

Introduction
The past decade has witnessed a burst of speculation and data about how astrocyte dysfunction contributes to the phenotypes of the well known neurodegenerative diseases such as Alzheimer's, Parkinson's, Huntington's, and amyotrophic lateral sclerosis, as well as other types of disorders such as epilepsies and multiple sclerosis (Rempe and Nedergaard, 2010). However, these are complex syndromes that likely represent combined abnormalities of neurons, glia, and immune cells. The clearest example of astrocytes acting as the primary culprit in disease is Alexander disease, which is caused by dominant gain-of-function mutations in the glial fibrillary acidic protein (GFAP) gene (for an extensive review, see Brenner et al., 2009). Although this disorder is quite rare, the extent to which we can understand how astrocyte function is impaired in Alexander disease, and the strategies we can devise to restore astrocyte function, will have significant implications for how we deal with the many more common neurological diseases that confront us. The purpose of this review is to introduce the wider neuroscience audience to the unique research opportunities posed by this disease.
The first recognized patient was a 16-month-old boy who died after a progressive course that included megalencephaly, hydrocephalus, and psychomotor delays (Alexander, 1949). Pathology revealed abundant astrocytic accumulations of eosinophilic cytoplasmic inclusions, recognized by neuropathologists as Rosenthal fibers (after the 19th century German pathologist who first described them in the context of an old astrocyte scar; Rosenthal, 1898) (for review, see Wippold et al., 2006) (Fig. 1). During the subsequent 15 years, additional individuals with similar pathology were reported, and in 1964 Friede suggested that these all represented a single disease, and recommended they be named after Alexander. Although the initial finding of prominent aggregates in astrocytes prompted Alexander himself to suggest that this might represent a primary disorder of astrocytes, it was the discovery of the genetic basis for the disease that established Alexander disease as a primary disorder of this major CNS cell type (Brenner et al., 2001).
Figure 1.
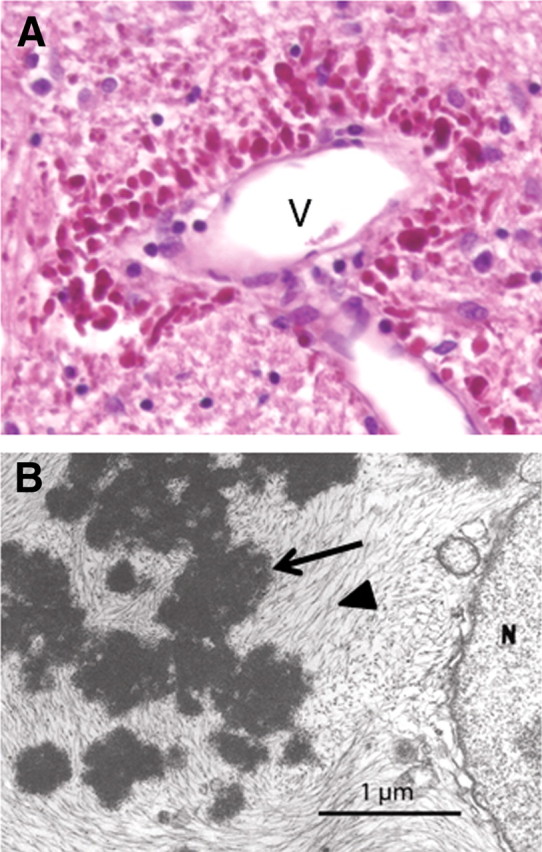
Morphological features of Rosenthal fibers. A, Rosenthal fibers concentrated in the astrocytic endfeet surrounding a blood vessel (V) in the brainstem of a 1-year-old child with Alexander disease. Hematoxylin and eosin stain, paraffin section. B, Rosenthal fibers (arrow) surrounded by intermediate filaments (arrowhead) in an astrocyte cell body from a 17-month-old child with Alexander disease, viewed by transmission electron microscopy [reprinted from Eng et al. (1998), their Fig. 5B, with permission of Wiley-Liss, a subsidiary of John Wiley & Sons].
Approximately 95% of patients harbor mutations in the GFAP gene, and no other genetic causes are known (Brenner et al., 2009). Only one population-based survey has been conducted, arriving at an incidence of ∼1:2.7 million (Yoshida et al., 2011), although this figure is likely an underestimate. The age of onset is quite variable, ranging from prenatal through the sixth decade. The most common classification divides patients into three categories based on age of onset, infantile (0–2 years), juvenile (2–12 years), and adult (>12 years) (Russo et al., 1976). More recently, a different classification has been proposed, with only two categories of type I and type II, hinging more on distribution of lesions and clinical presentation rather than age of onset (all type I cases being early onset, and type II cases occurring at all ages) (Prust et al., 2011). Early-onset patients predominate in the literature, but this likely reflects ascertainment bias as the adult-onset patients in particular are frequently misdiagnosed with other conditions. The early-onset patients typically present with seizures, spasticity, or developmental delays, whereas the later-onset patients more often have signs of hindbrain dysfunction such as ataxia, palatal myoclonus, and dysphagia or dysphonia (Russo et al., 1976). Diagnosis is usually suspected based on characteristic appearances on MRI, with a frontal leukodystrophy common in the younger patients and a hindbrain predominance of lesions, sometimes with atrophy of the medulla oblongata and cervical spinal cord, in the later-onset patients (Fig. 2) (van der Knaap et al., 2001, 2005, 2006; Namekawa et al., 2002). Lifespan is related to age of onset; type I patients have a median survival of 14 years, and type II patients a median survival of 25 years (Prust et al., 2011). Most of the initially discovered mutations occurred de novo, but with sharpening the tools for diagnoses of the more difficult patients to discern the later-onset form, increasing numbers of familial cases are being detected. When passed to progeny, the mutations have typical autosomal-dominant inheritance with nearly 100% penetrance (for further discussion on the topic of penetrance, see Stumpf et al., 2003; Messing et al., 2012). There is no predilection for any ethnic population, or any gender bias.
Figure 2.

Typical MRIs for Alexander disease. The two images on the left are from a juvenile-onset patient, illustrating the characteristic abnormalities of frontal white matter including increased signal on T2-weighted images and decreased signal on T1-weighted images (courtesy of Dr. M. S. van der Knaap, VU University Medical Center, Amsterdam, the Netherlands). The image on the right is a T2-weighted midline sagittal section from an adult-onset patient, illustrating the characteristic atrophy and signal abnormalities in the medulla oblongata (arrow) and atrophy of the cervical spinal cord [reprinted from Pareyson et al. (2008), their Fig. 1, by permission of Oxford University Press].
Concept of a primary astrocyte disease
The assignment of Alexander disease as an astrogliopathy—a primary disease of astrocytes—rests principally on the astrocyte specificity of expression of the GFAP gene. Multiple other cell types do express GFAP, such as Schwann cells, but at much lower levels (Su et al., 2004), and the only consistent symptoms are referable to the CNS (Brenner et al., 2009). Probably because of a requirement for a threshold level of GFAP, only astrocytes have been found to display the Rosenthal fibers that are the defining feature of this disorder. This is also consistent with Rosenthal fibers being particularly abundant in subpial and white matter CNS regions, which are areas of high GFAP content, compared with gray matter areas such as neocortex. Apart from lesions that have progressed to frank cavitation, there is no obvious decrease in astrocyte numbers. However, in white matter without cavitation there is a marked loss of myelin and a variable (but unquantitated) loss of oligodendrocyte lineage cells and axons. There is also a loss of neurons, particularly in the hippocampus and striatum (our unpublished observations). The oligodendrocyte (myelin) and neuronal deficits that produce the clinical signs of Alexander disease are likely consequences of astrocyte dysfunction rather than astrocyte loss. This dysfunction could involve the loss of a normal, supportive role (e.g., glutamate uptake), the gain of a new or increased toxic activity (e.g., TNFα release), or a combination of both.
Disease mechanisms
Alexander disease as a gain-of-function disorder of GFAP
Alexander disease is considered a gain-of-function disorder in the sense that the GFAP mutations produce consequences that differ dramatically from those caused by the absence of GFAP (for review, see Brenner et al., 2009). There are three observations that indicate that the primary effect of the GFAP mutations is a gain of function: (1) Gfap-null mice have a minimal phenotype that bears little resemblance to Alexander disease; (2) astrocytes of Alexander disease patients display abundant levels of normal-appearing, 10 nm GFAP filaments; and (3) all GFAP mutations discovered in Alexander disease patients allow production of essentially full-length mutant protein. Neither nonsense mutations nor major deletions have been found, and the only frameshifts are at the extreme C-terminal end of the protein (Fig. 3). The few internal deletions and insertions that have been reported are in-frame. In these respects, Alexander disease differs from diseases due to mutations in other cytoplasmic intermediate filament genes, such as keratins, which have a strong loss-of-function component (Omary, 2009). Nevertheless, these disorders share highly homologous hotspots for mutations that interfere with filament assembly.
Figure 3.

Locations of GFAP mutations in Alexander disease in relation to the protein structure. The four open rectangular boxes represent the helical coiled-coil rod domains of GFAP; these structural motifs are highly conserved among most intermediate filament proteins. The solid lines joining these segments are nonhelical linker regions, and the solid lines at either end are the nonconserved, random coil, N-terminal and C-terminal regions. The gray box just before segment 1A is a nonconserved prehelical sequence important for initiation of rod formation at the start of 1A; the gray box at the end of 2B represents a highly conserved sequence that includes the end of the coiled coil 2B segment. The wild-type amino acid is indicated next to the structure, and amino acid replacements within symbols on either side. Early-onset cases (first symptom before the age of 2 years) are on the left, shown as blue circles, and late-onset cases (first symptom after the age of 2 years) are on the right, shown as red circles. Each symbol represents a single patient, except that familial cases, including identical twins, are represented by a single symbol coded for the onset type of the proband. Adapted with permission from Brenner et al. (2009), their Figure 24.4.
A rationalization of this apparent paradox has been discussed in detail by Li et al. (2002), who proposed a model in which the effect of the mutation is to slow the rate of normal polymer formation rather than to block it completely. More recent findings indicate that mutant GFAP monomers form abnormally large, soluble oligomers (as discussed below in the context of effects on the proteasome) (Tang et al., 2010) that may in fact result from a slowing of the normal polymerization rate. In addition, astrocytes respond in a self-destructive fashion by increasing their expression of GFAP (Jany et al., 2011). The resultant elevation in GFAP monomers available for polymerization could explain why astrocytes are able to form normal-appearing filaments, whereas intermediate filament proteins with homologous mutations in other cell types do not. What stimulates this astrocyte response is not clear, but could involve an intercellular circuit in which other cell types are damaged by the dysfunctional astrocytes and then signal back to the astrocytes via any of several pathways (Buffo et al., 2010), or could occur entirely within the affected astrocyte by mechanisms not yet known. This increase in expression, coupled with decreased degradation (see The concept of GFAP toxicity, below), results in a positive feedback loop for disease progression. The presence of the feedback loop could also be the reason that only astrocytes, and not other GFAP-expressing cells, form Rosenthal fibers.
While the above model provides an explanation for why normal-appearing filaments are seen in Alexander disease but not in other intermediate filament disorders, it does raise the possibility that these other disorders may also have gain-of-function components in addition to their loss-of-function phenotypes. Indeed, it has been observed that disease due to keratin point mutations, in which aggregates form, may be more severe than that produced by its absence (Fuchs and Cleveland, 1998). The extent to which the toxic mechanisms are similar in these different intermediate filament disorders is yet to be explored. It is also interesting, but perhaps reflective of our lack of understanding of intermediate filament structures, that mutations throughout the rod and tail domains of GFAP cause the same disease.
The concept of GFAP toxicity
That GFAP accumulation beyond a toxic threshold is itself pathogenic came initially from studies of transgenic mice engineered to overexpress wild-type human GFAP constitutively. Mice in the highest expressing lines died within a few weeks after birth, and their brains contained copious quantities of Rosenthal fibers (Messing et al., 1998). Other mouse models have since been developed by homologous recombination of the endogenous mouse gene (the genes are highly conserved between human and mouse, with 92% identity at the amino acid level) to produce the equivalent of the common and particularly deleterious human R79H and R239H mutations (R76H and R236H in the mouse). The mice expressing the point mutant forms of GFAP also form Rosenthal fibers and spontaneously increase their levels of GFAP, although they have normal lifespans (Hagemann et al., 2006). However, when certain lines are crossed together to raise GFAP levels to a far higher degree, the double-transgenics consistently die by 3–5 weeks after birth, most likely due to seizures (Hagemann et al., 2006). Curiously, none of the mouse models made to date exhibit any apparent abnormalities of white matter (Hagemann et al., 2006; Tanaka et al., 2007), though detailed analysis of the murine white matter remains to be done. Hypomyelination and/or demyelination are severe in the early-onset form of the human disease, and milder or even undetectable in the adult-onset patients (Barkovich and Messing, 2006).
Rosenthal fibers also appear in many non-Alexander disease conditions, such as old gliotic scars in infarcts, multiple sclerosis, trauma, and in pilocytic astrocytomas (Chin and Goldman, 1996; Wippold et al., 2006), and are replicated by mice that simply overexpress wild-type human GFAP (Messing et al., 1998). This strongly suggests that high enough levels of wild-type GFAP can produce the astrocyte phenotype of Alexander disease. However, astrocytes in cultures that express mutant GFAP may accumulate GFAP more rapidly and to higher levels than astrocytes that express wild-type GFAP (Tang et al., 2006, 2010).
Although much attention has been focused on Rosenthal fibers, since they are the most striking pathology, astrocytes in Alexander disease show profound alterations in cell shape (Fig. 4A,B) and in function. One dramatic effect is the induction of cell stress, initially prompted by the finding that Alexander disease astrocytes contain massive amounts of the small heat shock protein, αB-crystallin (Iwaki et al., 1989). Subsequently, we have found upregulation of MAPK pathways, constitutive activation of JNK and p38 kinases, upregulation of small heat shock protein genes, and an increase in autophagy (Tang et al., 2006, 2008, 2010). The mouse models described above also exhibit upregulation of αB-crystallin (Hagemann et al., 2005). The downstream effects of cell stress may be many fold, but one that is particularly relevant to Alexander disease and the accompanying accumulation of GFAP is impairment of proteasomal activity (Tang et al., 2006, 2010; Cho and Messing, 2009; Chen et al., 2011). Accumulation of abnormal oligomers of mutant GFAP appears to be the cause of proteasomal dysfunction. In addition, these abnormal oligomers inhibit the degradation of artificial proteasomal substrates, suggesting that they may also prevent other proteins from being degraded. One prediction from these results is that protein degradation and turnover are slowed, a hypothesis that is currently being tested in the mouse models.
Figure 4.
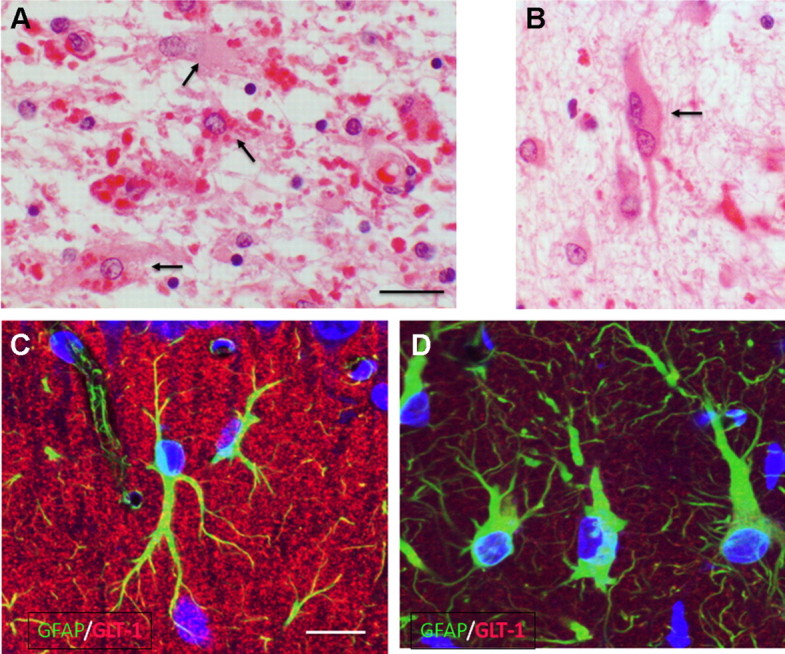
Astrocytes in Alexander disease. A, B, Enlarged, mis-shapen astrocytes in hemispheric white matter (arrows), some of which contain Rosenthal fibers (eosinophilic inclusions) in cell bodies (A). Other Rosenthal fibers lie in astrocyte processes. Multinucleated astrocytes are common (B). Scale bar, 20 μm. C, D, Astrocytes in the hippocampal CA1 stratum moleculare in wild-type (C) and knock-in (R236H) (D) mice. Immunofluorescence for GFAP (green) and GLT-1 (red) shows enlarged, irregular astrocytes in the knock-in and an appreciable loss of GLT-1 [reprinted from Tian et al. (2010), their Fig. 2A]. Scale bar, 20 μm.
Stress pathways and the machinery for protein degradation are linked in several ways that may contribute to the positive feedback loops that result in an ever-increasing accumulation of GFAP. For instance, inhibiting proteasome activity results in increased GFAP accumulation, which itself presumably causes further proteasome inhibition. In addition, there is activation of stress kinase pathways such as JNK and p38, the latter promoting the transcription of αB-crystallin and promoting autophagy (Tang et al., 2006, 2008). The increased autophagy may reflect an attempt, albeit unsuccessful, to degrade the accumulated GFAP and associated proteins. Proteasome inhibition and the consequent accumulation of short-lived proteins such as the transcription factor Nrf2 (known to be elevated in several neurodegenerative diseases associated with oxidative stress) may help to explain another sign of cell stress, the widespread activation of antioxidant genes found in the GFAP-overexpressing and R236H point mutant mice (Hagemann et al., 2005, 2006). This coordinated response is mediated through the binding of Nrf2 to a common antioxidant response element contained within the promoters of these genes.
Does GFAP toxicity cause loss of function in other pathways?
Although we present the view that Alexander disease reflects a gain-of-function disorder of GFAP, there could be downstream effects that are more properly viewed as the loss of function of other cellular components. An important consideration is that large accumulations of GFAP, as well as the Rosenthal fibers themselves, could function as substrates to which other molecules bind. In some cases, these molecules may be taken away from their normal cytosolic functions. For instance, several other proteins appear to be associated with GFAP and Rosenthal fibers, including the intermediate filament—actin linker protein, plectin (Tian et al., 2006), and the 20S proteasome subunit (Tang et al., 2010). Normally, plectin promotes a spread intermediate filament system in cells by linking filaments to other cytoskeletal elements and to the cell periphery (Wiche, 1998). Binding to Rosenthal fibers, which presumably contain tightly knit filaments, would remove plectin from its normal locations. We have found that overexpressing plectin in astrocytes that express an Alexander mutation will convert the GFAP coils and aggregates that form in these cells into a normal, spread filament system (Tian et al., 2006). Therefore, the ratio of plectin to filaments may be critical in promoting a spread organization. Since some of the proteasome subunits in cells normally associate with intermediate filaments (Tang et al., 2010), it is not clear whether the 20S binding to Rosenthal fibers would inhibit proteasome function. Since we have found that the mutant GFAP will inhibit proteasome function (see The concept of GFAP toxicity, above) the binding to abnormal filaments may well participate in the proteasome inhibition. Activated JNK also associates with Rosenthal fibers (Tang et al., 2006), perhaps binding to GFAP, as it has been shown to do with keratin 8 (He et al., 2002). Its association with keratin 8 may promote the phosphorylation of the keratin, but also inhibit JNK-dependent phosphorylation of other substrates, such as c-Jun (He et al., 2002).
Some of GFAP's normal binding partners, such as αB-crystallin, are also affected by the accumulation and aggregation taking place in Alexander disease. In normal astrocytes, αB-crystallin exists primarily in a soluble pool of cytosolic proteins, whereas in cells expressing mutant GFAP it shifts its localization to a cytoskeletal fraction, presumably because so much of it is bound to the increased GFAP (Tang et al., 2006; Perng et al., 2008). In one of the mouse models of Alexander disease, crosses to αB-crystallin nulls revealed a dose-dependent increase in mortality resulting from αB-crystallin deficiency (Hagemann et al., 2009). Hence, the shift in solubility noted above may produce a deficiency in the soluble pool that is critical for astrocyte function. One cannot blame the full Alexander phenotype on αB-crystallin deficiency, however, since mouse knockouts and humans with nearly complete deletions of this gene have predominantly skeletal muscle phenotypes with no evidence yet of neurological effects (Brady et al., 2001; Del Bigio et al., 2011). Even more striking is the finding that forcing overexpression of αB-crystallin above the natural induction caused by the disease results in complete rescue of the lethal phenotype observed when the knock-in point mutant is combined with the transgenic overexpressing wild-type GFAP (Hagemann et al., 2009). One mechanism for the rescue effects of αB-crystallin is its ability to reduce the levels of toxic GFAP oligomers to produce a largely monomeric population, which can then be degraded by the proteasome (Tang et al., 2010). Since αB-crystallin overexpression appears to have no obvious deleterious effects on astrocytes, this stress protein now becomes an interesting prospect as a therapeutic target.
Physiological consequences of GFAP toxicity—gray matter versus white matter
Seizures are one of the most common manifestations of Alexander disease among the type I patients. Thus, identifying abnormalities in physiological properties of gray matter astrocytes is an obvious area of interest. One of the most notable changes in the Alexander astrocyte is the diminution of GLT-1, the major glutamate transporter in astrocytes (Tian et al., 2010). Whole hippocampal lysates of the R236H mice reveal >75% reduction in GLT-1 as assayed by immunoblots, and astrocytes in human hippocampal CA1 show variable to complete loss of immunostaining (Tian et al., 2010). Reduction of GLT-1 immunoreactivity in the mouse models initially appears in a mosaic pattern. As the disease progresses, more astrocytes lose GLT-1 immunoreactivity and concurrently lose their protoplasmic shape (A. Sosunov, E. Guilfoyle, G. McKhann, J. E. Goldman, unpublished observations), and patch-clamping of the abnormal astrocytes shows a marked loss of glutamate transporter current (on average, a 50% loss; X. Wu, E. Guilfoyle, G. McKhann, J. E. Goldman, unpublished observations). Indeed, the loss of GLT-1 could put neurons at high risk for excitotoxic death, a phenomenon that has been observed in astrocyte-hippocampal neuron cocultures (Tian et al., 2010). A diminution of GLT-1 in the hippocampus of the R236H mice (Tian et al., 2010) could explain the increased severity of kainic acid seizures that result in pyramidal neuron death (Hagemann et al., 2006) (Fig. 3C,D). A loss of glutamate buffering could also play a role in oligodendrocyte toxicity and loss of myelin, since oligodendrocyte precursors are sensitive to glutamate toxicity (Deng et al., 2006). Whether the baseline glutamate concentration in this tissue has actually increased is not yet known.
Astrocytes in white matter, where GFAP expression is higher than in gray matter, may be affected differently by GFAP mutations than those in gray matter. Glutamate uptake is not a major role for white matter astrocytes, where glutamatergic transmission is likely considerably lower (Ziskin et al., 2007), and the maximal glutamate transporter current recorded from these fibrous astrocytes represents <10% of that recorded from cortical protoplasmic astrocytes (Regan et al., 2007). On the other hand, white matter astrocytes provide critical metabolic support to oligodendrocytes, which are dependent upon astroglial lactate for their homeostatic maintenance, and astrocytic exchangers for interstitial ion homeostasis (Nedergaard et al., 2003). Potassium buffering by white matter astrocytes may be critically important, as axonal conductance is linked to large-amplitude changes in interstitial ion concentrations (Ransom et al., 2000). Analysis from fibrous astrocytes derived from the spinal white matter suggests that these cells aggressively buffer extracellular potassium, via influx through potassium channels (Kir4.1) and concurrent active transport by the Na,K ATPase. On this basis, we speculate that an additional downstream effect of GFAP toxicity may be dysfunction of astrocytic potassium buffering, which can lead to a failure in both myelin formation and maintenance. Consistent with a supportive, normal astrocyte function for oligodendrocytes, two other leukodystrophies have also recently been ascribed to genetic mutations affecting astrocytes. Vanishing white matter disease is caused by mutations in the eukaryotic translation initiation factor 2B, and is characterized by dysmorphic astrocytes with abnormal intermediate filament architecture (Bugiani et al., 2011). A phenotypically similar but genotypically distinct disorder, megalencephalic leukoencephalopathy with subcortical cysts, is caused by mutations in MLC1, which codes for a protein that is almost exclusively present in astrocytic vascular endfeet (Blattner et al., 2003). Both of these diseases are similar to Alexander disease in that they are characterized by childhood abnormalities of white matter. Thus, convergent evidence points to astrocytic dysfunction as a common antecedent to white matter loss in multiple leukodystrophies.
New model systems
To address the many important questions about astrocyte biology and pathology posed by Alexander disease, there is a critical need for new model systems. Recently, we described one such model in Drosophila (Wang et al., 2011). When wild-type or disease-linked mutant versions of GFAP are expressed in fly glia, the resultant animals show many phenotypic features of authentic Alexander disease. Seizures, a common feature of affected infants, are increased in young transgenic flies. Neuropathologically, there is dysfunction and some apoptotic death of glia, which is accompanied by non-cell-autonomous degeneration of neurons. Perhaps most strikingly, transgenic GFAP aggregates into eosinophilic, beaded, and elongated inclusion bodies in fly glia. These inclusions bear strong similarity to authentic Rosenthal fibers at both the light and electron microscopic levels. Disease-associated mutant forms of GFAP, including R79H, R239C, R239H, L352P, and A364P, are significantly more toxic than wild-type human GFAP, consistent with the Drosophila model, reflecting toxicity relevant to the human disease.
Similarities to vertebrate systems are also apparent at the cell biological level. As in mouse models of Alexander disease, overexpression of the fly homolog of αB-crystallin strongly ameliorates the behavioral and neuropathological defects of GFAP transgenic flies. Additional evidence for a critical role for protein misfolding in toxicity is provided by suppression by multiple heat shock proteins, including HSP26, HSP27, and HSP70. Quantitative analysis of levels of aggregated GFAP supports a role for the heat shock proteins acting via amelioration of abnormal protein aggregation. Other pathways previously implicated in Alexander disease or vertebrate models of the disorder, including oxidative stress, autophagy, JNK signaling, and glial glutamate transport, also appear to play an important role in the Drosophila model. The facile genetics of the fly model has allowed us to investigate the relationships among these pathways. We find that both protein aggregration and oxidative stress act upstream of, and promote, autophagy in glia expressing mutant human GFAP. In contrast, dysregulation of glial glutamate transport appears to be a downstream consequence of altered proteostasis and oxidative metabolism, and is critical for non-cell-autonomous neurodegeneration.
The similarities between the vertebrate and invertebrate systems motivate further use of the fly model to delineate mechanisms of disease pathobiology. Large-scale, unbiased forward genetic screens have long been a major strength of invertebrate model systems. GFAP transgenic flies display phenotypes amenable to such screens, including increased seizure frequency. As the results of these genetic screens emerge, we will be able to determine the extent to which mechanisms controlling toxicity of human GFAP to Drosophila glial cells are conserved with vertebrate systems. Importantly, excellent vertebrate cell culture and transgenic mouse models are available in which to test the relevance of results derived from genetic screens in flies. The observation of non-cell-autonomous neurodegeneration in the Drosophila model, as is seen in the human disease (recall that neither neurons nor oligodendrocytes express GFAP), suggests that genetic definition of the molecules and pathways involved in astrocyte–neuron communication may be possible. If so, there may be broader implications of the work in Alexander disease model flies for a variety of functions and dysfunctions of the nervous system involving cross talk between glia and neurons.
Strategies for therapy
Despite the many gaps in our understanding of the mechanisms and impact of astrocyte dysfunction in Alexander disease, several strategies for therapy have been suggested (Messing et al., 2010). The most obvious approach is to reduce the expression or accumulation of GFAP, so as to avoid the initial insult that drives the entire process. One drug screen has already been completed using wild-type astrocytes in primary culture (Cho et al., 2010), and several of these compounds are now being investigated using the in vivo models. A second approach is to target downstream effects of GFAP toxicity, such as the deficit in GLT-1 expression discussed above. Ceftriaxone, an antibiotic previously identified through a drug screen for enhancers of GLT-1 expression, is already in clinical trials for ALS and may be a good candidate for such an approach (Rothstein et al., 2005). Manipulation of stress pathways such as those involving αB-crystallin or Nrf2 may also prove effective. The latter is under investigation for application in a wide spectrum of neurological disorders (Vargas and Johnson, 2010).
It should be noted that astrocytes upregulate GFAP in response to nearly all injuries and diseases of the CNS. Thus, Alexander disease could be viewed in part as a highly exaggerated form of gliosis, or astrocyte scarring. Indeed, Alexander astrocytes share characteristics with astrocytes in gliotic responses, including increases in endothelin B receptor (Hagemann et al., 2006), loss of GLT-1 (Sheldon and Robinson, 2007), and in some situations even formation of Rosenthal fibers (Wippold et al., 2006). Our knowledge of astrocyte dysfunction in Alexander disease will likely give us important insights into how astrocyte functions change in such diverse pathologies as epilepsy, neurodegenerative diseases, or hypoxia/ischemia.
Footnotes
Editor's Note: Disease Focus articles provide brief overviews of a neural disease or syndrome, emphasizing potential links to basic neural mechanisms. They are presented in the hope of helping researchers identify clinical implications of their research. For more information, see http://www.jneurosci.org/misc/ifa_minireviews.dtl.
This work has been supported by grants from the NIH (NS-22475, NS-42803, NS-060120, and HD-03352), the Juanma Fund, the Jack Palamaro Fund, and the Jelte Rijkaart Fund. We are extremely grateful to the patients, families, and clinicians who have participated in these studies over the years.
References
- Alexander WS. Progressive fibrinoid degeneration of fibrillary astrocytes associated with mental retardation in a hydrocephalic infant. Brain. 1949;72:373–381. doi: 10.1093/brain/72.3.373. [DOI] [PubMed] [Google Scholar]
- Barkovich AJ, Messing A. Alexander disease: not just a leukodystrophy anymore. Neurology. 2006;66:468–469. doi: 10.1212/01.wnl.0000200905.43191.4d. [DOI] [PubMed] [Google Scholar]
- Blattner R, Von Moers A, Leegwater PA, Hanefeld FA, Van Der Knaap MS, Köhler W. Clinical and genetic heterogeneity in megalencephalic leukoencephalopathy with subcortical cysts (MLC) Neuropediatrics. 2003;34:215–218. doi: 10.1055/s-2003-42210. [DOI] [PubMed] [Google Scholar]
- Brady JP, Garland DL, Green DE, Tamm ER, Giblin FJ, Wawrousek EF. αB-crystallin in lens development and muscle integrity: a gene knockout approach. Invest Ophthalmol Vis Sci. 2001;42:2924–2934. [PubMed] [Google Scholar]
- Brenner M, Johnson AB, Boespflug-Tanguy O, Rodriguez D, Goldman JE, Messing A. Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat Genet. 2001;27:117–120. doi: 10.1038/83679. [DOI] [PubMed] [Google Scholar]
- Brenner M, Goldman JE, Quinlan RA, Messing A. Alexander disease: a genetic disorder of astrocytes. In: Parpura V, Haydon PG, editors. Astrocytes in (patho)physiology of the nervous system. New York: Springer; 2009. pp. 591–648. [Google Scholar]
- Buffo A, Rolando C, Ceruti S. Astrocytes in the damaged brain: molecular and cellular insights into their reactive response and healing potential. Biochem Pharmacol. 2010;79:77–89. doi: 10.1016/j.bcp.2009.09.014. [DOI] [PubMed] [Google Scholar]
- Bugiani M, Boor I, van Kollenburg B, Postma N, Polder E, van Berkel C, van Kesteren RE, Windrem MS, Hol EM, Scheper GC, Goldman SA, van der Knaap MS. Defective glial maturation in vanishing white matter disease. J Neuropathol Exp Neurol. 2011;70:69–82. doi: 10.1097/NEN.0b013e318203ae74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YS, Lim SC, Chen MH, Quinlan RA, Perng MD. Alexander disease causing mutations in the C-terminal domain of GFAP are deleterious both to assembly and network formation with the potential to both activate caspase 3 and decrease cell viability. Exp Cell Res. 2011;317:2252–2266. doi: 10.1016/j.yexcr.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin SS, Goldman JE. Glial inclusions in CNS degenerative diseases. J Neuropathol Exp Neurol. 1996;55:499–508. doi: 10.1097/00005072-199605000-00001. [DOI] [PubMed] [Google Scholar]
- Cho W, Messing A. Properties of astrocytes cultured from GFAP over-expressing and GFAP mutant mice. Exp Cell Res. 2009;315:1260–1272. doi: 10.1016/j.yexcr.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho W, Brenner M, Peters N, Messing A. Drug screening to identify suppressors of GFAP expression. Hum Mol Genet. 2010;19:3169–3178. doi: 10.1093/hmg/ddq227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Bigio MR, Chudley AE, Sarnat HB, Campbell C, Goobie S, Chodirker BN, Selcen D. Infantile muscular dystrophy in Canadian aboriginals is an αB-crystallinopathy. Ann Neurol. 2011;69:866–871. doi: 10.1002/ana.22331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Yue Q, Rosenberg PA, Volpe JJ, Jensen FE. Oligodendrocyte excitotoxicity determined by local glutamate accumulation and mitochondrial function. J Neurochem. 2006;98:213–222. doi: 10.1111/j.1471-4159.2006.03861.x. [DOI] [PubMed] [Google Scholar]
- Eng LF, Lee YL, Kwan H, Brenner M, Messing A. Astrocytes cultured from transgenic mice carrying the added human glial fibrillary acidic protein gene contain Rosenthal fibers. J Neurosci Res. 1998;53:353–360. doi: 10.1002/(SICI)1097-4547(19980801)53:3<353::AID-JNR9>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Friede RL. Alexander's disease. Arch Neurol. 1964;11:414–422. doi: 10.1001/archneur.1964.00460220076010. [DOI] [PubMed] [Google Scholar]
- Fuchs E, Cleveland DW. A structural scaffolding of intermediate filaments in health and disease. Science. 1998;279:514–519. doi: 10.1126/science.279.5350.514. [DOI] [PubMed] [Google Scholar]
- Hagemann TL, Gaeta SA, Smith MA, Johnson DA, Johnson JA, Messing A. Gene expression analysis in mice with elevated glial fibrillary acidic protein and Rosenthal fibers reveals a stress response followed by glial activation and neuronal dysfunction. Hum Mol Genet. 2005;14:2443–2458. doi: 10.1093/hmg/ddi248. [DOI] [PubMed] [Google Scholar]
- Hagemann TL, Connor JX, Messing A. Alexander disease-associated glial fibrillary acidic protein mutations in mice induce Rosenthal fiber formation and a white matter stress response. J Neurosci. 2006;26:11162–11173. doi: 10.1523/JNEUROSCI.3260-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemann TL, Boelens WC, Wawrousek EF, Messing A. Suppression of GFAP toxicity by αB-crystallin in mouse models of Alexander disease. Hum Mol Genet. 2009;18:1190–1199. doi: 10.1093/hmg/ddp013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He T, Stepulak A, Holmström TH, Omary MB, Eriksson JE. The intermediate filament protein keratin 8 is a novel cytoplasmic substrate for c-Jun N-terminal kinase. J Biol Chem. 2002;277:10767–10774. doi: 10.1074/jbc.M111436200. [DOI] [PubMed] [Google Scholar]
- Iwaki T, Kume-Iwaki A, Liem RK, Goldman JE. αB-Crystallin is expressed in non-lenticular tissues and accumulates in Alexander's disease brain. Cell. 1989;57:71–78. doi: 10.1016/0092-8674(89)90173-6. [DOI] [PubMed] [Google Scholar]
- Jany P, Hagemann TL, Messing A. Biomarkers of CNS injury in mouse models of Alexander disease. Soc Neurosci Abstr. 2011;37 875.20/13. [Google Scholar]
- Li R, Messing A, Goldman JE, Brenner M. GFAP mutations in Alexander disease. Int J Dev Neurosci. 2002;20:259–268. doi: 10.1016/s0736-5748(02)00019-9. [DOI] [PubMed] [Google Scholar]
- Messing A, Head MW, Galles K, Galbreath EJ, Goldman JE, Brenner M. Fatal encephalopathy with astrocyte inclusions in GFAP transgenic mice. Am J Pathol. 1998;152:391–398. [PMC free article] [PubMed] [Google Scholar]
- Messing A, Daniels CM, Hagemann TL. Strategies for treatment in Alexander disease. Neurotherapeutics. 2010;7:507–515. doi: 10.1016/j.nurt.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messing A, Li R, Naidu S, Taylor JP, Silverman L, Flint D, van der Knaap MS, Brenner M. Archetypal and new families with Alexander disease and novel mutations in GFAP. Arch Neurol. 2012;69:208–214. doi: 10.1001/archneurol.2011.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namekawa M, Takiyama Y, Aoki Y, Takayashiki N, Sakoe K, Shimazaki H, Taguchi T, Tanaka Y, Nishizawa M, Saito K, Matsubara Y, Nakano I. Identification of GFAP gene mutation in hereditary adult-onset Alexander's disease. Ann Neurol. 2002;52:779–785. doi: 10.1002/ana.10375. [DOI] [PubMed] [Google Scholar]
- Nedergaard M, Ransom B, Goldman SA. New roles for astrocytes: Redefining the functional architecture of the brain. Trends Neurosci. 2003;26:523–530. doi: 10.1016/j.tins.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Omary MB. “IF-pathies”: a broad spectrum of intermediate filament-associated diseases. J Clin Invest. 2009;119:1756–1762. doi: 10.1172/JCI39894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pareyson D, Fancellu R, Mariotti C, Romano S, Salmaggi A, Carella F, Girotti F, Gattellaro G, Carriero MR, Farina L, Ceccherini I, Savoiardo M. Adult-onset Alexander disease: a series of eleven unrelated cases with review of the literature. Brain. 2008;131:2321–2331. doi: 10.1093/brain/awn178. [DOI] [PubMed] [Google Scholar]
- Perng MD, Wen SF, Gibbon T, Middeldorp J, Sluijs J, Hol EM, Quinlan RA. Glial fibrillary acidic protein filaments can tolerate the incorporation of assembly-compromised GFAP-d, but with consequences for filament organization and αB-crystallin association. Mol Biol Cell. 2008;19:4521–4533. doi: 10.1091/mbc.E08-03-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prust M, Wang J, Morizono H, Messing A, Brenner M, Gordon E, Hartka T, Sokohl A, Schiffmann R, Gordish-Dressman H, Albin R, Amartino H, Brockman K, Dinopoulos A, Dotti MT, Fain D, Fernandez R, Ferreira J, Fleming J, Gill D, et al. GFAP mutations, age of onset, and clinical sub-types in Alexander disease. Neurology. 2011;77:1287–1294. doi: 10.1212/WNL.0b013e3182309f72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransom CB, Ransom BR, Sontheimer H. Activity-dependent extracellular K+ accumulation in rat optic nerve: the role of glial and axonal Na+ pumps. J Physiol. 2000;522:427–442. doi: 10.1111/j.1469-7793.2000.00427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan MR, Huang YH, Kim YS, Dykes-Hoberg MI, Jin L, Watkins AM, Bergles DE, Rothstein JD. Variations in promoter activity reveal a differential expression and physiology of glutamate transporters by glia in the developing and mature CNS. J Neurosci. 2007;27:6607–6619. doi: 10.1523/JNEUROSCI.0790-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rempe DA, Nedergaard M. Targeting glia for treatment of neurological disease. Neurotherapeutics. 2010;7:335–337. doi: 10.1016/j.nurt.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal W. Über eine eigenthümliche, mit syringomyelie complicirte geschwulst des rückenmarks. Bietr Pathol Anat. 1898;23:111–143. [Google Scholar]
- Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes Hoberg M, Vidensky S, Chung DS, Toan SV, Bruijn LI, Su ZZ, Gupta P, Fisher PB. β-Lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- Russo LS, Jr, Aron A, Anderson PJ. Alexander's disease: a report and reappraisal. Neurology. 1976;26:607–614. doi: 10.1212/wnl.26.7.607. [DOI] [PubMed] [Google Scholar]
- Sheldon AL, Robinson MB. The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem Int. 2007;51:333–355. doi: 10.1016/j.neuint.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpf E, Masson H, Duquette A, Berthelet F, McNabb J, Lortie A, Lesage J, Montplaisir J, Brais B, Cossette P. Adult Alexander disease with autosomal dominant transmission: a distinct entity caused by mutation in the glial fibrillary acid protein gene. Arch Neurol. 2003;60:1307–1312. doi: 10.1001/archneur.60.9.1307. [DOI] [PubMed] [Google Scholar]
- Su M, Hu H, Lee Y, d'Azzo A, Messing A, Brenner M. Expression specificity of GFAP transgenes. Neurochem Res. 2004;29:2075–2093. doi: 10.1007/s11064-004-6881-1. [DOI] [PubMed] [Google Scholar]
- Tanaka KF, Takebayashi H, Yamazaki Y, Ono K, Naruse M, Iwasato T, Itohara S, Kato H, Ikenaka K. The murine model of Alexander disease: analysis of GFAP aggregate formation and its pathological significance. Glia. 2007;55:617–631. doi: 10.1002/glia.20486. [DOI] [PubMed] [Google Scholar]
- Tang G, Xu Z, Goldman JE. Synergistic effects of the SAPK/JNK and the proteasome pathway on glial fibrillary acidic protein (GFAP) accumulation in Alexander disease. J Biol Chem. 2006;281:38634–38643. doi: 10.1074/jbc.M604942200. [DOI] [PubMed] [Google Scholar]
- Tang G, Yue Z, Talloczy Z, Hagemann T, Cho W, Messing A, Sulzer DL, Goldman JE. Autophagy induced by Alexander disease-mutant GFAP accumulation is regulated by p38/MAPK and mTOR signaling pathways. Hum Mol Genet. 2008;17:1540–1555. doi: 10.1093/hmg/ddn042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Perng MD, Wilk S, Quinlan R, Goldman JE. Oligomers of mutant glial fibrillary acidic protein (GFAP) inhibit the proteasome system in Alexander disease astrocytes, and the small heat shock protein, αB-crystallin, reverses the inhibition. J Biol Chem. 2010;285:10527–10537. doi: 10.1074/jbc.M109.067975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian R, Gregor M, Wiche G, Goldman JE. Plectin regulates the organization of glial fibrillary acidic protein in Alexander disease. Am J Pathol. 2006;168:888–897. doi: 10.2353/ajpath.2006.051028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian R, Wu X, Hagemann TL, Sosunov AA, Messing A, McKhann GM, Goldman JE. Alexander disease mutant GFAP compromises glutamate transport in astrocytes. J Neuropathol Exp Neurol. 2010;69:335–345. doi: 10.1097/NEN.0b013e3181d3cb52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Knaap MS, Naidu S, Breiter SN, Blaser S, Stroink H, Springer S, Begeer JC, van Coster R, Barth PG, Thomas NH, Valk J, Powers JM. Alexander disease: diagnosis with MR imaging. Am J Neuroradiol. 2001;22:541–552. [PMC free article] [PubMed] [Google Scholar]
- van der Knaap MS, Salomons GS, Li R, Franzoni E, Gutiérrez-Solana LG, Smit LM, Robinson R, Ferrie CD, Cree B, Reddy A, Thomas N, Banwell B, Barkhof F, Jakobs C, Johnson A, Messing A, Brenner M. Unusual variants of Alexander disease. Ann Neurol. 2005;57:327–338. doi: 10.1002/ana.20381. [DOI] [PubMed] [Google Scholar]
- van der Knaap MS, Ramesh V, Schiffmann R, Blaser S, Kyllerman M, Gholkar A, Ellison DW, van der Voorn JP, van Dooren SJ, Jakobs C, Barkhof F, Salomons GS. Alexander disease: ventricular garlands and abnormalities of the medulla and spinal cord. Neurology. 2006;66:494–498. doi: 10.1212/01.wnl.0000198770.80743.37. [DOI] [PubMed] [Google Scholar]
- Vargas MR, Johnson JA. Astrogliosis in amyotrophic lateral sclerosis: role and therapeutic potential of astrocytes. Neurotherapeutics. 2010;7:471–481. doi: 10.1016/j.nurt.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LQ, Colodner KJ, Feany MB. Protein misfolding and oxidative stress promote glial-mediated neurodegeneration in an Alexander disease model. J Neurosci. 2011;31:2868–2877. doi: 10.1523/JNEUROSCI.3410-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiche G. Role of plectin in cytoskeleton organization and dynamics. J Cell Sci. 1998;111:2477–2486. doi: 10.1242/jcs.111.17.2477. [DOI] [PubMed] [Google Scholar]
- Wippold FJ, 2nd, Perry A, Lennerz J. Neuropathology for the neuroradiologist: Rosenthal fibers. Am J Neuroradiol. 2006;27:958–961. [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Sasaki M, Yoshida M, Namekawa M, Okamoto Y, Tsujino S, Sasayama H, Mizuta I, Nakagawa M. Nationwide survey of Alexander disease in Japan and proposed new guidelines for diagnosis. J Neurol. 2011;258:1998–2008. doi: 10.1007/s00415-011-6056-3. [DOI] [PubMed] [Google Scholar]
- Ziskin JL, Nishiyama A, Rubio M, Fukaya M, Bergles DE. Vesicular release of glutamate from unmyelinated axons in white matter. Nat Neurosci. 2007;10:321–330. doi: 10.1038/nn1854. [DOI] [PMC free article] [PubMed] [Google Scholar]