

Abstract
We have further defined mechanism(s) by which the drug OSU-03012 (OSU) kills tumor cells. OSU lethality was suppressed by knock down of PERK and enhanced by knock down of ATF6 and IRE1α. OSU treatment suppressed expression of the chaperone, BiP/GRP78, and did so through reduced stability of the protein. Knock down of BiP/GRP78 further enhanced OSU lethality. Overexpression of BiP/GRP78 abolished OSU toxicity. Pre-treatment of cells with OSU enhanced radiosensitivity to a greater extent than concomitant or sequential drug treatment with radiation exposure. Expression of a mutant active p110 PI3K, or mutant active forms of the EGFR in GBM cells did not differentially suppress OSU killing. In contrast loss of PTEN function reduced OSU lethality, without altering AKT, p70 S6K or mTOR activity, or the drug's ability to radiosensitize GBM cells. Knock down of PTEN protected cells from OSU and radiation treatment whereas re-expression of PTEN facilitated drug lethality and radiosensitization. In a dose-dependent fashion OSU prolonged the survival of mice carrying GBM tumors and interacted with radiotherapy to further prolong survival. Collectively, our data show that reduced BiP/GRP78 levels play a key role in OSU-3012 toxicity in GBM cells, and that this drug has in vivo activity against an invasive primary human GBM isolate.
Keywords: OSU-03012, BiP/GRP78, ER stress, PERK, ionizing radiation, ceramide
Introduction
Inflammatory immune responses are part of the biology of many chronic human diseases, such as rheumatoid arthritis and multiple sclerosis. Cyclooxygenase inhibitors were developed to suppress the function of immune cells for treatment of such maladies.1,2 At the same time, it was discovered that COX2 was overexpressed in many tumor cell types and COX2 inhibitors such as Celecoxib could, at levels at or near their plasma Cmax, cause apoptosis and suppress tumor cell growth.3–6 Individuals with prolonged exposure to COX2 inhibitors were also shown to often have a lesser risk of developing cancer, notably colon cancer, suggestive that COX2 inhibitors had cancer preventative effects.7,8 As the biology of COX2 inhibitors was further investigated it was discovered that COX2 levels did not simply correlate with tumor cell sensitivity to COX2 inhibitors.9,10 Hence COX2 inhibitors had another target that played a role in their actions against cancer cells.
The agent OSU-03012, termed also AR-12, was developed as an anticancer agent, with Celecoxib as the chemical backbone.11–13 In vitro OSU-03012, lacks COX2 inhibitory activity. Based on these preliminary observations, OSU-03012 has been licensed and a Phase I trial has been initiated by Arno Pharmaceuticals. We have shown that OSU-03012 caused death through a form of endoplasmic reticulum (ER) stress, mitochondrial dysfunction and loss of HSP70 expression, and that was a caspase-independent form of cell death.12,13 The lethality of OSU-03012 in tumor cells in several initial reports was argued to be due to inhibition of the enzyme PDK-1, part of the PI3 kinase pathway, and OSU-03012, in the ∼10 µM range, has been shown to suppress AKT phosphorylation and showed measurable inhibition of PDK-1 activity. OSU-03012 interacts with multiple kinase inhibitors to kill tumor cells in a manner that is caspase independent.12–17
In the present studies, we have utilized primary human glioma and established colon cancer cell lines that lack or have restored ceramide synthase activity. We have further examined the impact of OSU-03012 on cell viability, and additionally defined the molecular mechanisms by which OSU-03012 enhances tumor cell death. We have discovered that a major mediator of OSU-03012 biology is through inhibition of the chaperone protein BiP/GRP78. OSU-03012 suppresses BiP/GRP78 levels and causes excessive toxic forms of ER stress that lead to cell killing. This results in GBM cell death in vitro and in vivo. Our data argue that use of OSU-03012 as an anti-glioblastoma drug for patient studies is warranted.12,13
Results
Treatment of primary human glioma cells with OSU-03012 increased the levels of LC3 and LC3II in a PERK dependent fashion (Fig. 1A). OSU-03012 treatment also decreased expression of GRP78 and enhanced protein kinase RNA-like endoplasmic reticulum kinase (PERK) phosphorylation. In agreement with OSU-03012 stimulating LC3 processing/autophagy, cell killing was suppressed by transient siRNA knock down of Beclin 1 or ATG5 (Fig. 1B). Knock down of mediators of ER stress signaling—inositol-requiring protein 1α (IRE1α), activating transcription factor 6 (ATF6) and PERK—differentially altered the cell death response. Knock down of IRE1α or of ATF6 enhanced OSU-03012 lethality whereas knock down of PERK reduced drug toxicity (Fig. 1C). A similar pattern of survival/killing was observed when OSU-03012 was combined with ionizing radiation exposure. Expression of dominant negative PERK generated a similar response to that of knock down of PERK (Fig. 1D).
Figure 1.

OSU-03012 toxicity in transformed cells is regulated by ER stress signaling proteins. (A) Upper: GBM12 cells were treated with 2 µM OSU-03012 and the expression of LC3I and LC3II determined over a 24 h time-course. Lower: GBM12 cells were transfected with empty vector plasmid (CMV) or a plasmid to express dominant negative PERK. Twenty-four hours after transfection cells were treated with vehicle (DMSO) or with OSU-03012 (2 µM). Cells were isolated after 6 h and the levels of LC3II determined. (B) GBM6 cells were transfected with scrambled siRNA (siSCR) or siRNAs to knock down ATG5 or Beclin1. Twenty-four hours after transfection cells were treated with vehicle (DMSO) or 2 µM OSU-03012. Viability was determined 48 h later (n = 3, +/- SEM). (C) GBM6 cells were transfected with scrambled siRNA (siSCR) or siRNAs to knock down PERK, ATF6 or IRE1α. Twenty-four hours after transfection cells were treated with vehicle (DMSO) or 2 µM OSU-03012. Cells were then irradiated (4 Gy). Viability was determined 48 h later (n = 3, +/- SEM). (D) GBM6 cells were transfected with empty vector plasmid (CMV) or a plasmid to express dominant negative PERK. Twenty-four hours after transfection cells were treated with vehicle (DMSO) or with OSU-03012 (2 µM). Viability was determined 48 h later (n = 3, +/- SEM).
In short-term viability and in colony formation assays OSU-03012 radiosensitized GBM cells (Fig. 2A–C). Based on these findings we then determined whether prior, concomitant or sequential drug exposure differentially affected radiosensitivity. In GBM6 and GBM12 cells, prior treatment with OSU-03012 radiosensitized cells to a greater extent than concomitant or sequential drug exposure (Fig. 2D).
Figure 2.
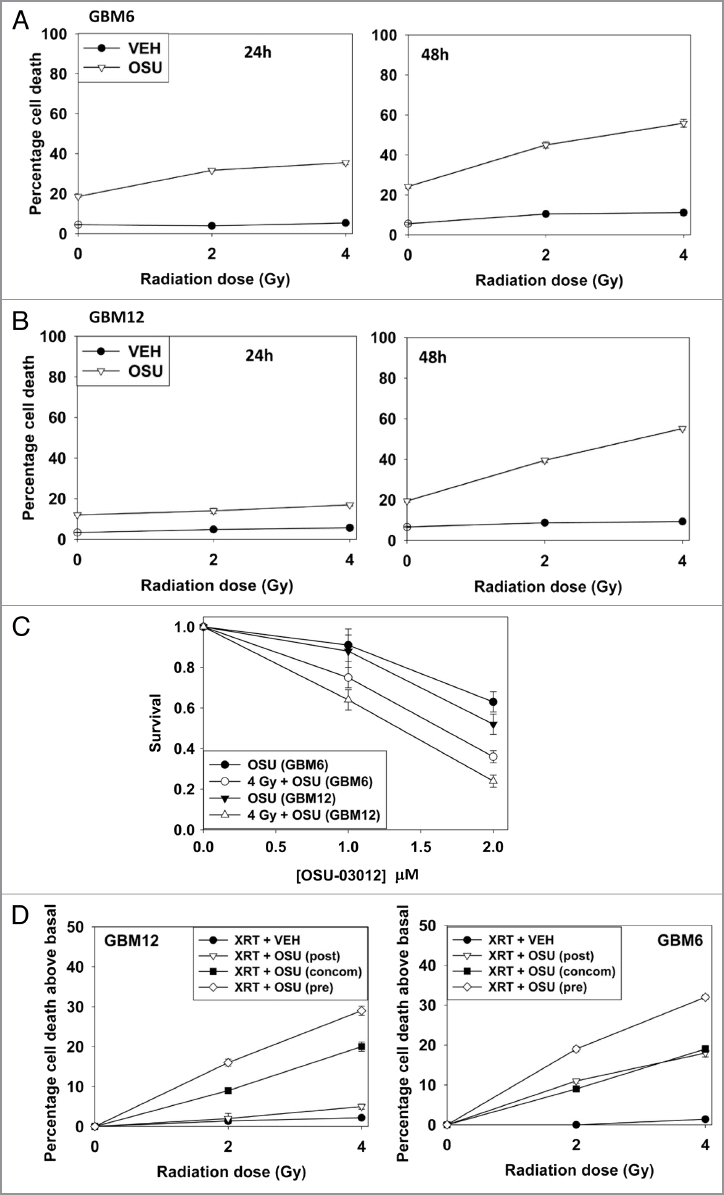

OSU-03012 radiosensitizes GBM cells. (A) GBM6 cells were treated with vehicle (DMSO) or with OSU-03012 (2 µM) and 30 min later irradiated (2 Gy, 4 Gy). Viability was determined 24 and 48 h later (n = 3, +/- SEM). (B) GBM12 cells were treated with vehicle (DMSO) or with OSU-03012 (2 µM) and 30 min later irradiated (2 Gy, 4 Gy). Viability was determined 24 and 48 h later (n = 3, +/- SEM). (C) GBM6 and GBM12 cells were plated as single cells. Twelve hours after plating cells were treated with vehicle or OSU-03012 (2 µM) and 30 min later irradiated (4 Gy). Forty-eight hours later the media was replaced with drug free media. Colonies were permitted to form over the following ∼14 d. Colonies of > 50 cells were counted (n = 3, +/- SEM). (D) GBM6 and GBM12 cells were treated with vehicle (DMSO) or with 2 µM OSU-03012 for 24 h (pre-treatment: “pre”); after which time portions of the vehicle treated cells were treated with OSU-03012 (concomitant: “concom”). Cells were irradiated (2 Gy, 4 Gy). In parallel sets, cells were incubated with vehicle for 48 h, irradiated and OSU-03012 added 24 h after irradiation. All cells were isolated for viability 24 h after radiation exposure. Data are presented as the true percentage increase above control values for each condition (n = 3, +/- SEM). (E) Left: SW480 and SW620 cells were treated with increasing doses of OSU-03012 (0–3 µM). Cells were isolated 48 h after drug treatment and viability determined (n = 3, +/- SEM); Right: SW480 and SW620 cells were treated with OSU-03012 (2 µM). Cells were irradiated and then isolated 48 h after drug treatment and viability determined (n = 3, +/- SEM). (F) Wild-type SW620 and SW620 cells with restored LASS6 expression were treated with OSU-03012 (2 µM). Cells were irradiated and then isolated 48 h after drug treatment and viability determined (n = 3, +/- SEM).
The bio-active lipid ceramide can play a role in many toxic processes and we next tested whether actions of the de novo ceramide synthesis pathway played a role in OSU-03012 lethality. To perform these assays we made use of isogenic colon cancer cell lines that lack or have restored activity of ceramide synthase 6 (LASS6). In a manner similar to our data in GBM cells, in SW480 cells, but not in isogenic SW620 cells that lack ceramide synthase 6 expression, OSU-03012 caused a dose-dependent increase in cell killing, and radiosensitized the cells (Fig. 2E). Stable transfection of SW620 cells to express ceramide synthase 6 enhanced OSU-3012 lethality as a single agent and restored the radiosensitization effects of OSU-03012 (Fig. 2F). Knock down of LASS6 in GBM cells also reduced OSU-03012 toxicity and the radiosensitizing effects of the drug (Fig. 2G). Collectively our data argue that ceramide plays an important role in the lethal effects of OSU-03012.
OSU-03012 is derived from Celecoxib, and we next determined the relative abilities of Celecoxib and OSU-03012 to kill cells, as well as to alter BiP/GRP78 levels. Treatment with OSU-03012 reduced BiP/GRP78 expression whereas Celecoxib increased BiP/GRP78 levels in GBM cells (Fig. 3A, upper blots). In side-by-side dose response analyses we noted that OSU-03012 was approximately an order of magnitude more potent at killing GBM cells than Celecoxib (Fig. 3A, lower graphs). We then determined, using Celecoxib and OSU-03012 doses that have similar single agent lethality, whether both agents radiosensitized GBM cells. Celecoxib had a very weak radiosensitizing effect compared with OSU-03012 (Fig. 3B). Prior treatment of cells with Celecoxib, unlike OSU-03012, did not further enhance radiosensitivity (data not shown).
Figure 3.
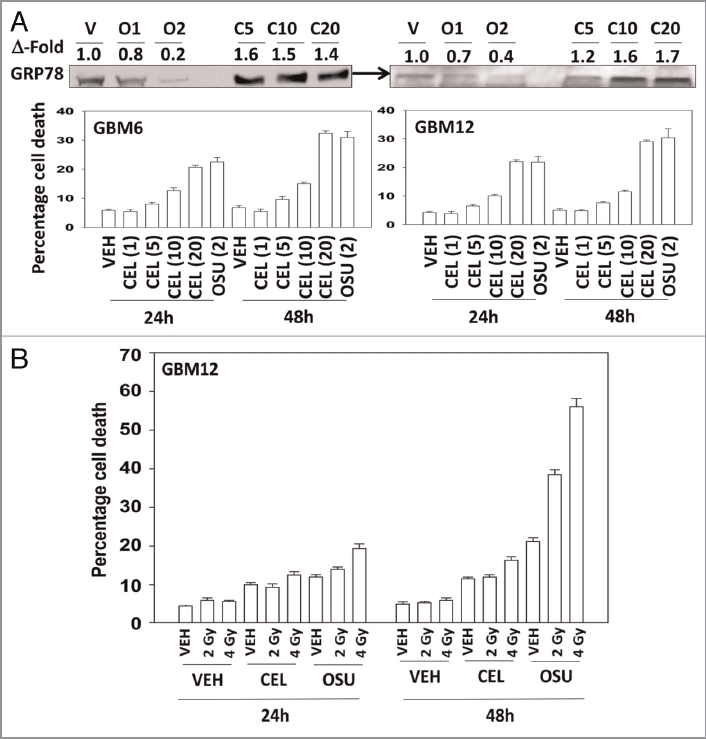
OSU-03012 is a more effective anticancer drug than Celecoxib. Panel (A) Lower: GBM6 and GBM12 cells were treated with increasing concentrations of Celecoxib (0–20 µM) or with OSU-03012 (2 µM). Cells were isolated 24 and 48 h later and viability determined (n = 3, +/- SEM). Upper: OSU-03012 reduces BiP/GRP78 expression whereas Celecoxib increases BiP/GRP78 levels. Cells were treated with OSU-03012 (1–2 µM) or Celecoxib (5–20 µM) and cells isolated 24 h after drug treatment. (B) GBM12 cells were treated with vehicle (DMSO), OSU-03012 (2 µM) or Celecoxib (10 µM). Cells were irradiated 30 min after drug treatment and portions of cells isolated 24 and 48 h later for viability determination (n = 3, +/- SEM).
We next determined, in multiple GBM lines, whether loss of BiP/GRP78 expression reduced the ability of OSU-03012 as a single agent to further stimulate cell killing and whether overexpression of BiP/GRP78 abolished OSU-03012 toxicity. Knock down of BiP/GRP78 significantly enhanced OSU-03012 lethality in all cell lines tested (Fig. 4A–C). Conversely, over-expression of BiP/GRP78, particularly at early time points (∼24 h) abolished OSU-03012 killing (Fig. 5A–C). We then examined the role of BiP/GRP78 on the radiosensitizing ability of OSU-03012. Knock down of BiP/GRP78 radiosensitized tumor cells (Fig. 6A–C). Nonetheless, in cells with reduced BiP/GRP78 expression, OSU-03012 still retained capability to enhance radiation toxicity and to a similar extent as in cells that expressed the protein (Fig. 6A–C; note the arrows and values for the percentage increase in cell death). Overexpression of BiP/GRP78 largely abolished OSU-03012 lethality as a single agent and profoundly reduced the ability of OSU-03012 to radiosensitize tumor cells (Fig. 7A–C; note the values for the percentage change in cell death). This argues that BiP/GRP78 is one of the key targets of OSU-03012 in causing cell death.
Figure 4.

Knock down of BiP/GRP78 enhances OSU-03012 toxicity. (A) GBM12 cells were transfected with scrambled siRNA or with an siRNA to knock down BiP/GRP78. Twenty-four hours after transfection cells were treated with vehicle (DMSO) or OSU-03012 (2 µM). Portions of cells were isolated 24 and 48 h later for viability determination (n = 3, +/- SEM). (B) GBM6 cells were transfected with scrambled siRNA or with an siRNA to knock down BiP/GRP78. Twenty-four hours after transfection cells were treated with vehicle (DMSO) or OSU-03012 (2 µM). Portions of cells were isolated 24 h and 48 h later for viability determination (n = 3, +/- SEM). (C) GBM14 cells were transfected with scrambled siRNA or with an siRNA to knock down BiP/GRP78. Twenty-four hours after transfection cells were treated with vehicle (DMSO) or OSU-03012 (2 µM). Portions of cells were isolated 24 and 48 h later for viability determination (n = 3, +/- SEM).
Figure 5.
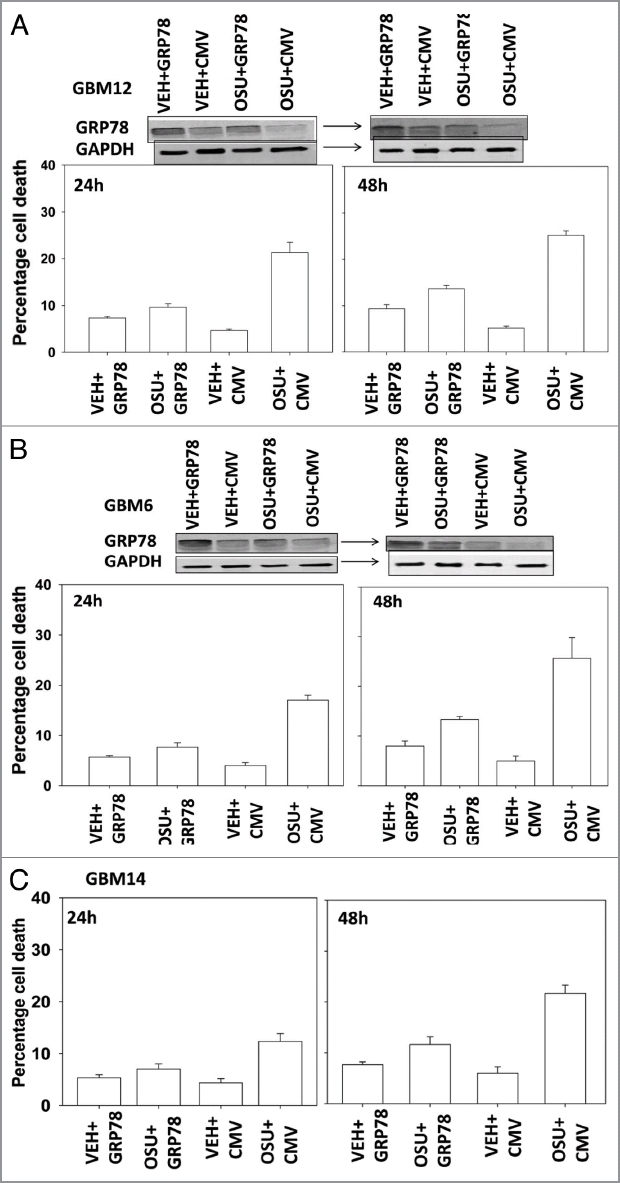
OSU-03012 lethality is suppressed by overexpression of BiP/GRP78. GBM cells, as indicated, were transfected with empty vector plasmid (CMV) or with a plasmid to express BiP/GRP78. Twenty-four hours after transfection cells were treated with vehicle (DMSO) or OSU-03012 (2 µM). Portions of cells were isolated 24 and 48 h later for viability determination (n = 3, +/- SEM). Upper blots in (A and B) show that expression of exogenous plasmid derived BiP/GRP78 is inhibited by OSU-03012.
Figure 6.
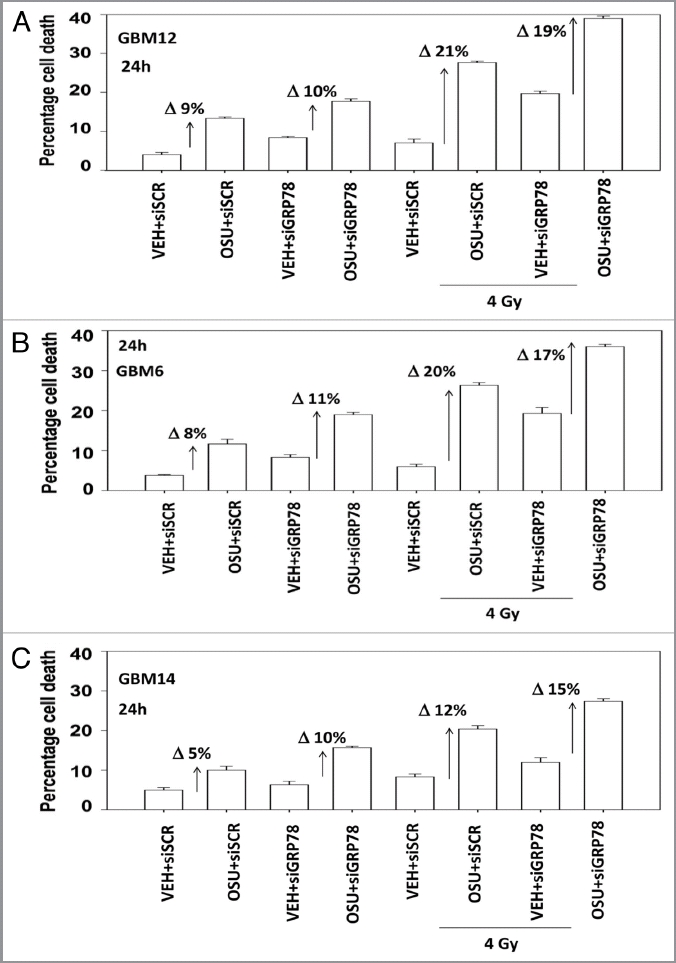
Loss of BiP/GRP78 does not further radiosensitize GBM cells above the sensitization effect caused by OSU-03012. GBM cells as indicated were transfected with scrambled siRNA or with an siRNA to knock down BiP/GRP78. Twenty-four hours after transfection cells were treated with vehicle (DMSO) or OSU-03012 (2 µM). Cells were irradiated (4 Gy). Cells were isolated 24 h later for viability determination (n = 3, +/- SEM). The arrows with numeric value indicate the percentage increase in death above each corresponding vehicle control.
Figure 7.
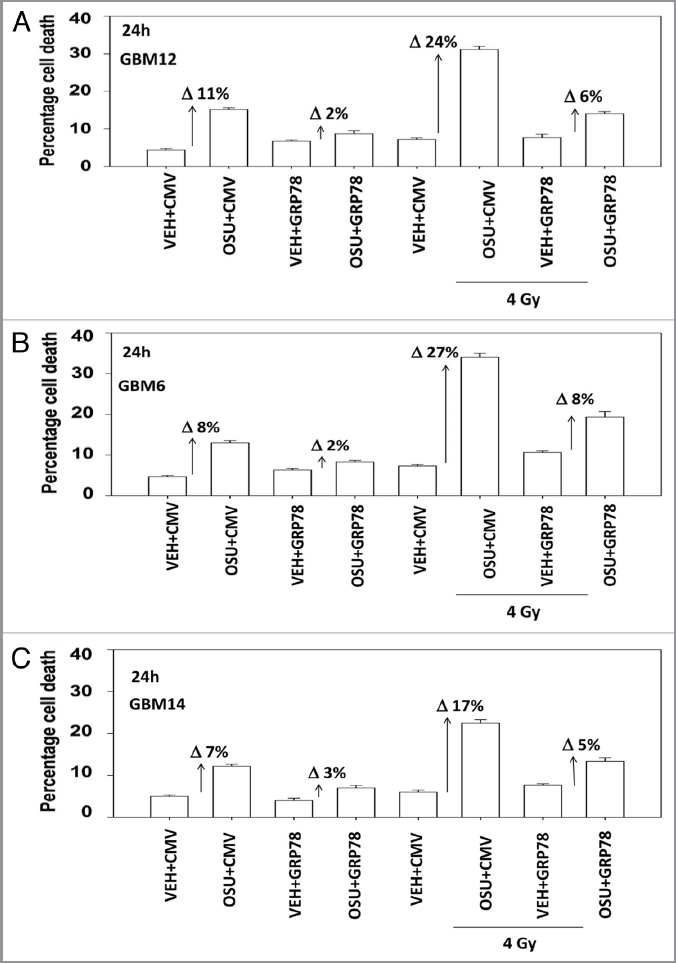
Overexpression of BiP/GRP78 suppresses OSU-03012-induced radiosensitization. GBM cells, as indicated, were transfected with empty vector plasmid (CMV) or with a plasmid to express BiP/GRP78. Twenty-four hours after transfection cells were treated with vehicle (DMSO) or OSU-03012 (2 µM). Cells were then irradiated. Cells were isolated 24 h later for viability determination (n = 3, +/- SEM). The arrows with numeric value indicate the percentage increase in death above each corresponding vehicle control.
OSU-03012 was initially proposed to be an inhibitor of PDK1 and based on this fact, and that loss of Phosphatase and tensin homolog (PTEN) is often observed in GBM, we defined the role of PTEN in the ability of OSU-03012 to cause cell killing. In GBM14 cells that lack PTEN, expression of PTEN enhanced OSU-03012 toxicity (Fig. 8A, lower graphs). Of note, although expression of PTEN reduced basal levels of mTOR, p70 S6K and AKT activity, this effect was not further lowered in the presence of OSU-03012, i.e., this argues against OSU-03012 at this concentration being a PDK1 inhibitor (Fig. 8A, upper blots). We next performed similar analyses in GBM6 cells that have had their PTEN expression knocked down. Knock down of PTEN in PTEN wild type GBM cells decreased the ability of OSU-03012 to kill (Fig. 8B, lower graphs). Knock down of PTEN increased basal activity levels of mTOR, p70 S6K and AKT that were not altered by OSU-03012 treatment (Fig. 8B, upper blots).
Figure 8.

Modulation of PTEN function changes OSU-03012 lethality. (A) GBM14 cells were transfected with control plasmid (GFP) or a plasmid to express PTEN (PTEN-GFP). Twenty-four hours after transfection cells were treated with OSU-03012 (2 µM). Cells were isolated 24 and 48 h later for viability determination (n = 3, +/- SEM). Upper blots: portions of cells were isolated 24 and 48 h after OSU-03012 treatment and lysates subjected to blotting to determine the phosphorylation and expression of the indicated proteins. (B) GBM6 cells were transfected with control plasmid (shSCR) or a plasmid to knock down expression of PTEN (shPTEN). Twenty-four after transfection cells were treated with OSU-03012 (2 µM). Cells were isolated 24 and 48 h later for viability determination (n = 3, +/- SEM). Upper blots: portions of cells were isolated 24 and 48 h after OSU-03012 treatment and lysates subjected to blotting to determine the phosphorylation and expression of the indicated proteins.
We next investigated the mechanism(s) by which OSU-03012 was reducing BiP/GRP78 levels. Treatment of cells with OSU-03012 caused a significant reduction in BiP/GRP78 protein levels that increased with time (Fig. 9A, upper; cf induction of LC3 processing in Fig. 1A). The relative level of BiP/GRP78 mRNA, did not significantly alter compared with vehicle control over this time course (Fig. 9A, lower). We then determined whether OSU-03012 reduced the stability of BiP/GRP78 protein or the stability of mRNA levels. We found that OSU-03012 did not alter mRNA stability (data not shown). In contrast, we noted that OSU-03012 decreased BiP/GRP78 protein stability (Fig. 9B).
Figure 9.

OSU-03012 reduces the half-life of BiP/GRP78. (A) Upper: GBM12 cells were treated with vehicle or OSU-03012 (2 µM). Cell lysates were isolated at the indicated times to determine the expression of BiP/GRP78 and densitometry performed. Lower: GBM12 cells were treated with vehicle or OSU-03012 (2 µM). Total RNA was isolated at the indicated time points and quantitative RT-PCR performed to determine BiP/GRP78 levels (n = 3, +/- SEM). (B) GBM12 cells were treated with vehicle or cycloheximide (20 µg/ml), followed by OSU-03012 (2 µM). Cells were lysed to determine the protein expression of BiP/GRP78 compared with GAPDH and the relative intensity of protein levels presented graphically (n = 3, +/- SEM).
Finally, we determined whether OSU-03012 could prolong animal survival and cause tumor radiosensitization in vivo using our primary human GBM isolate that expresses ERBB1 vIII, GBM6, and which has been stably transfected to express luciferase. In GBM6 tumors, treatment with OSU-03012 significantly suppressed tumor growth, an effect that was enhanced further by radiation exposure (Fig. 10, luciferase upper images). OSU-03012 prolonged animal survival in a dose-dependent fashion and interacted with radiation to promote additional animal survival (Fig. 10, lower graph).
Figure 10.
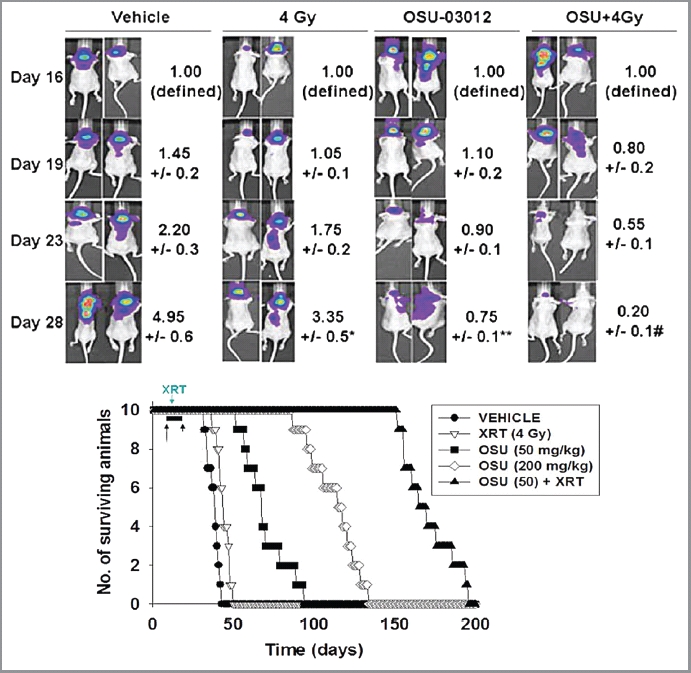
OSU-03012 prolongs animal survival using an orthotopic model and invasive primary human glioma cells. Upper: GBM6-luciferase cells (1 x 106) were implanted into the brains of mice (45 total). Tumors were permitted to form for 16 d after which animals were grouped based on tumor luminosity. The panel shows that over the following 12 d treatment with 50 mg/kg OSU-03012 significantly reduced tumor mass (**p < 0.05) and interacted with radiation to further suppress tumor mass (#p < 0.05)
Discussion
The present studies were initiated to further elucidate the molecular mechanisms by which the Celecoxib derivative OSU-03012 kills transformed cells in vitro and in vivo. Our analyses have demonstrated that OSU-03012 kills glioblastoma and colon cancer cells in a dose-dependent fashion and that OSU-03012 toxicity was linked to the induction of a form of ER stress signaling that correlated with increased levels of ceramide through the de novo synthesis pathway.
Our studies examined several possible proteins and signaling pathways that could play a role in modulating OSU-03012 lethality. One possible mode of OSU-03012 biology was originally claimed that the compound acted as an inhibitor of PDK-1. In our present studies, and in agreement with prior data in other tumor isolates, toxic doses of OSU-03012 did not significantly inhibit phosphorylation of the downstream target of PDK1: AKT, and of a novel nature herein, of mTOR and p70 S6K. These findings would seem to rule out the PI3K-PDK1 pathway as a major target of OSU-03012-induced killing in GBM cells.
The ER resident, HSP70 family member, BiP/GRP78 has been extensively examined as a response marker for endoplasmic reticulum stress.18,19 Increased expression of BiP/GRP78 after treatment with ER stress inducing agents such as thapsigargin and Celecoxib is well known and is thought to be a compensatory survival response in stressed cells.18–21 We had postulated previously that OSU-030120-induced HSP70 expression was also a compensatory survival reaction. In our present studies with OSU-03012 we discovered that drug exposure suppressed BiP/GRP78 levels, primarily by reducing the half-life of BiP/GRP78 protein. Reduced BiP/GRP78 protein levels partially explained some of the changes in cell viability seen after OSU-03012 exposure, i.e., siRNA knock down of already drug-reduced BiP/GRP78 levels further enhanced OSU-03012 toxicity as a single agent or combined with radiation. Overexpression of BiP/GRP78 abolished and/or significantly reduced drug lethality. Collectively these findings argue that at least two cellular targets exist for OSU-03012, with one primary target being BiP/GRP78.
Prior studies from this laboratory examining the actions of OSU-03012 had focused on the observation that the drug modestly decreased HSP90 levels and modestly increased the levels of HSP70.13 Further inhibition of HSP70 function markedly enhanced OSU-03012 lethality.13 In these studies we also discovered that the increase in HSP70 levels was dependent upon PERK signaling. This is of note because the ER-localized chaperone and inhibitor of PERK is BiP/GRP78. Collectively, with our present findings this argues that OSU-03012 promoted PERK-dependent increases in HSP70 levels through inhibition of BiP/GRP78.
There are three main sensors of ER stress: inositol-requiring protein 1α (IRE1α), activating transcription factor 6 (ATF6) and protein kinase RNA-like endoplasmic reticulum kinase (PERK). Signals by IRE1α and ATF6 are generally thought to have, based on the form of stress, either a protective or a toxic role based on the intensity and duration of the signal.22–28 Signaling by PERK is generally thought to mediate toxic ER stress signals. BiP/GRP78 acts as a chaperone for all three proteins and over-expression of this protein will act to suppress activation of all three ER stress sensors and is generally thought to be protective. Loss of BiP/GRP78 will therefore prolong and increase the intensity of ER stress signaling that acts to cause tumor cell death.
Glioblastoma is one of the most lethal malignancies with survival times measured in months. At present standard modalities for GBM therapy include surgery followed by radiotherapy and the use of several toxic chemotherapy drugs (Temodar and Gliadel wafers). A number of additional novel approaches are being developed based on the use of therapeutic agents which act upon inhibit growth factor receptors; receptors present both on the glioma cell and the tumor endothelial cells.29,30 However, no Phase I trial as yet has shown a highly significant prolongation of patient survival using novel signal modulatory drugs. We performed animal studies using an invasive GBM isolate which expresses the constitutively active form of the EGF receptor (ERBB1 vIII). Expression of ERBB1 vIII is normally associated with a poorer patient survival. In animals carrying GBM tumors we noted that OSU-03012 cause a dose-dependent increase in animal survival, as would be expected based on our in vitro data. OSU-03012 also interacted with ionizing radiation to further prolong survival. OSU-03012 is still undergoing evaluation in a Phase I trial in a variety of tumor types, though not in GBM. The evidence in this manuscript strongly argues that, based on our in vitro and in vivo data, the antitumor properties of OSU-03012 should be explored in GBM patients.
Materials and Methods
Reagents and techniques were as described in references 12 and 13.
Culture and in vitro exposure of cells to drugs.
In vitro cell treatments, microscopy, SDS-PAGE and western blot analysis.
For in vitro analyses of short-term cell death effects, cells were treated with Vehicle or OSU-03012 for the indicated times in the Figure legends.12,13 For SDS PAGE and immunoblotting, cells were plated and treated as described in references 12 and 13. All immunoblots were visualized using fluorescent secondary antibodies and an Odyssey Infrared imaging machine.12,13
Infection of cells with recombinant adenoviruses.
Cells were infected with recombinant adenoviruses as described in references 12 and 13.
Transfection of cells with siRNA or with plasmids.
For Plasmids. Plasmids were transfected according to details in references 12 and 13.
For BiP/GRP78 promoter-luciferase assays. Assays were as described in references 12 and 13.
Transfection with siRNA. Transfections were as described in references 12 and 13.
Data analysis.
Comparison of the effects of various treatments was performed following ANOVA using the Student's t-test. Differences with a p value of < 0.05 were considered statistically significant. Experiments shown are the means of multiple individual points (± SEM).
Acknowledgments
This work was funded to P.D. from PHS grants (R01-DK52825, P01-CA104177, R01-CA141188), Department of Defense Awards (DAMD17-03-1-0262); to S.G. from PHS grants (R01-CA63753; R01-CA77141) and a Leukemia Society of America grant 6405-97. PD is the holder of the Universal Inc. Professorship in Signal Transduction Research.
Abbreviations
- ERK
extracellular regulated kinase
- MEK
mitogen activated extracellular regulated kinase
- EGF
epidermal growth factor
- OSU
OSU-03012
- PARP
poly ADP ribosyl polymerase
- PI3K
phosphatidyl inositol 3 kinase
- -/-
null / gene deleted
- ERK
extracellular regulated kinase
- MAPK
mitogen activated protein kinase
- MEK
mitogen activated extracellular regulated kinase
- PTEN
phosphatase and tensin homolog
- R
receptor
- JNK
c-Jun NH2-terminal kinase
- dn
dominant negative
- COX
cyclooxygenase
- P
phospho-
- ca
constitutively active
- WT
wild type
- PERK
PKR like endoplasmic reticulum kinase
- HSP
heat shock protein
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Cathcart MC, O'Byrne KJ, Reynolds JV, O'Sullivan J, Pidgeon GP. COX-derived prostanoid pathways in gastrointestinal cancer development and progression: Novel targets for prevention and intervention. Biochim Biophys Acta. 2011;1825:49–63. doi: 10.1016/j.bbcan.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 2.Meyerhardt JA. Beyond standard adjuvant therapy for colon cancer: role of nonstandard interventions. Semin Oncol. 2011;38:533–541. doi: 10.1053/j.seminoncol.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kang SG, Kim JS, Park K, Kim JS, Groves MD, Nam DH. Combination celecoxib and temozolomide in C6 rat glioma orthotopic model. Oncol Rep. 2006;15:713. [PubMed] [Google Scholar]
- 4.Khan Z, Khan N, Tiwari RP, Sah NK, Prasad GB, Bisen PS. Biology of Cox-2: an application in cancer therapeutics. Curr Drug Targets. 2011;12:1082–1093. doi: 10.2174/138945011795677764. [DOI] [PubMed] [Google Scholar]
- 5.Moreira L, Castells A. Cyclooxygenase as a Target for Colorectal Cancer Chemoprevention. Curr Drug Targets. 2010 doi: 10.2174/138945011798184218. In press. [DOI] [PubMed] [Google Scholar]
- 6.Cerella C, Sobolewski C, Dicato M, Diederich M. Targeting COX-2 expression by natural compounds: a promising alternative strategy to synthetic COX-2 inhibitors for cancer chemoprevention and therapy. Biochem Pharmacol. 2010;80:1801–1815. doi: 10.1016/j.bcp.2010.06.050. [DOI] [PubMed] [Google Scholar]
- 7.Romagnolo DF, Papoutsis AJ, Selmin O. Nutritional targeting of cyclooxygenase-2 for colon cancer prevention. Inflamm Allergy Drug Targets. 2010;9:181–191. doi: 10.2174/187152810792231922. [DOI] [PubMed] [Google Scholar]
- 8.Ghosh N, Chaki R, Mandal V, Mandal SC. COX-2 as a target for cancer chemotherapy. Pharmacol Rep. 2010;62:233–244. doi: 10.1016/s1734-1140(10)70262-0. [DOI] [PubMed] [Google Scholar]
- 9.Patel MI, Subbaramaiah K, Du B, Chang M, Newman RA, Cordon-Cardo C, Thaler HT, Dannenberg AJ. (2005) Celecoxib inhibits prostate cancer growth: evidence of a cyclooxygenase-2-independent mechanism. Clin Cancer Res. 2005;11:1999–2007. doi: 10.1158/10780432.CCR-04-1877. [DOI] [PubMed] [Google Scholar]
- 10.Chakraborti AK, Garg SK, Kumar R, Motiwala HF, Jadhavar PS. Progress in COX-2 inhibitors: a journey so far. Curr Med Chem. 2010;17:1563–1593. doi: 10.2174/092986710790979980. [DOI] [PubMed] [Google Scholar]
- 11.Zhu J, Huang JW, Tseng PH, Yang YT, Fowble JW, Shiau CW, et al. From the cyclooxygenase-2 inhibitor celecoxib to a novel class of 3-phosphoinositide-dependent protein kinase-1 inhibitors. Cancer Res. 2004;64:4309–4318. doi: 10.1158/0008-5472.CAN-03-4063. [DOI] [PubMed] [Google Scholar]
- 12.Park MA, Yacoub A, Rahmani M, Zhang G, Hart L, Hagan MP, et al. OSU-03012 stimulates PKR-like endoplasmic reticulum-dependent increases in 70-kDa heat shock protein expression, attenuating its lethal actions in transformed cells. Mol Pharmacol. 2008;73:1168–1184. doi: 10.1124/mol.107.042697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yacoub A, Park MA, Hanna D, Hong Y, Mitchell C, Pandya AP, Harada H, Powis G, Chen CS, Koumenis C, Grant S, Dent P. OSU-03012 promotes caspase-independent but PERK-, cathepsin B-, BID-, and AIF-dependent killing of transformed cells. Mol Pharmacol. 2006;70:589–603. doi: 10.1124/mol.106.025007. [DOI] [PubMed] [Google Scholar]
- 14.Johnson AJ, Smith LL, Zhu J, Heerema NA, Jefferson S, Mone A, et al. A novel celecoxib derivative, OSU-03012, induces cytotoxicity in primary CLL cells and transformed B-cell lymphoma cell line via a caspase-and Bcl-2-independent mechanism. Blood. 2005;105:2504–2509. doi: 10.1182/blood-2004-05-1957. [DOI] [PubMed] [Google Scholar]
- 15.Tseng PH, Lin HP, Zhu J, Chen KF, Hade EM, Young DC, et al. Synergistic interactions between imatinib mesylate and the novel phosphoinositide-dependent kinase-1 inhibitor OSU-03012 in overcoming imatinib mesylate resistance. Blood. 2005;105:4021–4027. doi: 10.1182/blood-2004-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tseng PH, Wang YC, Weng SC, Weng JR, Chen CS, Brueggemeier RW, et al. Overcoming trastuzumab resistance in HER2-overexpressing breast cancer cells by using a novel celecoxib-derived phosphoinositide-dependent kinase-1 inhibitor. Mol Pharmacol. 2006;70:1534–1541. doi: 10.1124/mol.106.023911. [DOI] [PubMed] [Google Scholar]
- 17.To K, Zhao Y, Jiang H, Hu K, Wang M, Wu J, et al. The phosphoinositide-dependent kinase-1 inhibitor, OSU-03012, prevents Y-box binding protein-1 (YB-1) from inducing epidermal growth factor receptor (EGFR) Mol Pharmacol. 2007;72:641–652. doi: 10.1124/mol.107.036111. [DOI] [PubMed] [Google Scholar]
- 18.Ni M, Zhang Y, Lee AS. Beyond the endoplasmic reticulum: atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem J. 2011;434:181–188. doi: 10.1042/BJ20101569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bánhegyi G, Baumeister P, Benedetti A, Dong D, Fu Y, Lee AS, et al. Endoplasmic reticulum stress. Ann N Y Acad Sci. 2007;1113:58–71. doi: 10.1196/annals.1391.007. [DOI] [PubMed] [Google Scholar]
- 20.McLaughlin M, Vandenbroeck K. The endoplasmic reticulum protein folding factory and its chaperones: new targets for drug discovery? Br J Pharmacol. 2011;162:328–345. doi: 10.1111/j.1476-5381.2010.01064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ishihara T, Tanaka K, Tashiro S, Yoshida K, Mizushima T. Protective effect of rebamipide against celecoxib-induced gastric mucosal cell apoptosis. Biochem Pharmacol. 2010;79:1622–1633. doi: 10.1016/j.bcp.2010.01.030. [DOI] [PubMed] [Google Scholar]
- 22.Ye J, Koumenis C. ATF4, an ER stress and hypoxiainducible transcription factor and its potential role in hypoxia tolerance and tumorigenesis. Curr Mol Med. 2009;9:411–416. doi: 10.2174/156652409788167096. [DOI] [PubMed] [Google Scholar]
- 23.Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- 24.Sjöström J, Blomqvist C, von Boguslawski K, Bengtsson NO, Mjaaland I, Malmström P, et al. The predictive value of bcl-2, bax, bcl-xL, bag-1, fas, and fasL for chemotherapy response in advanced breast cancer. Clin Cancer Res. 2002;8:811–816. [PubMed] [Google Scholar]
- 25.Diehl JA, Fuchs SY, Koumenis C. The cell biology of the unfolded protein response. Gastroenterology. 2011;141:38–41. doi: 10.1053/j.gastro.2011.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bobrovnikova-Marjon E, Grigoriadou C, Pytel D, Zhang F, Ye J, Koumenis C, et al. PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene. 2010;29:3881–3895. doi: 10.1038/onc.2010.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chakrabarti A, Chen AW, Varner JD. A review of the mammalian unfolded protein response. Biotechnol Bioeng. 2011;108:2777–2793. doi: 10.1002/bit.23282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Ridder G, Ray R, Misra UK, Pizzo SV. Modulation of the unfolded protein response by GRP78 in prostate cancer. Methods Enzymol. 2011;489:245–257. doi: 10.1016/B978-012-385116-1.00014-5. [DOI] [PubMed] [Google Scholar]
- 29.Reardon DA, Vredenburgh JJ, Desjardins A, Peters K, Gururangan S, Sampson JH, et al. Effect of CYP3A-inducing anti-epileptics on sorafenib exposure: results of a phase II study of sorafenib plus daily temozolomide in adults with recurrent glioblastoma. J Neurooncol. 2011;101:57–66. doi: 10.1007/s11060-010-0217-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rokes CA, Remke M, Guha-Thakurta N, Witt O, Korshunov A, Pfister S, et al. Sorafenib plus valproic acid for infant spinal glioblastoma. J Pediatr Hematol Oncol. 2010;32:511–514. doi: 10.1097/MPH.0b013e3181d74702. [DOI] [PubMed] [Google Scholar]