

Abstract
Axonal degeneration is a molecular self-destruction cascade initiated following traumatic, toxic, and metabolic insults. Its mechanism underlies a number of disorders including hereditary and diabetic neuropathies and the neurotoxic side effects of chemotherapy drugs. Molecules that promote axonal degeneration could represent potential targets for therapy. To identify such molecules, we designed a screening platform based on intoxication of Drosophila larvae with paclitaxel (taxol), a chemotherapeutic agent that causes neuropathy in cancer patients. In Drosophila, taxol treatment causes swelling, fragmentation, and loss of axons in larval peripheral nerves. This axonal loss is not due to apoptosis of neurons. Taxol-induced axonal degeneration in Drosophila shares molecular execution mechanisms with vertebrates, including inhibition by both NMNAT (nicotinamide mononucleotide adenylyltransferase) expression and loss of wallenda/DLK (dual leucine zipper kinase). In a pilot RNAi-based screen we found that knockdown of retinophilin (rtp), which encodes a MORN (membrane occupation and recognition nexus) repeat-containing protein, protects axons from degeneration in the presence of taxol. Loss-of-function mutants of rtp replicate this axonal protection. Knockdown of rtp also delays axonal degeneration in severed olfactory axons. We demonstrate that the mouse ortholog of rtp, MORN4, promotes axonal degeneration in mouse sensory axons following axotomy, illustrating conservation of function. Hence, this new model can identify evolutionarily conserved genes that promote axonal degeneration, and so could identify candidate therapeutic targets for a wide-range of axonopathies.
Introduction
Injury to axons occurs in a wide variety of pathological contexts, including diabetic neuropathy, exposure to chemotherapeutic agents during cancer treatment, and hereditary neuropathies such as Charcot-Marie-Tooth disease (Coleman, 2005). In recent years, axonal injury has been appreciated as an early event in CNS degenerative diseases such as Alzheimer's disease (Stokin et al., 2005) as well as traumatic brain injuries (Xu et al., 2007). These diverse mechanical, toxic, and metabolic insults initiate a stereotyped set of cellular changes beginning with axonal swellings that progress into breaches of membrane integrity and axonal fragmentation (Coleman and Freeman, 2010).
Axon degeneration occurs through a molecular pathway that is evolutionarily conserved. Evidence for this molecular cascade came initially from the remarkable protective effects of expression of nicotinamide mononucleotide adenylyltransferase (NMNAT), the critical component of the Wallerian degeneration slow (Wlds) fusion protein (Mack et al., 2001; Araki et al., 2004). Using a mechanism that is still mysterious, NMNAT gain-of-function in the axon blocks axonal degeneration in Drosophila and mouse from a wide range of insults (Mack et al., 2001; Hoopfer et al., 2006; MacDonald et al., 2006; Press and Milbrandt, 2008; Zhai et al., 2008; Sasaki et al., 2009a). Endogenous pathways also promote axonal degeneration following injury. The dual leucine zipper kinase DLK, or its Drosophila ortholog wallenda, is required for efficient axonal degeneration in response to axotomy or chemotherapy treatment in vitro, and in vivo following sciatic nerve injury (Miller et al., 2009). That the roles of NMNAT and DLK are functionally conserved across species suggests that an ancient cascade executes the removal of irreparably damaged axons. Identification of important players in this cascade may enable strategies to block axon degeneration and could slow or prevent progression of many neurological diseases.
In human patients, the administration of chemotherapeutic agents such as vincristine and paclitaxel (taxol) causes a dose-limiting peripheral neuropathy, such that very aggressive cancers cannot be adequately controlled by these drugs (Malik and Stillman, 2008; Park et al., 2008). Since the timing of chemotherapy treatment is known, strategies to protect axons before and/or during treatment have great potential to minimize this toxic side effect (Windebank, 1999). The identification of genes that promote axon degeneration after injury is a first step toward realizing this therapeutic potential.
To identify molecules underlying axonal degeneration, we devised a novel paradigm for injuring axons in the fruit fly Drosophila melanogaster using the chemotherapy drug paclitaxel (taxol). In this model, axons swell, fragment, and are lost, and this loss is not caused by apoptosis. Axons injured in this paradigm can be protected by both NMNAT overexpression and loss of wallenda/DLK, demonstrating that the conserved axonal degeneration program mediates this taxol-induced neuropathy. To test the utility of this new model as a gene discovery platform, we performed a pilot RNAi-based screen and identified the MORN (membrane occupation and recognition nexus) family member retinophilin as a gene promoting degeneration. Importantly, the mouse ortholog of retinophilin, Morn4, also promotes mammalian axonal degeneration. Our work introduces a novel and clinically relevant axonal injury paradigm in Drosophila and has identified a role for MORN4 as a conserved component of the axonal degeneration program.
Materials and Methods
Fly strains.
The following strains were used in this study: Canton S (wild-type), elav-GAL4 (Yao and White, 1994), pickpocket5-EGFP (referred to as ppkEGFP; Grueber et al., 2003), pickpocket-CD8:3xGFP (referred to as ppkCD8GFP; Shimono et al., 2009), ppk-GAL4 (Grueber et al., 2007), wnd2 and wnd3 mutant alleles (Collins et al., 2006), UAS-dmNMNAT (MacDonald et al., 2006), retin1 (Venkatachalam et al., 2010), and rtp1 (Mecklenburg et al., 2010). All other strains are from the Bloomington Stock Center. All RNAi lines used are from the Vienna Drosophila RNAi Consortium (Retinophilin RNAi, 109000).
The UAS-cytNMNAT1 expression construct was made using mouse cytNMNAT1, a mutant version of NMNAT1 with the nuclear localization disrupted (Sasaki et al., 2006). The full-length cytNMNAT1 was inserted into the pUAST expression vector and injected into w− fly embryos to create transgenic lines. A viable second chromosome insertion of this construct was used for experiments.
Genetics.
For screening, females with homozygous UAS-Dicer2, elavGAL4, and ppkEGFP were crossed to homozygous UAS-RNAi males to generate progeny of either sex for analysis. For olfactory axotomies, females of a strain containing GAL4 driven by the promoter region of olfactory receptor 82a (Or82a-GAL4; Fishilevich and Vosshall, 2005), UAS-CD8:GFP, and UAS-Dicer2 were crossed to UAS-RNAi males, and male progeny were analyzed. Other genotypes are as noted in figure legends.
Taxol treatment.
Embryos from crosses were collected on grape plates, and first instar larvae of both sexes were washed off and resuspended in water with 30 μm paclitaxel (Tocris Bioscience) or an equivalent amount of DMSO (vehicle; Tissue Culture Support Center, Washington University School of Medicine). This liquid was then used to make up instant Drosophila media (“blue food;” Carolina Biological Supply). For large-scale screening, 12 well plates were used and plugged with Flugs (Genesee Scientific). Animals on taxol-treated food were incubated at 25°C for 4 d before dissection and analysis.
Immunocytochemistry.
GFP signal was enhanced with Alexa Fluor 488-rabbit-anti-GFP (1:1000, Invitrogen). Other antibodies used were Cy3-HRP (1:1000, Jackson ImmunoResearch) and Rabbit anti-cleaved-Caspase 3 (1:200, Cell Signaling Technology). Secondary antibodies were obtained from Jackson ImmunoResearch and used at 1:1000 dilution.
Mouse DRG cultures.
Embryonic day 12.5 mouse DRG neurons were cultured as described previously (Sasaki et al., 2009b) using embryos of both sexes. Lentiviral particles containing Morn4-targeting (5′-TGACCTTCTCAGATGGCTCAA-3′) or control shRNA sequences were packaged as described previously (Araki et al., 2004) and added to DRG cultures at 2 DIV. In vitro axotomy was performed at 7–8 DIV. Quantitative reverse transcriptase-PCR analysis of neuronal mRNA was performed using an ABI Prism 7900HT sequence detection instrument (Applied Biosystems) as described previously (Gerdts et al., 2011). Following insult, axonal degeneration was quantified from four phase-contrast images per well by ImageJ software that measures fragmentation of axon segments (Sasaki et al., 2009b). Briefly, axon images are binarized, and the ratio of fragmented to total axonal area axonal area is computed by using circularity as an indicator of fragmentation.
Microscopy.
Images of Drosophila larval axons and adult brains were taken on a Nikon Eclipse E600 confocal microscope and are maximal Z-projections. Phase contrast images of DRG axons (20× objective) were taken using an inverted microscope (Eclipse TE 300; Nikon).
To analyze Drosophila axons, we took images of nerves between segments A3 and A4 of vehicle-treated third instar larvae or equivalently timed taxol-treated larvae. We counted the number of intact GFP-positive axons per nerve from each image (∼100 μm length of nerve per picture). The average number of GFP+ axons/nerve in vehicle-treated control (wild-type) animals labeled with ppkEGFP was 1.97 ± 0.06 (SEM) (n = 71 nerves) and was not significantly different in animals labeled with ppkCD8GFP, or between wild-type and rtp mutant strains. For presentation, we normalized data to the average of the wild-type vehicle control of the relevant experiment. All analysis of peripheral sensory axons and olfactory axons was done blind to genotype. One-way ANOVA was used for statistical analysis unless otherwise specified. All error bars are SEM.
For ultrastructural analysis of the Drosophila peripheral nerves, samples were fixed in 2% paraformaldehyde/2.5% glutaraldehyde (Polysciences Inc.) in 100 mm phosphate buffer, pH 7.2 overnight at 4°C. Samples were washed in phosphate buffer and postfixed in 0.5% osmium tetroxide (Polysciences Inc.)/0.08% potassium ferricyanide (Electron Microscopy Sciences)/100 mm phosphate buffer for 1 h, and subsequently in 1% tannic acid (Electron Microscopy Sciences, Fort Washington, PA)/100 mm phosphate buffer for 1 h. Samples were then rinsed extensively in dH2O before en bloc staining with 1% aqueous uranyl acetate (Ted Pella Inc., Redding, CA) for 1 h. Following several rinses in dH2O, samples were dehydrated in a graded series of ethanol and embedded in Eponate 12 resin (Ted Pella Inc.). Sections of 100 nm were cut with a Leica Ultracut UCT ultramicrotome (Leica Microsystems Inc., Bannockburn, IL), stained with uranyl acetate and lead citrate. Electron micrographs were taken on a transmission electron microscope (H-7500; Hitachi). Quantification of axon number was performed on 10 nonconsecutive sections of nerves from at least three independent larvae per treatment condition.
Results
Paclitaxel causes axonal injury and loss in Drosophila
Because only a few genes have been identified that regulate axonal degeneration, we sought to develop a model of axonal degeneration in Drosophila amenable to screens for genes that promote axonal demise. Loss-of-function of a protein participating in the axonal degeneration cascade would be predicted to cause axonal protection following an insult and would be a putative therapeutic target. To maximize throughput, we wanted to devise an axonal injury paradigm that did not depend on mechanically injuring each animal individually. In addition, we wanted to use a clinically relevant insult. Drosophila has been used as a model of neurodegeneration following cisplatin toxicity (Podratz et al., 2011), and so may respond to chemotherapeutics in a manner analogous to mammals. We therefore attempted to mimic the neuropathy caused in human patients by paclitaxel (taxol) administration. To do this, we fed larval-stage animals food laced with taxol. By beginning treatment after animals have reached the larval stage, we avoid disrupting neurogenesis or axon pathfinding, events that are complete at the time of hatching (for review, see Prokop, 1999). Following treatment, we examined larval peripheral axons using a genetically encoded GFP that labels class IV sensory neurons and their projections (ppkEGFP) (Grueber et al., 2003). Because this marker highlights only a few neurons per hemisegment, it allows single axon resolution within the segmental nerves leading to the ventral nerve cord.
We find that treatment with taxol causes axonal destruction and loss (Fig. 1). Axonal injury following taxol treatment was apparent after 3 d, with maximal injury after 4 d. In the majority of nerves, GFP is severely fragmented or lost following taxol treatment (Fig. 1C). Interestingly, in nerves where taxol has not caused complete GFP-positive axon loss, we see axonal swellings and debris, hallmarks of mammalian axonal degeneration (Coleman, 2005) (Fig. 1B,I). HRP labeling occasionally shows axonal swellings (Fig. 1B) or overall staining intensity decreases (Fig. 1C), likely due to the degeneration of non-GFP-labeled nerves. Examination of nerves using electron microscopy reveals an approximately twofold decrease in axon number (vehicle, 80.9 ± 3.9 axons per nerve; taxol, 42.7 ± 5.1 axons per nerve; p < 0.001, Student's t test) (Fig. 1D,E), suggesting that the GFP loss often corresponds to the frank loss of axons. Nerves in taxol-treated animals have large holes (Fig. 1E), and free organelles are occasionally seen in these areas. However, remaining axons appear relatively unaffected and contain microtubule profiles (Fig. 1F,G). In addition to the phenotypes in GFP-labeled axons, we also observe peripheral nerve defasciculation. In these defasciculated nerves, swellings of single axons that accumulate the microtubule-associated protein Futsch are apparent (Fig. 1H,I). Sensory axons are still present within the neuropil of the ventral nerve cord following taxol treatment, however the ladder pattern of sensory endings is often disrupted and individual axons are fragmented (Fig. 1J,K). Using the same ppkEGFP label, we also examined the effects of taxol on the dendrites of multidendritic sensory neurons in the larval cuticle. Similar to its effects on axons, taxol causes sensory dendrite swelling and loss (Fig. 1L–N). To assess whether taxol treatment caused “dying back”-like retraction, we examined larval neuromuscular synapses. Unlike genetic models in Drosophila that disrupt the cytoskeleton (Massaro et al., 2009), we observe normal apposition of presynaptic and postsynaptic markers following taxol treatment, indicating that our model of taxol-induced degeneration does not cause motoneuron synaptic instability (data not shown). Together, these results highlight the range of degenerative phenotypes resulting from paclitaxel treatment in flies and reveal morphological similarities to degenerating mammalian axons.
Figure 1.
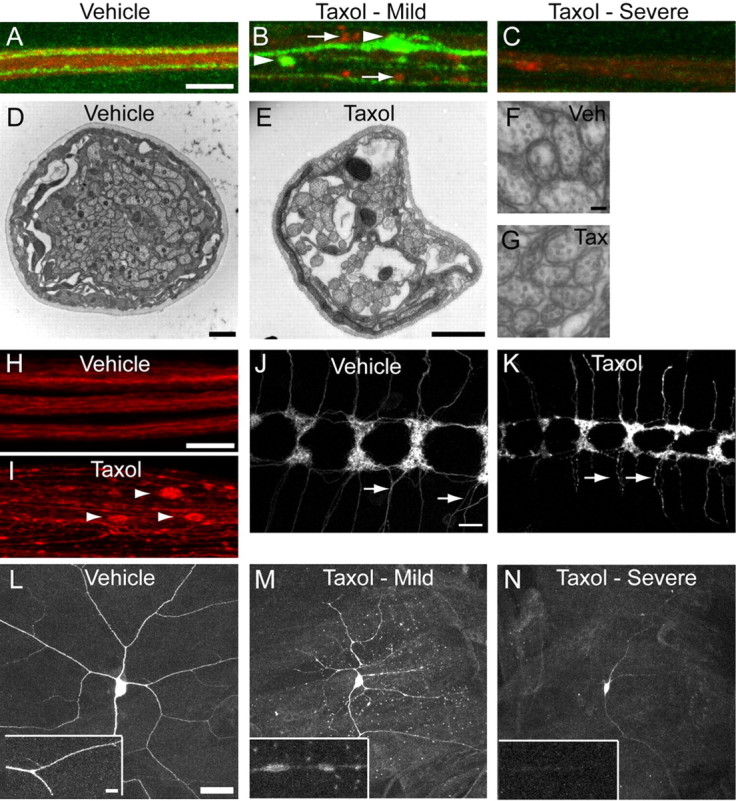
Drosophila peripheral axons are damaged or lost following taxol treatment. A–C, The segmental nerve between segment A3 and A4 is visualized by Cy3-HRP (labels neuronal membranes, red). ppkEGFP labels a subset of sensory neurons. A, Vehicle (DMSO) treated. B, C, 30 μm taxol treated. Axon degeneration was assessed 4 d after treatment initiation. In B, an intermediate phenotype of swellings and fragmentation is visible with both EGFP (arrowheads) and with HRP (arrows). In C, a more severe phenotype of EGFP and HRP loss is shown. Scale bar: (in A) A–D, 5 μm. D, E, EM cross-section images of nerves from wild-type (Canton S) vehicle- and taxol-treated animals. Scale bars: D, E, 1 μm. F, G, Zoomed-in images of axons from D and E. Scale bar (in F) F, G, 200 nm. H, I, Peripheral nerves stained with antibody to Futsch (22C10). Three nerves are shown in the vehicle; one defasciculated nerve is shown in the taxol-treated animal. Arrowheads point to prominent swellings in individual axons. Scale bar (in H), H, I, 10 μm. J, K, Class IV sensory axons (ppkgal4/+; UAS-CD8:GFP/+) in the ventral nerve cord in vehicle (left) and taxol treated (right). Arrows point to intact axons (J) or swollen and fragmenting axons (K). Scale bar: (in J) J, K, 10 μm. L–N, Dendrites and cell body of ddaC sensory neurons in vehicle-treated (L) or 30 μm taxol-treated (M, N) animals. Scale bar: (in L) L–N, 30 μm. Insets show portions of a dendrite from the larger images. Scale bar, 6 μm.
Taxol-Induced Axonal Degeneration is Not Secondary to Apoptosis
In patients, neuronal cell death is not a major contributor to paclitaxel-induced neuropathy (Lee and Swain, 2006; Windebank and Grisold, 2008). To test whether this is also true in our Drosophila model, we overexpressed the baculoviral anti-apoptotic protein p35 in sensory neurons (Hay et al., 1994). Expression of p35 with the ppkGAL4 driver (Grueber et al., 2007) effectively blocks activation of Caspase 3 following induction of apoptosis in sensory neurons via the forced expression of the proapoptotic genes hid and reaper (Fig. 2F). However, when p35-expressing animals are treated with taxol, their peripheral sensory axons are not preserved (Fig. 2A–E), indicating that axonal loss in this model is not secondary to apoptosis in the sensory cell bodies. In addition, cell bodies of taxol-treated animals fail to label with anti-activated Caspase-3 antibodies (Fig. 2F). These results are consistent with studies that have found a role for caspases in dendrite pruning but not in injury-induced degeneration (Schoenmann et al., 2010; Tao and Rolls, 2011). These data highlight the independence of the mechanism of taxol-induced axonal degeneration from that of apoptosis.
Figure 2.

Axonal degeneration following taxol treatment is independent of apoptosis. A–D, The viral anti-apoptotic protein p35 is unable to rescue sensory axon degeneration. Confocal images of single representative nerves labeled with CD8:GFP (green) and HRP (red). Genotype is ppkgal4/+;UAS-CD8:GFP/+ (Ctrl, A and C) or ppkgal4/UAS-p35;UAS-CD8:GFP/+ (p35, B and D). Animals were fed food containing vehicle (DMSO) (A, B) or taxol (C, D) for 4 d. Scale bar (A), 2 μm. E, Quantification of GFP-labeled axons per nerve remaining after treatment from the genotypes shown in A–D. N = 21, 20, 18, and 16 for the genotypes quantified. NS, Not significant (p > 0.1). F, GFP (left column) and activated caspase 3 (right column) in control larvae (ppkGAL4/+;UAS-CD8:GFP/+) that are untreated (top) or treated with 30 μm taxol (second from top). Bottom two sets of images are controls showing caspase 3 activation following ppkGAL4-driven expression of UAS-hid and UAS-reaper (second from bottom), and loss of caspase 3 activation in flies expressing UAS-hid, UAS-reaper, and UAS-p35 (bottom). Scale bar (F), 3 μm.
NMNAT overexpression can block paclitaxel-induced axonal degeneration
To determine whether paclitaxel-induced axonal degeneration in flies shares molecular mechanisms with axonal degeneration in mammalian sensory neurons, we asked whether molecules known to block axonal degeneration in mice also protect axons in our Drosophila model. First, we tested whether overexpression of NMNAT could protect fly sensory axons from degeneration. NMNAT is protective for both sensory axons and dendrites in other injury paradigms in the fly (Tao and Rolls, 2011; Wen et al., 2011). We made transgenic flies that allow for the targeted expression of cytoplasmically targeted mouse NMNAT1 (cyt-mNMNAT1) (Sasaki et al., 2009a). Following taxol treatment, axons from neurons overexpressing cyt-mNMNAT1 were preserved, whereas control axons were lost (Fig. 3). cyt-mNMNAT1 expression also blocked dendrite degeneration following taxol treatment (Fig. 3F,G). These results illustrate conservation of the protective effects of NMNAT for taxol-induced neuropathy between invertebrates and vertebrates (Wang et al., 2002).
Figure 3.

Overexpression of mouse NMNAT protects axons and dendrites from taxol-induced degeneration. A–D, Confocal images of single representative nerves labeled with ppkEGFP. Genotypes are elavgal4, ppkEGFP/+ (Ctrl, A and C), or UAS-cytNMNAT1/+;elavgal4, ppkEGFP/+ (NMNAT, B and D). Animals were treated with DMSO (A, B) or taxol (C, D) for 4 d and then analyzed. Scale bar (A), 2 μm. E, Quantification of GFP-labeled axons per nerve following taxol treatment in the genotypes shown in A–D. N = 10, 17, 17, and 22 for the genotypes quantified. **p < 0.01. F, G, Dendrites and cell bodies of ddaC neurons treated with taxol from control (same genotype as A and C) and UAS-cytNMNAT1-expressing (same genotype as B and D) animals. Scale bar (F), 10 μm.
Wallenda/DLK is required for taxol-induced axonal degeneration in Drosophila
To examine whether endogenous genes required for axonal degeneration in mammals also participate in taxol-induced degeneration in flies, we examined the effect of loss of the MAP3K Wallenda (DLK in mammals). Wallenda participates in the degeneration cascade following Drosophila olfactory axotomy or mouse axonal injury (Miller et al., 2009) and is required for injury signaling following Drosophila nerve crush (Xiong et al., 2010). Following taxol treatment, wallenda mutants displayed robust protection of peripheral axons (Fig. 4). Similar protection is observed when transgenic RNAi is used to knockdown wallenda exclusively in neurons (data not shown). These data demonstrate that known pathways used in mammalian axonal degeneration are conserved members of the taxol-induced degeneration cascade in flies. Furthermore, it illustrates that loss-of-function analysis in taxol-treated animals could be useful to identify novel players in axonal degeneration that may be relevant to mammalian axonal degeneration.
Figure 4.

Wallenda is required for taxol-induced axonal degeneration in Drosophila. A–D, Confocal images of single representative nerves labeled with ppkEGFP. Animals were treated with DMSO (A, C) or taxol (B, D) for 4 d and then analyzed. WT genotype is heterozygous wnd3/+; wnd genotype is wnd3/wnd2. A, Wild-type vehicle; B, wnd mutant vehicle; C, wild-type taxol; D, wnd mutant taxol. Scale bar (A), 2 μm. E, Quantification of axonal preservation in the genotypes shown in A–D. N = 54, 54, 58, and 77 for genotypes quantified. **p < 0.01.
RNAi interference identifies a role For retinophilin in axonal degeneration
We undertook a pilot RNAi screen looking for genes that, similar to wallenda, would block axonal degeneration when knocked down. We used a collection of inducible, transgenic RNAi strains available from an international consortium (Dietzl et al., 2007). These RNAi lines were expressed under the control of UAS elements using the pan-neuronal driver elav-GAL4 to express both the RNAi and Dicer2, which enhances efficacy of RNAi knockdown. Under these conditions, ∼80% of genes with known phenotypes in the nervous system give the expected phenotype when knocked down (Dietzl et al., 2007).
We limited our analysis to Drosophila genes with clear mammalian orthologs as determined by Homologene NCBI software. A subset of 490 RNAi strains was chosen for our pilot screen that was enriched for proteins with enzymatic functions and for genes known to function in the nervous system; this selection was done using Flybase QueryBuilder (www.flybase.org). These strains were screened for degeneration or preservation of ppkEGFP-labeled axons following taxol intoxication. From this collection we identified the MORN family member retinophilin [rtp, also called undertaker (uta)]. Retinophilin participates in termination of phototransduction in the fly retina (Mecklenburg et al., 2010; Venkatachalam et al., 2010) and in store-operated calcium release and phagocytosis in fly embryonic macrophages (Cuttell et al., 2008). It contains four MORN repeats, regions that can bind to phospholipid membranes (Ma et al., 2006). Knockdown of retinophilin protected axons from chemotherapy-induced peripheral neuropathy (Fig. 5A). The RNAi protection is quantitatively modest, but consistent (Fig. 5B). To test whether loss of Retinophilin is responsible for this phenotype, we generated a trans-heterozygote between two previously characterized loss-of-function mutant alleles of rtp (Mecklenburg et al., 2010; Venkatachalam et al., 2010). Axons in this rtp mutant are preserved following taxol treatment (Fig. 5C,D), confirming that removal of retinophilin is indeed axoprotective.
Figure 5.

Retinophilin promotes axonal degeneration. A, Comparison of GFP-labeled axonal maintenance with knockdown of retinophilin in vehicle-treated (top two images) or taxol-treated animals (bottom two images). Genotypes are UAS-DCR/+;elavGAL4, ppkEGFP/+ (Ctrl) and UAS-DCR/+;UAS-rtpRNAi/+;elavGAL4, ppkEGFP/+ (rtp RNAi). Scale bar, 2 μm. B, Quantification of rtp RNAi and control axons as shown in A. For the graph, N = 23, 26, 38, and 33 nerves for each genotype and treatment. *p < 0.05 between taxol-treated groups, Student's t test. C, Images of GFP-labeled axons in vehicle-treated (top two images) or taxol-treated animals (bottom two images) that are of the genotypes ppkCD8GFP/+ (Ctrl) or ppkCD8GFP/+;rtp1/retin1 (rtp). D, Quantification of rtp mutant and control axons as shown in C. For the graph, N = 58, 37, 51, and 40 nerves for each genotype and treatment. **p < 0.01. E, Representative images of olfactory axons. All are labeled with Or82a-gal4,UAS-CD8:GFP. Top, Uncut control (no RNAi); middle, cut control; bottom, rtp RNAi expressed by Or82a-gal4. Scale bar, 15 μm. F, Schematic of quantification scheme for olfactory axotomies. A score of 0 indicates the lack of any detectable axons outside of glomeruli; a score of 4 indicates that axons reach and cross the central commissure. Scores of 1–3 rank axons by number and length of remaining axons or axonal fragments. G, Axotomy scores of the genotypes shown in C. N = 18, 12, and 10 for genotypes quantified. **p < 0.01.
Retinophilin could be specifically involved in chemotherapy-induced neuropathy in the peripheral nervous system, or could play a broader role in injury-induced axonal degeneration. To test whether retinophilin participates in other types of degeneration, we turned to the well established model of olfactory axotomy in Drosophila (MacDonald et al., 2006; Doherty et al., 2009). The cell bodies of olfactory neurons are located in the antenna, and their projections extend into the brain, cross a central commissure, and synapse onto bilateral glomeruli (Fig. 5E, top). By removing the third antennal segment, olfactory axons are severed (MacDonald et al., 2006). We labeled a subset of olfactory axons via expression of GFP driven by the Or82a-GAL4 line (Fishilevich and Vosshall, 2005). Twenty-four hours after axotomy, wild-type axons have degenerated almost completely, with only a few axonal fragments remaining outside the glomeruli (Fig. 5E, middle). In contrast, axons in which retinophilin has been knocked down exclusively within the labeled neurons show a delay in this degeneration, with many axons still extending near the commissure (Fig. 5E, bottom). Using a scoring system to rank the number and length of remaining axons, we find that axon loss is inhibited following rtp knockdown, but much less so than via expression of cyt-mNMNAT1 (NMNAT score = 3.8 ± 0.1) (Fig. 5F,G). Together, these results suggest that Retinophilin promotes the normal axonal degeneration program initiated by both traumatic and toxic axonal injury, and that it functions in a cell-autonomous manner.
The mammalian rtp ortholog Morn4 is required for axonal degeneration
The closest mammalian ortholog to Drosophila retinophilin is Morn4 (50% identity at the amino acid level) (Mecklenburg, 2007). Morn4 is an uncharacterized protein in mouse and human that also contains four MORN repeats. To test whether Morn4 functions like Retinophilin in promoting axonal degeneration, we knocked down Morn4 expression in cultured mouse embryonic DRG neurons and then cut their axons. Control axons swell and then degenerate within the first 12 h, a process that can be quantified using image analysis software that detects axon fragments as circular particles (Sasaki et al., 2009b; Gerdts et al., 2011). Knockdown of Morn4 was very efficient, with only ∼1% of mRNA remaining by quantitative PCR analysis (1.3 ± 0.7%). Knockdown of Morn4 significantly reduced axonal degeneration following axotomy (Fig. 6A,B). Protection was evident for at least 72 h, with knockdown cultures displaying signs of early degeneration (swellings) but no fragmentation. Thus Morn4 may participate in an intermediate step of axonal degeneration, following axonal swelling but preceding fragmentation. The involvement of mouse Morn4 in axonal degeneration highlights the ability of this new Drosophila chemotherapy-induced neuropathy model to identify genes that participate in axonal degeneration in mammals.
Figure 6.

Mouse Morn4 participates in axonal degeneration. A, Representative images of axons from cultured mouse DRG neurons infected with shRNAs targeting luciferase (ctrl) or Morn4. Axons are uncut (top images) or 24 h post-axotomy (bottom images). Scale bar, 100 μm. B, Quantification of degeneration index of control shRNA (red trace)- or Morn4 shRNA (blue trace)-infected axons at 0, 9, 24, 48, and 72 h post-axotomy, and uncut control shRNA-infected axons (black trace). Double asterisks indicate p < 0.001 between cut control shRNA and Morn4 shRNA.
Discussion
Here we describe a novel Drosophila model of chemotherapy-induced peripheral neuropathy and demonstrate that the process shares molecular components with the mammalian axonal degeneration program. We then use this platform to identify the Retinophilin/Morn4 protein as a component of an evolutionarily conserved axonal self-destruction cascade.
A Drosophila model of chemotherapy-induced peripheral neuropathy
Chemotherapy drugs such as paclitaxel (taxol) and vincristine cause a dose-limiting peripheral neuropathy in humans. Taxol causes sensory neuropathy which manifests as pain and loss of sensation in extremities, while vincristine causes both sensory and motor dysfunction. These drugs are used widely in both pediatric and adult patients to treat aggressive metastatic cancer (Sparano, 2000; Bradley and Hussain, 2008; Marsh, 2009; Moore and Pinkerton, 2009; Grossi et al., 2010). Because we know so little about the mechanisms causing this severe side effect, we currently have no way to prevent axonal degeneration in these patients.
Axonal degeneration is not only a component of chemotherapy-induced neuropathy but is also a hallmark of many neurological diseases including hereditary neuropathies, neurodegenerative diseases such as Alzheimer's Disease, and trauma including spinal cord injury. Hence, the need is great to identify therapeutic strategies that could slow or block axon loss and thereby prevent progression of neurological symptoms in these and other disorders. To identify such targets, we have developed a model of toxic neuropathy in Drosophila. Treating larval-stage Drosophila with paclitaxel causes axonal degeneration that is anatomically and molecularly similar to that occurring in rodents and, potentially, in human patients undergoing chemotherapy treatment. In Drosophila, as in humans, paclitaxel preferentially damages sensory rather than motor axons (Park et al., 2011). This conservation provides a platform to identify molecules that could serve as future therapeutic targets.
Retinophilin/Morn4 participates in axonal degeneration
We identified Rtp/Morn4 as an evolutionarily conserved component of the axonal degeneration cascade. Furthermore, we observe that Rtp/Morn4 must be present for efficient degeneration following both neurotoxicity (i.e., chemotherapy administration) and traumatic injury (i.e., axotomy). Hence, while the screen uses a neurotoxin as the axonal damaging agent, it can identify components that function downstream of multiple stimuli. Importantly, our screen was able to identify a subtle effect of rtp on the timing of axonal demise. When using RNA interference to implicate a gene in a process, the ability to detect small changes is useful since gene expression is not completely eliminated. Importantly, we were able to confirm this finding using a true loss-of-function mutant in flies. While identification of a Drosophila gene that promotes axonal degeneration is important, we are most interested in genes with an evolutionarily conserved role in the process. Lentiviral-mediated gene knockdown in mouse sensory neurons (Gerdts et al., 2011) is a powerful secondary screen allowing us to identify such genes. Using these methods in combination, we demonstrate that the role of Rtp/Morn4 in the progression of axonal demise is evolutionarily conserved.
While we do not know the mechanism of action of Morn4 in axonal degeneration, Retinophilin has been implicated in both localization of signaling molecules required for termination of phototransduction (Mecklenburg et al., 2010; Venkatachalam et al., 2010) and regulating cytoplasmic calcium levels preceding phagocytosis (Cuttell et al., 2008). Morn4 contains four membrane occupation and recognition nexus (MORN) repeats that can target proteins to lipid membranes (Ma et al., 2006). Potentially, these domains could bring Morn4 to a membrane surface to modulate lipid activity or second messenger generation. Interestingly, Morn4 has recently been implicated in the neuroprotective process of preconditioning following oxygen-glucose deprivation (Dai et al., 2010). Further work to clarify the molecular role of Morn4 in these two seemingly opposite cascades will be necessary.
Axonal degeneration mechanisms and implications for neurological disease
While we know a great deal about the molecular control of the apoptotic process and how it can contribute to disease, we know comparatively little about axonal degeneration despite its importance in a wide variety of pathological states. Many activating insults ultimately use a common pathway (Vohra et al., 2010) that includes the mitochondrial permeability transition pore (Forte et al., 2007; Barrientos et al., 2011) and can be blocked by axonal expression of NMNAT isoforms (Araki et al., 2004; Conforti et al., 2009; Sasaki et al., 2009a; Yahata et al., 2009; Babetto et al., 2010; Sasaki and Milbrandt, 2010). In addition, kinases including DLK, JNK, IKK, and GSK3 promote axonal degeneration (Miller et al., 2009; Gerdts et al., 2011). The relationships among these proteins, as well as the identity of upstream and downstream components of the axonal degeneration program, are still mysterious.
Screens in both Drosophila and mouse (Gerdts et al., 2011) have the potential to reveal molecular targets for therapeutic intervention to block axonal degeneration in the early stages of neurodegenerative diseases, hereditary neuropathies, or before chemotherapeutic treatment. Here we have used a clinically relevant insult in Drosophila to identify a role for Retinophilin/Morn4 in promoting axonal degeneration. Future screening should yield additional candidates whose inhibition could disable this degenerative process.
Footnotes
This work was supported by NIH Grants NS070053, NS078007, and NS065053 (J.M., A.D.), and NS065615 (M.R.C.B.), as well as the Hope Center for Neurological Disorders (M.R.C.B.) and the Craig H. Neilson Foundation (J.M.). We thank all members of their laboratories for critical discussion and suggestions. In particular, we thank Drs. Bradley Miller and Craig Press for helpful advice, and Gurpreet Seehra for her contributions to the Drosophila screen. We are grateful to Drs. Wesley Grueber, Hugo Bellen, Craig Montell, Joe O'Tousa, Tadashi Uemura, and John Carlson for providing fly stocks and reagents used in this work, Kelli Simburger for cloning assistance, Li-Wei Chang for bioinformatics, and Wandy Beatty for preparation of EM samples. We would also like to thank the Bloomington Stock Center and the Vienna Drosophila RNAi Center for providing numerous stocks.
The authors declare no competing financial interests.
References
- Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- Babetto E, Beirowski B, Janeckova L, Brown R, Gilley J, Thomson D, Ribchester RR, Coleman MP. Targeting NMNAT1 to axons and synapses transforms its neuroprotective potency in vivo. J Neurosci. 2010;30:13291–13304. doi: 10.1523/JNEUROSCI.1189-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos SA, Martinez NW, Yoo S, Jara JS, Zamorano S, Hetz C, Twiss JL, Alvarez J, Court FA. Axonal degeneration is mediated by the mitochondrial permeability transition pore. J Neurosci. 2011;31:966–978. doi: 10.1523/JNEUROSCI.4065-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley DA, Hussain M. Promising novel cytotoxic agents and combinations in metastatic prostate cancer. Cancer J. 2008;14:15–19. doi: 10.1097/PPO.0b013e31816220b7. [DOI] [PubMed] [Google Scholar]
- Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci. 2005;6:889–898. doi: 10.1038/nrn1788. [DOI] [PubMed] [Google Scholar]
- Coleman MP, Freeman MR. Wallerian degeneration, wld(s), and nmnat. Annu Rev Neurosci. 2010;33:245–267. doi: 10.1146/annurev-neuro-060909-153248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins CA, Wairkar YP, Johnson SL, DiAntonio A. Highwire restrains synaptic growth by attenuating a MAP kinase signal. Neuron. 2006;51:57–69. doi: 10.1016/j.neuron.2006.05.026. [DOI] [PubMed] [Google Scholar]
- Conforti L, Wilbrey A, Morreale G, Janeckova L, Beirowski B, Adalbert R, Mazzola F, Di Stefano M, Hartley R, Babetto E, Smith T, Gilley J, Billington RA, Genazzani AA, Ribchester RR, Magni G, Coleman M. Wld S protein requires Nmnat activity and a short N-terminal sequence to protect axons in mice. J Cell Biol. 2009;184:491–500. doi: 10.1083/jcb.200807175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuttell L, Vaughan A, Silva E, Escaron CJ, Lavine M, Van Goethem E, Eid JP, Quirin M, Franc NC. Undertaker, a Drosophila Junctophilin, links Draper-mediated phagocytosis and calcium homeostasis. Cell. 2008;135:524–534. doi: 10.1016/j.cell.2008.08.033. [DOI] [PubMed] [Google Scholar]
- Dai C, Liang D, Li H, Sasaki M, Dawson TM, Dawson VL. Functional identification of neuroprotective molecules. PLoS One. 2010;5:e15008. doi: 10.1371/journal.pone.0015008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, Fellner M, Gasser B, Kinsey K, Oppel S, Scheiblauer S, Couto A, Marra V, Keleman K, Dickson BJ. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007;448:151–156. doi: 10.1038/nature05954. [DOI] [PubMed] [Google Scholar]
- Doherty J, Logan MA, Taşdemir OE, Freeman MR. Ensheathing glia function as phagocytes in the adult Drosophila brain. J Neurosci. 2009;29:4768–4781. doi: 10.1523/JNEUROSCI.5951-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishilevich E, Vosshall LB. Genetic and functional subdivision of the Drosophila antennal lobe. Curr Biol. 2005;15:1548–1553. doi: 10.1016/j.cub.2005.07.066. [DOI] [PubMed] [Google Scholar]
- Forte M, Gold BG, Marracci G, Chaudhary P, Basso E, Johnsen D, Yu X, Fowlkes J, Rahder M, Stem K, Bernardi P, Bourdette D. Cyclophilin D inactivation protects axons in experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis. Proc Natl Acad Sci U S A. 2007;104:7558–7563. doi: 10.1073/pnas.0702228104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J, Sasaki Y, Vohra B, Marasa J, Milbrandt J. Image-based screening identifies novel roles for I{kappa}B kinase and glycogen synthase kinase 3 in axonal degeneration. J Biol Chem. 2011;286:28011–28018. doi: 10.1074/jbc.M111.250472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossi F, Kubota K, Cappuzzo F, de Marinis F, Gridelli C, Aita M, Douillard JY. Future scenarios for the treatment of advanced non-small cell lung cancer: focus on taxane-containing regimens. Oncologist. 2010;15:1102–1112. doi: 10.1634/theoncologist.2010-0322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueber WB, Ye B, Moore AW, Jan LY, Jan YN. Dendrites of distinct classes of Drosophila sensory neurons show different capacities for homotypic repulsion. Curr Biol. 2003;13:618–626. doi: 10.1016/s0960-9822(03)00207-0. [DOI] [PubMed] [Google Scholar]
- Grueber WB, Ye B, Yang CH, Younger S, Borden K, Jan LY, Jan YN. Projections of Drosophila multidendritic neurons in the central nervous system: links with peripheral dendrite morphology. Development. 2007;134:55–64. doi: 10.1242/dev.02666. [DOI] [PubMed] [Google Scholar]
- Hay BA, Wolff T, Rubin GM. Expression of baculovirus P35 prevents cell death in Drosophila. Development. 1994;120:2121–2129. doi: 10.1242/dev.120.8.2121. [DOI] [PubMed] [Google Scholar]
- Hoopfer ED, McLaughlin T, Watts RJ, Schuldiner O, O'Leary DDM, Luo L. Wlds protection distinguishes axon degeneration following injury from naturally occurring developmental pruning. Neuron. 2006;50:883–895. doi: 10.1016/j.neuron.2006.05.013. [DOI] [PubMed] [Google Scholar]
- Lee JJ, Swain SM. Peripheral neuropathy induced by microtubule-stabilizing agents. J Clin Oncol. 2006;24:1633–1642. doi: 10.1200/JCO.2005.04.0543. [DOI] [PubMed] [Google Scholar]
- Ma H, Lou Y, Lin WH, Xue HW. MORN motifs in plant PIPKs are involved in the regulation of subcellular localization and phospholipid binding. Cell Res. 2006;16:466–478. doi: 10.1038/sj.cr.7310058. [DOI] [PubMed] [Google Scholar]
- MacDonald JM, Beach MG, Porpiglia E, Sheehan AE, Watts RJ, Freeman MR. The Drosophila cell corpse engulfment receptor Draper mediates glial clearance of severed axons. Neuron. 2006;50:869–881. doi: 10.1016/j.neuron.2006.04.028. [DOI] [PubMed] [Google Scholar]
- Mack TG, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, Fernando FS, Tarlton A, Andressen C, Addicks K, Magni G, Ribchester RR, Perry VH, Coleman MP. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci. 2001;4:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- Malik B, Stillman M. Chemotherapy-induced peripheral neuropathy. Curr Neurol Neurosci Rep. 2008;8:56–65. doi: 10.1007/s11910-008-0010-5. [DOI] [PubMed] [Google Scholar]
- Marsh S. Pharmacogenomics of taxane/platinum therapy in ovarian cancer. Int J Gynecol Cancer. 2009;19(Suppl 2):S30–S34. doi: 10.1111/IGC.0b013e3181c10513. [DOI] [PubMed] [Google Scholar]
- Massaro CM, Pielage J, Davis GW. Molecular mechanisms that enhance synapse stability despite persistent disruption of the spectrin/ankyrin/microtubule cytoskeleton. J Cell Biol. 2009;187:101–117. doi: 10.1083/jcb.200903166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecklenburg KL. Drosophila retinophilin contains MORN repeats and is conserved in humans. Mol Genet Genomics. 2007;277:481–489. doi: 10.1007/s00438-007-0211-7. [DOI] [PubMed] [Google Scholar]
- Mecklenburg KL, Takemori N, Komori N, Chu B, Hardie RC, Matsumoto H, O'Tousa JE. Retinophilin is a light-regulated phosphoprotein required to suppress photoreceptor dark noise in Drosophila. J Neurosci. 2010;30:1238–1249. doi: 10.1523/JNEUROSCI.4464-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BR, Press C, Daniels RW, Sasaki Y, Milbrandt J, DiAntonio A. A dual leucine kinase-dependent axon self-destruction program promotes Wallerian degeneration. Nat Neurosci. 2009;12:387–389. doi: 10.1038/nn.2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore A, Pinkerton R. Vincristine: Can its therapeutic index be enhanced? Pediatr Blood Cancer. 2009;53:1180–1187. doi: 10.1002/pbc.22161. [DOI] [PubMed] [Google Scholar]
- Park SB, Krishnan AV, Lin CSY, Goldstein D, Friedlander M, Kiernan MC. Mechanisms underlying chemotherapy-induced neurotoxicity and the potential for neuroprotective strategies. Curr Med Chem. 2008;15:3081–3094. doi: 10.2174/092986708786848569. [DOI] [PubMed] [Google Scholar]
- Park SB, Lin CSY, Krishnan AV, Friedlander ML, Lewis CR, Kiernan MC. Early, progressive, and sustained dysfunction of sensory axons underlies paclitaxel-induced neuropathy. Muscle Nerve. 2011;43:367–374. doi: 10.1002/mus.21874. [DOI] [PubMed] [Google Scholar]
- Podratz JL, Staff NP, Froemel D, Wallner A, Wabnig F, Bieber AJ, Tang A, Windebank AJ. Drosophila melanogaster: a new model to study cisplatin-induced neurotoxicity. Neurobiol Dis. 2011;43:330–337. doi: 10.1016/j.nbd.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Press C, Milbrandt J. Nmnat delays axonal degeneration caused by mitochondrial and oxidative stress. J Neurosci. 2008;28:4861–4871. doi: 10.1523/JNEUROSCI.0525-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokop A. Integrating bits and pieces: synapse structure and formation in Drosophila embryos. Cell Tissue Res. 1999;297:169–186. doi: 10.1007/s004410051345. [DOI] [PubMed] [Google Scholar]
- Sasaki Y, Milbrandt J. Axonal degeneration is blocked by nicotinamide mononucleotide adenylyltransferase (Nmnat) protein transduction into transected axons. J Biol Chem. 2010;285:41211–41215. doi: 10.1074/jbc.C110.193904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Araki T, Milbrandt J. Stimulation of nicotinamide adenine dinucleotide biosynthetic pathways delays axonal degeneration after axotomy. J Neurosci. 2006;26:8484–8491. doi: 10.1523/JNEUROSCI.2320-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Vohra BPS, Baloh RH, Milbrandt J. Transgenic mice expressing the Nmnat1 protein manifest robust delay in axonal degeneration in vivo. J Neurosci. 2009a;29:6526–6534. doi: 10.1523/JNEUROSCI.1429-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Vohra BPS, Lund FE, Milbrandt J. Nicotinamide mononucleotide adenylyl transferase-mediated axonal protection requires enzymatic activity but not increased levels of neuronal nicotinamide adenine dinucleotide. J Neurosci. 2009b;29:5525–5535. doi: 10.1523/JNEUROSCI.5469-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenmann Z, Assa-Kunik E, Tiomny S, Minis A, Haklai-Topper L, Arama E, Yaron A. Axonal degeneration is regulated by the apoptotic machinery or a NAD+-sensitive pathway in insects and mammals. J Neurosci. 2010;30:6375–6386. doi: 10.1523/JNEUROSCI.0922-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimono K, Fujimoto A, Tsuyama T, Yamamoto-Kochi M, Sato M, Hattori Y, Sugimura K, Usui T, Kimura K, Uemura T. Multidendritic sensory neurons in the adult Drosophila abdomen: origins, dendritic morphology, and segment- and age-dependent programmed cell death. Neural Dev. 2009;4:37. doi: 10.1186/1749-8104-4-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparano JA. Taxanes for breast cancer: an evidence-based review of randomized phase II and phase III trials. Clin Breast Cancer. 2000;1:32–40. doi: 10.3816/CBC.2000.n.002. discussion 41–42. [DOI] [PubMed] [Google Scholar]
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LSB. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- Tao J, Rolls MM. Dendrites have a rapid program of injury-induced degeneration that is molecularly distinct from developmental pruning. J Neurosci. 2011;31:5398–5405. doi: 10.1523/JNEUROSCI.3826-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatachalam K, Wasserman D, Wang X, Li R, Mills E, Elsaesser R, Li HS, Montell C. Dependence on a retinophilin/myosin complex for stability of PKC and INAD and termination of phototransduction. J Neurosci. 2010;30:11337–11345. doi: 10.1523/JNEUROSCI.2709-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vohra BPS, Sasaki Y, Miller BR, Chang J, DiAntonio A, Milbrandt J. Amyloid precursor protein cleavage-dependent and -independent axonal degeneration programs share a common nicotinamide mononucleotide adenylyltransferase 1-sensitive pathway. J Neurosci. 2010;30:13729–13738. doi: 10.1523/JNEUROSCI.2939-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MS, Davis AA, Culver DG, Glass JD. Wlds mice are resistant to paclitaxel (taxol) neuropathy. Ann Neurol. 2002;52:442–447. doi: 10.1002/ana.10300. [DOI] [PubMed] [Google Scholar]
- Wen Y, Parrish JZ, He R, Zhai RG, Kim MD. Nmnat exerts neuroprotective effects in dendrites and axons. Mol Cell Neurosci. 2011;48:1–8. doi: 10.1016/j.mcn.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windebank AJ. Chemotherapeutic neuropathy. Curr Opin Neurol. 1999;12:565–571. doi: 10.1097/00019052-199910000-00010. [DOI] [PubMed] [Google Scholar]
- Windebank AJ, Grisold W. Chemotherapy-induced neuropathy. J Peripher Nerv Syst. 2008;13:27–46. doi: 10.1111/j.1529-8027.2008.00156.x. [DOI] [PubMed] [Google Scholar]
- Xiong X, Wang X, Ewanek R, Bhat P, DiAntonio A, Collins CA. Protein turnover of the Wallenda/DLK kinase regulates a retrograde response to axonal injury. J Cell Biol. 2010;191:211–223. doi: 10.1083/jcb.201006039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Rasmussen IA, Lagopoulos J, Håberg A. Diffuse axonal injury in severe traumatic brain injury visualized using high-resolution diffusion tensor imaging. J Neurotrauma. 2007;24:753–765. doi: 10.1089/neu.2006.0208. [DOI] [PubMed] [Google Scholar]
- Yahata N, Yuasa S, Araki T. Nicotinamide mononucleotide adenylyltransferase expression in mitochondrial matrix delays Wallerian degeneration. J Neurosci. 2009;29:6276–6284. doi: 10.1523/JNEUROSCI.4304-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao KM, White K. Neural specificity of elav expression: defining a Drosophila promoter for directing expression to the nervous system. J Neurochem. 1994;63:41–51. doi: 10.1046/j.1471-4159.1994.63010041.x. [DOI] [PubMed] [Google Scholar]
- Zhai RG, Zhang F, Hiesinger PR, Cao Y, Haueter CM, Bellen HJ. NAD synthase NMNAT acts as a chaperone to protect against neurodegeneration. Nature. 2008;452:887–891. doi: 10.1038/nature06721. [DOI] [PMC free article] [PubMed] [Google Scholar]