

“You, too, were once a cyst.” Anonymous
Autosomal dominant polycystic kidney disease (ADPKD) affects millions of individuals worldwide, is one of the most common, potentially lethal single-gene disorders, is the fourth leading cause of kidney failure in the United States, and annually costs over one billion dollars to treat. ADPKD research is now seeing an intense effort to define the functions of polycystin-1 and -2, the proteins encoded by the PKD1 and PKD2 genes, and there is now a clear target in sight. In this issue of PNAS, a report by Gonzalez-Perrett et al. (1) presents evidence that polycystin-2, the protein product of the PKD2 gene, functions as a nonspecific Ca2+-permeable cation channel. In this respect, PKD research appears to be taking a page from the notebook on cystic fibrosis, an inherited disease in which pulmonary and pancreatic epithelial dysfunction is attributed to a defect in transepithelial Cl− transport (2).
Polycystic kidneys have a striking appearance (Fig. 1). Typically, both kidneys are massively enlarged as a consequence of the growth of hundreds or thousands of cysts scattered throughout the renal cortex and medulla. The gross anatomical distortion caused by these cysts is thought to be responsible for the progressive loss of kidney function associated with this disease and the renal failure experienced by approximately 50% of affected individuals.
Figure 1.
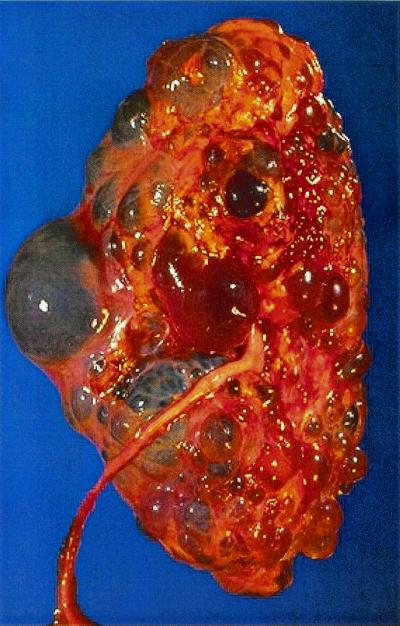
Gross pathology of polycystic kidney. Closeup of surface at autopsy. Image provided by Dr. Edwin P. Ewing, Center for Disease Control and Prevention (1972).
It is common to think of cysts as abnormal, but in a more considered view they are perhaps the most primeval of all epithelial structures. Epithelial cysts, which also include the very blastocysts from which we were all derived, are relatively simple structures comprised of a monolayer of cells enclosing a cavity filled with liquid (3). Blastocysts form through the conjoint acts of epithelial cell proliferation and transepithelial secretion of solutes and water (4, 5). They assume a roughly spherical anatomic configuration and implant into the uterine lining, whereon embryogenesis supervenes. Renal cysts, in contrast to blastocysts, derive from proximal and distal tubules and fill initially with unabsorbed glomerular filtrate passing into them from the upstream tubule segment. Later, after the tubules have expanded to many times their original diameter, the cysts disengage from the parent tubules to become isolated sacs of liquid, whereon filling depends exclusively on transepithelial fluid secretion, as in blastocysts. There being no other source of cavitary liquid, the progressive enlargement of these isolated renal cysts requires this fluid secretion together with the sustained proliferation of the mural epithelial cells.
Mutations affecting polycystin-1 or -2 give rise to virtually identical diseases. Although in both cases the germline mutations are present in all renal tubule cells, cysts develop in only a tiny fraction of them (6). This may be because a somatic mutation or “second hit” in the normal allele is required to initiate cystic transformation (7). Polycystin-1 and -2 are highly conserved (8) ubiquitous (9, 10) proteins that are thought to have a fundamental role in embryonic development and in the maintenance of adult tissues (11, 12). Polycystin-1 is a 4,302-aa protein (13, 14) with 7–11 transmembrane-spanning elements, an extensive extracellular stretch loaded with potential protein–protein interaction domains, a receptor for egg jelly domain (15), and an intracellular carboxyl terminal “business end” harboring serine and tyrosine phosphorylation sites (16, 17) and a G-protein-binding domain (18). The carboxyl terminal tail of polycystin-1 interacts by means of a coiled-coil domain (19, 20) with polycystin-2, a 968-aa polypeptide with 6 transmembrane segments and homology to transient receptor potential and voltage-gated K+, Na+, and Ca2+ channels (21). On the basis of these findings, polycystin-1 and -2 are thought to be interacting signaling partners. Subsequently, a homolog of polycystin-2, polycystin-L, was discovered and demonstrated to have properties of a Ca2+-regulated cation channel permeable to Ca2+ (22) and, more recently, the carboxyl terminal portion of polycystin-1 was shown to regulate an endogenous Ca2+-permeable cation channel in frog oocytes (23). In the current comprehensive study (1), lipid bilayer reconstitution of polycystin-2-containing apical membranes from human placental syncytiotrophoblasts, patch–clamp studies by using Sf9 insect cells expressing heterologous human polycystin-2, and channel measurements by using recombinant and in vitro translated human polycystin-2 provide evidence that this protein is a Ca2+-permeable nonselective cation channel. The similarities among the reconstituted polycystin-2 proteins of different sources are a major strength of the current study. Human polycystin-2, polycystin-L, and the polycystin-1-induced oocyte channel share several common features, including nonselective cation permeability, with relatively greater permeability to Ca2+ and inhibition of conductance by La3+, Gd3+ by reduction of pH and by amiloride. These properties distinguish polycystin-2 from transient receptor potential and voltage-gated Ca2+ channels.
Because of their unique and complex structures, the polycystins are candidates for a variety of regulatory roles, including those related to ion transport. In this regard, the propensity of polycystic epithelial cells to secrete solutes and fluid rather than absorb them (24) has directed attention to the regulation of ion permeability as possibly the misstep that initiates cystogenesis in patients with ADPKD. However, because cyst growth starts in existing tubules, abnormal cell proliferation must be one of the earliest processes that initiate cystogenesis. It seems unlikely that mutated polycystin proteins drive cyst formation in patent renal tubules by increasing the rate of net fluid secretion into the tubule lumens, because the transported liquid would simply escape downstream into the extrarenal collecting system. In this regard, it seems more likely that the polycystins will be found to be involved in the abnormal regulation of tubular epithelial cell proliferation rather than the transepithelial transport of electrolytes and water. In the later stages of the disease, fluid accumulation within the expanding cyst cavities would be an active process, involving the primary transepithelial transport of Cl− through cAMP-regulated channels in the apical membranes of the cyst-lining cells (24, 25). This process is likely to be important in cyst expansion and progression to renal failure.
…it seems more likely that the polycystins will be found to be involved in the abnormal regulation of tubular epithelial cell proliferation rather than the transepithelial transport of electrolytes and water.
So what observations suggest that the polycystins regulate cell proliferation? Homozygous knockout mice develop polycystic kidneys late in fetal development but usually die in utero of cardiovascular abnormalities (26, 27), reflecting the generalized distribution of the encoded proteins and their importance in organ development. In fact, the kidneys of these homozygous knockout mice display normal tubulogenesis and do not become cystic until the final stages of tubule maturation (28, 29). Thus, the polycystins appear to be essential only relatively late in renal tubulogenesis, perhaps as a means to forestall further cell proliferation. Madin–Darby canine kidney cell lines expressing exogenous full-length polycystin-1 exhibit decreased proliferation and enhanced tubulogenesis in comparison to cyst-forming parental cells (30). In renal tubules, there must be sufficient polycystin function to enable renal cells to achieve terminal differentiation. In ADPKD, haploinsufficiency and/or loss of the normal allele through a somatic second hit may then compromise polycystin function to the extent that cells revert to a more immature state where they are more likely to proliferate (3, 31). As such, the polycystins may act as “growth suppressors” akin to the tumor suppressors associated with many other kinds of neoplastic cell growth.
Polycystin-1 and -2 have a number of functions that potentially relate to the regulation of cell proliferation. The carboxyl terminal tail of polycystin-1 has been shown to modulate Wnt signaling by the inhibition of glycogen synthase kinase-3β and the stabilization of β-catenin, with a resultant activation of T cell factor/lymphoid enhancer factor (32). Polycystin-1 has also been found in membrane complexes containing E-cadherin and the α-, β-, and γ-catenins (33). Both polycystin-1 and -2 activate the activator protein-1 transcription factor in a c-Jun N-terminal kinase-dependent fashion (34, 35). The downstream functional effects of these pathways are unknown, but it is not unreasonable to suppose that these processes regulate both cell proliferation and transepithelial solute and fluid reabsorption. When polycystin function is abrogated, cells appear to transform into a less mature phenotype in which cAMP activates the extracellular signal-regulated protein kinase 1/2 pathway, leading to an increase in the rate of cell proliferation, together with a change from net NaCl absorption to net NaCl secretion within the affected tubular segment (24, 36, 37). At this juncture, it is not known whether the putative alteration in cation flux may signal directly to the proliferative apparatus by an alteration in the function of one (or several) of the mitogen-activated protein kinases (36) or indirectly by altering adhesion complexes between adjacent cells or the extracellular matrix (33, 38).
Additional clues to the initial downstream steps in cyst pathogenesis can perhaps be found in the functions of polycystin homologs. The sea urchin receptor for egg jelly protein is a membrane receptor that functions in the sperm–egg interaction by triggering the Ca2+ influx that leads to the acrosome reaction and the subsequent union of the gametes (15). In a similar vein, the Caenorhabditis elegans location of vulva (LOV)-1 protein serves as a mechanosensory receptor enabling male worms to find the vulva of hermaphrodites during the consummation of their relationship (39). Thus, on the basis of developments reported here (1), it is conceivable that polycystin-1 may function as an extracellular sensor that modulates Ca2+ flux through polycystin-2 in a way that controls cell proliferation, cell polarity, and cell morphology. Loss of their intimate interactions through the inappropriate behavior of either polycystin-1 or -2 could set the course for abnormal tubule cell proliferation, abnormal growth stimulation by cAMP, and ultimately the wayward growth and development of cysts from renal tubules.
It is becoming clear that PKD research is drawing from a wide spectrum of seemingly unrelated fields, some potentially salacious. By embracing the insights provided by male mating behavior (LOV-1), sperm–egg interactions (receptor for egg jelly), frog oocyte injection systems, blastocyst development, and placental syncytiotrophoblasts, let us hope that this diversity of approaches will clarify the functions of the polycystins and the molecular pathogenesis of PKD.
Acknowledgments
We thank Billy Hudson, Greg Vanden Heuvel, Robin Maser, and Stephen Parnell for helpful suggestions. This work was supported in part by grants from the National Institutes of Health and the Polycystic Kidney Research Foundation.
Note Added in Proof.
Hanaoka et al. (40) report that full-length polycystin-1 and polycystin-2, transfected into Chinese hamster ovary cells, must interact at the plasma membrane to produce new calcium-permeable nonselective cation currents.
Footnotes
See companion article on page 1182.
References
- 1.Gonzalez-Perret S, Kim K, Ibarra C, Damiano A E, Zotta E, Batelli M, Harris P C, Reisin I L, Arnaout M A, Cantiello H F. Proc Natl Acad Sci USA. 2001;98:1182–1187. doi: 10.1073/pnas.98.3.1182. . (First Published December 26, 2000; 10.1073/pnas.021456598) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwiebert E M, Benos D J, Egan M E, Stutts M J, Guggino W B. Physiol Rev. 1999;79:S145–S166. doi: 10.1152/physrev.1999.79.1.S145. [DOI] [PubMed] [Google Scholar]
- 3.Grantham J J. J Am Soc Nephrol. 1993;3:1841–1857. doi: 10.1681/ASN.V3121841. [DOI] [PubMed] [Google Scholar]
- 4.Borland R M, Biggers J D, Lechene C P. J Reprod Fertil. 1977;51:131–135. doi: 10.1530/jrf.0.0510131. [DOI] [PubMed] [Google Scholar]
- 5.Watson A J. Mol Reprod Dev. 1992;33:492–504. doi: 10.1002/mrd.1080330417. [DOI] [PubMed] [Google Scholar]
- 6.Grantham J J, Geiser J L, Evan A P. Kidney Int. 1987;31:1145–1152. doi: 10.1038/ki.1987.121. [DOI] [PubMed] [Google Scholar]
- 7.Qian F, Germino G G. Am J Hum Genet. 1997;61:1000–1005. doi: 10.1086/301618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sandford R, Sgotto B, Aparicio S, Brenner S, Vaudin M, Wilson R K, Chissoe S, Pepin K, Bateman A, Chothia C, et al. Hum Mol Genet. 1997;6:1483–1489. doi: 10.1093/hmg/6.9.1483. [DOI] [PubMed] [Google Scholar]
- 9.Griffin M D, Torres V E, Grande J P, Kumar R. Proc Assoc Am Physicians. 1996;108:185–197. [PubMed] [Google Scholar]
- 10.Geng L, Segal Y, Pavlova A, Barros E J, Lohning C, Lu W, Nigam S K, Frischauf A M, Reeders S T, Zhou J. Am J Physiol. 1997;272:F451–F459. doi: 10.1152/ajprenal.1997.272.4.F451. [DOI] [PubMed] [Google Scholar]
- 11.Guillaume R, D'Agati V, Daoust M, Trudel M. Dev Dyn. 1999;214:337–348. doi: 10.1002/(SICI)1097-0177(199904)214:4<337::AID-AJA6>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 12.Guillaume R, Trudel M. Mech Dev. 2000;93:179–183. doi: 10.1016/s0925-4773(00)00257-4. [DOI] [PubMed] [Google Scholar]
- 13.Ward C J, Turley H, Ong A C, Comley M, Biddolph S, Chetty R, Ratcliffe P J, Gattner K, Harris P C. Proc Natl Acad Sci USA. 1996;93:1524–1528. doi: 10.1073/pnas.93.4.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ibraghimov-Beskrovnaya O, Dackowski W R, Foggensteiner L, Coleman N, Thiru S, Petry L R, Burn T C, Connors T D, Van Raay T, Bradley J, et al. Proc Natl Acad Sci USA. 1997;94:6397–6402. doi: 10.1073/pnas.94.12.6397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moy G W, Mendoza L M, Schulz J R, Swanson W J, Glabe C G, Vacquier V D. J Cell Biol. 1996;133:809–817. doi: 10.1083/jcb.133.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parnell S C, Magenheimer B S, Maser R L, Calvet J P. Biochem Biophys Res Commun. 1999;259:539–543. doi: 10.1006/bbrc.1999.0810. [DOI] [PubMed] [Google Scholar]
- 17.Li H P, Geng L, Burrow C R, Wilson P D. Biochem Biophys Res Commun. 1999;259:356–363. doi: 10.1006/bbrc.1999.0780. [DOI] [PubMed] [Google Scholar]
- 18.Parnell S C, Magenheimer B S, Maser R L, Rankin C A, Smine A, Okamoto T, Calvet J P. Biochem Biophys Res Commun. 1998;251:625–631. doi: 10.1006/bbrc.1998.9514. [DOI] [PubMed] [Google Scholar]
- 19.Qian F, Germino F J, Cai Y, Zhang X, Somlo S, Germino G G. Nat Genet. 1997;16:179–183. doi: 10.1038/ng0697-179. [DOI] [PubMed] [Google Scholar]
- 20.Tsiokas L, Kim E, Arnould T, Sukhatme V P, Walz G. Proc Natl Acad Sci USA. 1997;94:6965–6970. doi: 10.1073/pnas.94.13.6965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mochizuki T, Wu G, Hayashi T, Xenophontos S L, Veldhuisen B, Saris J J, Reynolds D M, Cai Y, Gabow P A, Pierides A, et al. Science. 1996;272:1339–1342. doi: 10.1126/science.272.5266.1339. [DOI] [PubMed] [Google Scholar]
- 22.Chen X Z, Vassilev P M, Basora N, Peng J B, Nomura H, Segal Y, Brown E M, Reeders S T, Hediger M A, Zhou J. Nature (London) 1999;401:383–386. doi: 10.1038/43907. [DOI] [PubMed] [Google Scholar]
- 23.Vandorpe, D. H., Chernova, M. N., Jiang, L., Sellin, L. K., Wilhelm, S., Stuart-Tilley, A. K., Walz, G. & Alper, S. L. (2001) J. Biol. Chem., in press. [DOI] [PubMed]
- 24.Sullivan L P, Wallace D P, Grantham J J. Physiol Rev. 1998;78:1165–1191. doi: 10.1152/physrev.1998.78.4.1165. [DOI] [PubMed] [Google Scholar]
- 25.Hanaoka K, Devuyst O, Schwiebert E M, Wilson P D, Guggino W B. Am J Physiol. 1996;270:C389–C399. doi: 10.1152/ajpcell.1996.270.1.C389. [DOI] [PubMed] [Google Scholar]
- 26.Kim K, Drummond I, Ibraghimov-Beskrovnaya O, Klinger K, Arnaout M A. Proc Natl Acad Sci USA. 2000;97:1731–1736. doi: 10.1073/pnas.040550097. . (First Published February 4, 2000, 10.1073/pnas.040550097) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu G, Markowitz G S, Li L, D'Agati V D, Factor S M, Geng L, Tibara S, Tuchman J, Cai Y, Park J H, et al. Nat Genet. 2000;24:75–78. doi: 10.1038/71724. [DOI] [PubMed] [Google Scholar]
- 28.Lu W, Fan X, Basora N, Babakhanlou H, Law T, Rifai N, Harris P C, Perez-Atayde A R, Rennke H G, Zhou J. Nat Genet. 1999;21:160–161. doi: 10.1038/5944. [DOI] [PubMed] [Google Scholar]
- 29.Wu G, D'Agati V, Cai Y, Markowitz G, Park J H, Reynolds D M, Maeda Y, Le T C, Hou H, Jr, Kucherlapati R, et al. Cell. 1998;93:177–188. doi: 10.1016/s0092-8674(00)81570-6. [DOI] [PubMed] [Google Scholar]
- 30.Boletta A, Qian F, Onuchic L F, Bhunia A K, Phakdeekitcharoen B, Hanaoka K, Guggino W, Monaco L, Germino G G. Mol Cell. 2000;6:1267–1273. doi: 10.1016/s1097-2765(00)00123-4. [DOI] [PubMed] [Google Scholar]
- 31.Calvet J P. Curr Opin Nephrol Hypertens. 1994;3:340–348. doi: 10.1097/00041552-199405000-00017. [DOI] [PubMed] [Google Scholar]
- 32.Kim E, Arnould T, Sellin L K, Benzing T, Fan M J, Gruning W, Sokol S Y, Drummond I, Walz G. J Biol Chem. 1999;274:4947–4953. doi: 10.1074/jbc.274.8.4947. [DOI] [PubMed] [Google Scholar]
- 33.Huan Y, van Adelsberg J. J Clin Invest. 1999;104:1459–1468. doi: 10.1172/JCI5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arnould T, Kim E, Tsiokas L, Jochimsen F, Gruning W, Chang J D, Walz G. J Biol Chem. 1998;273:6013–6018. doi: 10.1074/jbc.273.11.6013. [DOI] [PubMed] [Google Scholar]
- 35.Arnould T, Sellin L, Benzing T, Tsiokas L, Cohen H T, Kim E, Walz G. Mol Cell Biol. 1999;19:3423–3434. doi: 10.1128/mcb.19.5.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamaguchi T, Pelling J C, Ramaswamy N T, Eppler J W, Wallace D P, Nagao S, Rome L A, Sullivan L P, Grantham J J. Kidney Int. 2000;57:1460–1471. doi: 10.1046/j.1523-1755.2000.00991.x. [DOI] [PubMed] [Google Scholar]
- 37.Hanaoka K, Guggino W B. J Am Soc Nephrol. 2000;11:1179–1187. doi: 10.1681/ASN.V1171179. [DOI] [PubMed] [Google Scholar]
- 38.Charron A J, Nakamura S, Bacallao R, Wandinger-Ness A. J Cell Biol. 2000;149:111–124. doi: 10.1083/jcb.149.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barr M M, Sternberg P W. Nature (London) 1999;401:386–389. doi: 10.1038/43913. [DOI] [PubMed] [Google Scholar]
- 40.Hanaoka K, Qian F, Boletta A, Bhunia A K, Piontek K, Tsiokas L, Sukhatme V P, Giggino W B, Germino G G. Nature (London) 2000;408:990–994. doi: 10.1038/35050128. [DOI] [PubMed] [Google Scholar]