knowledge might be useful to uncover the mechanisms that lead to disease. It also follows that should interaction between loci be established, this interaction may provide clues as to the gene responsible for an observed association that remains unresolved. Thus, in our case, examination of IBD5-IL23R interaction may help localize the “true” IBD5 disease gene from that of the multiple risk genes within the IBD5 haplotype. For example, the IBD5 haplotype has been found to have equivalent association over 4 candidate genes, from prolyl 4-hydroxylase (P4HA2) through SLC22A4 and SLC22A5 to interferon regulatory factor 1 (IRF1),47 with only SLC22A4 and SLC22A5 demonstrating haplotype associated functional variants. IRF-1, which is activated by interferon, binds to promoter elements in both the IL12 p35 and IL12 p40 genes, and this binding has been shown to have significant effects on expression of both the IL12 p35 subunit and the p40 subunit common to both IL12 and IL23.48 Hence, one may speculate that should IL23R indeed require presence of the IBD5 risk haplotype as suggested by our findings and that of Cummings et al,31 IL23R CD risk variants may only be present when risk alleles on IRF-1 allow enough IRF-1 activity necessary to produce adequate p40 to interact with IL23R. Hence, functionally, of the genes within the key IBD5 haplotype, IRF-1 has the most overt relevance to IL23R, and with evidence in 2 studies for potential IBD5 and IL23R genetic interaction, the functional relationship of variations in these 2 genes may deserve specific study.
Because both our cases and controls were recruited similarly and are from a population-based cohort, our study, albeit limited in power, is more ideal to investigate gene–gene interaction, gene– environment interaction, and the effect of multiple risk alleles, in the context of other risk factors, on developing CD. Evidence for significant interactions tends to require very large sample sizes. Thus, epigenesist methods like MDR will be increasingly important to find support for interactions in smaller sample sets. The addition of multiple risk alleles is a powerful step in developing informative models for predicting IBD risk. With the discovery of numerous CD risk genes and loci, population-based cohorts will be critical to accurately determine the risks of CD-predisposing genes, and their relevance to each other and to other personal (e.g., family history or appendectomy) and environmental risk factors. These assessments will be especially useful for genetic counseling and disease risk modeling, initiation of preventive measures, and reliable public health estimates of the burden of disease genes. With the presence of numerous genes, different analytic strategies such as “reverse phenotyping”49 will be necessary to identify sets of variations in multiple subphenotypes that correlate with single or multiple disease genes, and to find complex interactions between multiple disease genes to predict disease risk and course. Such discoveries will also assist in disease pathway determinations and hopefully development of novel therapeutics. However, all of these important measures will be best achieved through development of very large population-based cohorts (with several thousand affected individuals). Planning should begin now for the practical development of such cohorts, perhaps through strategies such as coordinated recruitment for several different but perhaps related complex-genetic disorders (i.e., immune diseases) and 1 very large comparison (HC) cohort.
Acknowledgments
Supported in part by National Institutes of Health USA grant R01DK58189 (to S.R.B.), the Stuart M. Bainum Family (to S.R.B.), the Harvey M. and Lyn P. Meyerhoff Inflammatory Bowel Disease Center (to S.R.B., L.W.D., and M.H.W.), the Canadian Institutes of Health Research Investigator Award (to C.N.B.), and the Crohn’s and Colitis Foundation of Canada Research Scientist Award (to C.N.B.).
We thank Dr. Ingo Ruczinski, Johns Hopkins Bloomberg School of Public Health, for assistance with the statistical analysis for gene– gene interactions. We thank Yu-qiong Wu and Amnber Iqbal for genotyping assistance.
References
- 1.Brant SR, Shugart YY. Inflammatory bowel disease gene hunting by linkage analysis: rationale, methodology, and present status of the field. Inflamm Bowel Dis. 2004;10:300–311. doi: 10.1097/00054725-200405000-00019. [DOI] [PubMed] [Google Scholar]
- 2.Hugot JP, Chamaillard M, Zouali H, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 3.Ogura Y, Bonen DK, Inohara N, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 4.Economou M, Trikalinos TA, Loizou KT, et al. Differential effects of NOD2 variants on Crohn’s disease risk and phenotype in diverse populations: a metaanalysis. Am J Gastroenterol. 2004;99:2393–2404. doi: 10.1111/j.1572-0241.2004.40304.x. [DOI] [PubMed] [Google Scholar]
- 5.Rioux JD, Daly MJ, Silverberg MS, et al. Genetic variation in the 5q31 cytokine gene cluster confers susceptibility to Crohn disease. Nat Genet. 2001;29:223–228. doi: 10.1038/ng1001-223. [DOI] [PubMed] [Google Scholar]
- 6.Peltekova VD, Wintle RF, Rubin LA, et al. Functional variants of OCTN cation transporter genes are associated with Crohn disease. Nat Genet. 2004;36:471–475. doi: 10.1038/ng1339. [DOI] [PubMed] [Google Scholar]
- 7.Daly MJ, Rioux JD. New approaches to gene hunting in IBD. Inflamm Bowel Dis. 2004;10:312–317. doi: 10.1097/00054725-200405000-00020. [DOI] [PubMed] [Google Scholar]
- 8.Duerr RH, Taylor KD, Brant SR, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franke A, Hampe J, Rosenstiel P, et al. Systematic association mapping identifies NELL1 as a novel IBD disease gene. PLoS ONE. 2007;2:e691. doi: 10.1371/journal.pone.0000691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rioux JD, Xavier RJ, Taylor KD, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Inoue N, Tamura K, Kinouchi Y, et al. Lack of common NOD2 variants in Japanese patients with Crohn’s disease. Gastroenterology. 2002;123:86–91. doi: 10.1053/gast.2002.34155. [DOI] [PubMed] [Google Scholar]
- 12.Croucher PJ, Mascheretti S, Hampe J, et al. Haplotype structure and association to Crohn’s disease of CARD15 mutations in two ethnically divergent populations. Eur J Hum Genet. 2003;11:6–16. doi: 10.1038/sj.ejhg.5200897. [DOI] [PubMed] [Google Scholar]
- 13.Guo QS, Xia B, Jiang Y, et al. NOD2 3020insC frameshift mutation is not associated with inflammatory bowel disease in Chinese patients of Han nationality. World J Gastroenterol. 2004;10:1069–1071. doi: 10.3748/wjg.v10.i7.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamazaki K, Onouchi Y, Takazoe M, et al. Association analysis of genetic variants in IL23R, ATG16L1 and 5p13.1 loci with Crohn’s disease in Japanese patients. J Hum Genet. 2007;52:575–583. doi: 10.1007/s10038-007-0156-z. [DOI] [PubMed] [Google Scholar]
- 15.Brant SR, Wang MH, Rawsthorne P, et al. A population-based case-control study of CARD15 and other risk factors in Crohn’s disease and ulcerative colitis. Am J Gastroenterol. 2007;102:313–323. doi: 10.1111/j.1572-0241.2006.00926.x. [DOI] [PubMed] [Google Scholar]
- 16.Bernstein CN, Blanchard JF, Rawsthorne P, et al. Epidemiology of Crohn’s disease and ulcerative colitis in a central Canadian province: a population-based study. Am J Epidemiol. 1999;149:916–924. doi: 10.1093/oxfordjournals.aje.a009735. [DOI] [PubMed] [Google Scholar]
- 17.Lewontin RC, Feldman MW. A general asymptotic property of two-locus selection models. Theor Popul Biol. 1988;34:177–193. doi: 10.1016/0040-5809(88)90041-x. [DOI] [PubMed] [Google Scholar]
- 18.Devlin B, Risch N. A comparison of linkage disequilibrium measures for fine-scale mapping. Genomics. 1995;29:311–322. doi: 10.1006/geno.1995.9003. [DOI] [PubMed] [Google Scholar]
- 19.Schaid DJ, Rowland CM, Tines DE, et al. Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am J Hum Genet. 2002;70:425–434. doi: 10.1086/338688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lake SL, Lyon H, Tantisira K, et al. Estimation and tests of haplotype-environment interaction when linkage phase is ambiguous. Hum Hered. 2003;55:56–65. doi: 10.1159/000071811. [DOI] [PubMed] [Google Scholar]
- 21.Hampe J, Cuthbert A, Croucher PJ, et al. Associations between insertion mutation in NOD2 gene and Crohn’s disease in German and British populations. Lancet. 2001;357:1925–8. doi: 10.1016/S0140-6736(00)05063-7. [DOI] [PubMed] [Google Scholar]
- 22.Schlesselman J. Case-control studies. Oxford: Oxford University Press; 1982. [Google Scholar]
- 23.Blanchard JF, Bernstein CN, Wajda A, et al. Small-area variations and sociodemographic correlates for the incidence of Crohn’s disease and ulcerative colitis. Am J Epidemiol. 2001;154:328–335. doi: 10.1093/aje/154.4.328. [DOI] [PubMed] [Google Scholar]
- 24.Ritchie MD, Hahn LW, Roodi N, et al. Multifactor-dimensionality reduction reveals high-order interactions among estrogen-metabolism genes in sporadic breast cancer. Am J Hum Genet. 2001;69:138–147. doi: 10.1086/321276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hahn LW, Ritchie MD, Moore JH. Multifactor dimensionality reduction software for detecting gene-gene and gene-environment interactions. Bioinformatics (Oxford, UK) 2003;19:376–382. doi: 10.1093/bioinformatics/btf869. [DOI] [PubMed] [Google Scholar]
- 26.Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parkes M, Barrett JC, Prescott NJ, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat Genet. 2007;39:830–832. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mirza MM, Fisher SA, King K, et al. Genetic evidence for interaction of the 5q31 cytokine locus and the CARD15 gene in Crohn disease. Am J Hum Genet. 2003;72:1018–1022. doi: 10.1086/373880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Armuzzi A, Ahmad T, Ling KL, et al. Genotype-phenotype analysis of the Crohn’s disease susceptibility haplotype on chromosome 5q31. Gut. 2003;52:1133–1139. doi: 10.1136/gut.52.8.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hampe J, Franke A, Rosenstiel P, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 31.Cummings JR, Ahmad T, Geremia A, et al. Contribution of the novel inflammatory bowel disease gene IL23R to disease susceptibility and phenotype. Inflamm Bowel Dis. 2007;13:1063–1068. doi: 10.1002/ibd.20180. [DOI] [PubMed] [Google Scholar]
- 32.Raelson JV, Little RD, Ruether A, et al. Genome-wide association study for Crohn’s disease in the Quebec Founder Population identifies multiple validated disease loci. Proc Natl Acad Sci U S A. 2007;104:14747–14752. doi: 10.1073/pnas.0706645104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tremelling M, Cummings F, Fisher SA, et al. IL23R variation determines susceptibility but not disease phenotype in inflammatory bowel disease. Gastroenterology. 2007;132:1657–1664. doi: 10.1053/j.gastro.2007.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weersma RK, Zhernakova A, Nolte IM, et al. ATG16L1 and IL23R are associated with inflammatory bowel diseases but not with celiac disease in The Netherlands. Am J Gastroenterol. 2008;103:621–627. doi: 10.1111/j.1572-0241.2007.01660.x. [DOI] [PubMed] [Google Scholar]
- 35.Glas J, Konrad A, Schmechel S, et al. The ATG16L1 gene variants rs2241879 and rs2241880 (T300A) are strongly associated with susceptibility to Crohn’s disease in the German population. Am J Gastroenterol. 2008;103:682–691. doi: 10.1111/j.1572-0241.2007.01694.x. [DOI] [PubMed] [Google Scholar]
- 36.Giallourakis C, Stoll M, Miller K, et al. IBD5 is a general risk factor for inflammatory bowel disease: replication of association with Crohn disease and identification of a novel association with ulcerative colitis. Am J Hum Genet. 2003;73:205–211. doi: 10.1086/376417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Waller S, Tremelling M, Bredin F, et al. Evidence for association of OCTN genes and IBD5 with ulcerative colitis. Gut. 2006;55:809–814. doi: 10.1136/gut.2005.084574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prescott NJ, Fisher SA, Franke A, et al. A nonsynonymous SNP in ATG16L1 predisposes to ileal Crohn’s disease and is independent of CARD15 and IBD5. Gastroenterology. 2007;132:1665–1671. doi: 10.1053/j.gastro.2007.03.034. [DOI] [PubMed] [Google Scholar]
- 39.Negoro K, McGovern DP, Kinouchi Y, et al. Analysis of the IBD5 locus and potential gene-gene interactions in Crohn’s disease. Gut. 2003;52:541–546. doi: 10.1136/gut.52.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Noble CL, Nimmo ER, Drummond H, et al. The contribution of OCTN1/2 variants within the IBD5 locus to disease susceptibility and severity in Crohn’s disease. Gastroenterology. 2005;129:1854–1864. doi: 10.1053/j.gastro.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 41.Roberts RL, Gearry RB, Hollis-Moffatt JE, et al. IL23R R381Q and ATG16L1 T300A are strongly associated with Crohn’s disease in a study of New Zealand Caucasians with inflammatory bowel disease. Am J Gastroenterol. 2007;102:2754–2761. doi: 10.1111/j.1572-0241.2007.01525.x. [DOI] [PubMed] [Google Scholar]
- 42.Cummings JR, Cooney R, Pathan S, et al. Confirmation of the role of ATG16L1 as a Crohn’s disease susceptibility gene. Inflamm Bowel Dis. 2007;13:941–946. doi: 10.1002/ibd.20162. [DOI] [PubMed] [Google Scholar]
- 43.Onnie CM, Fisher SA, Prescott NJ, et al. Diverse effects of the CARD15 and IBD5 loci on clinical phenotype in 630 patients with Crohn’s disease. Eur J Gastroenterol Hepatol. 2008;20:37–45. doi: 10.1097/MEG.0b013e3282f1622b. [DOI] [PubMed] [Google Scholar]
- 44.Vermeire S, Pierik M, Hlavaty T, et al. Association of organic cation transporter risk haplotype with perianal penetrating Crohn’s disease but not with susceptibility to IBD. Gastroenterology. 2005;129:1845–1853. doi: 10.1053/j.gastro.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 45.Shugart YY, Silverberg MS, Duerr RH, et al. An SNP linkage scan identifies significant Crohn’s disease loci on chromosomes 13q13.3 and, in Jewish families, on 1p35. 2 and 3q29. Genes Immun. 2008;9:161–167. doi: 10.1038/sj.gene.6364460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Daly MJ on Behalf of Crohn’s Disease GWA Meta-analysis Working Group. Joint genome-wide analysis of 3200 Crohn disease patients documents more than 20 significant associations. Am J Hum Genet Abstr Book. 2007:35. [Google Scholar]
- 47.Silverberg MS, Duerr RH, Brant SR, et al. Refined genomic localization and ethnic differences observed for the IBD5 association with Crohn’s disease. Eur J Hum Genet. 2007;15:328–335. doi: 10.1038/sj.ejhg.5201756. [DOI] [PubMed] [Google Scholar]
- 48.Liu J, Guan X, Tamura T, et al. Synergistic activation of interleukin-12 p35 gene transcription by interferon regulatory factor-1 and interferon consensus sequence-binding protein. J Biol Chem. 2004;279:55609–55617. doi: 10.1074/jbc.M406565200. [DOI] [PubMed] [Google Scholar]
- 49.Schulze TG, McMahon FJ. Defining the phenotype in human genetic studies: forward genetics and reverse phenotyping. Hum Hered. 2004;58:131–138. doi: 10.1159/000083539. [DOI] [PubMed] [Google Scholar]