

Abstract
The aryl hydrocarbon receptor (AhR), a ligand-activated member of the basic helix-loop-helix (bHLH)/PER-ARNT-SIM (PAS) transcription superfamily, is known to regulate the toxicity of polyaromatic halogenated hydrocarbon environmental chemicals, most notably dioxin. However, the AhR has also been implicated in multiple stages of tumorigenesis. Medulloblastoma (MB), a primary cerebellar brain tumor arising in infants and children, is thought to originate from abnormally proliferating cerebellar granule neuron precursors (GNPs). GNPs express high levels of the AhR in the external germinal layer of the developing cerebellum. Moreover, our laboratory has previously reported that either abnormal activation or deletion of the AhR leads to dysregulation of GNP cell cycle activity and maturation. These observations led to the hypothesis that the AhR promotes the growth of MB. Therefore, this study evaluated whether the AhR serves a pro-proliferative role in an immortalized MB tumor cell line (DAOY). We produced a stable AhR knockdown DAOY cell line [AhR short hairpin RNA (shRNA)], which exhibited a 70% reduction in AhR protein levels. Compared with wild-type DAOY cells, AhR shRNA DAOY cells displayed an impaired G1-to-S cell cycle transition, decreased DNA synthesis, and reduced proliferation. Furthermore, these cell cycle perturbations were correlated with decreased levels of the pro-proliferative gene Hes1 and increased levels of the cell cycle inhibitor p27kip1. Supplementation experiments with human AhR restored the proliferative activity in AhR shRNA DAOY cells. Taken together, our data show that the AhR promotes proliferation of MB cells, suggesting that this pathway should be considered as a potential therapeutic target for MB treatment.
Introduction
The aryl hydrocarbon receptor (AhR), a ligand-activated member of the bHLH-PER-ARNT-SIM (PAS) transcription factor family, regulates genes associated with cell growth and differentiation (Puga et al., 2005). Polycyclic aromatic hydrocarbons, most notably the potent environmental contaminant 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), mediate their developmental toxicity through binding of AhR, which leads to the activation of gene programs that disrupt normal cell growth. The AhR has also been associated with various stages of carcinogenesis, such as initiation, progression, and migration (Gasiewicz et al., 2008). AhR activation up-regulates xenobiotic-metabolizing enzymes, which have been linked to cancer development through altered metabolic clearing of carcinogenic xenobiotics that may also bioactivate procarcinogens (Gasiewicz et al., 2008). In addition, AhR modulates the expression of growth-associated genes such as c-Myc, Hes1, p21cip1, and p27kip1 (Thomsen et al., 2004; Yang et al., 2005).
Increasing evidence suggests an endogenous role for the AhR in controlling the cell cycle (Puga et al., 2002). For example, mouse embryonic fibroblasts from AhR(−/−) mice exhibit slower growth and accumulation in the G2/M phase of the cell cycle (Elizondo et al., 2000). In addition, stable knockdown of the AhR in human keratinocytes induces expression of p27kip1 and cell cycle arrest (Kalmes et al., 2011). Moreover, AhR expression is elevated in cycling fibroblasts, compared with nondividing fibroblasts (Vaziri et al., 1996). The AhR-regulated signaling pathways responsible for modulating the cell cycle are, in most cases, unknown. Although these investigations provide much evidence that the AhR serves to promote cell growth in certain tissues, considerable data indicate that the effects are likely to be cell- and differentiation stage-specific.
Several studies in tumor cells describe AhR up-regulation and/or activity in the absence of exogenous ligands. For example, AhR is elevated in several rodent and human tumors, including leukemias and mammary tumor cells (Abdelrahim et al., 2003; Hayashibara et al., 2003). Inhibition of AhR also reduced 5-bromo-2′-deoxyuridine incorporation and clonogenic survival in human glioblastoma cells (Gramatzki et al., 2009). Moreover, ectopic expression of AhR in mammary epithelial cells resulted in malignant transformation (Brooks and Eltom, 2011). These studies indicate that AhR has a role in promoting the growth and survival of tumor cells.
Medulloblastoma (MB), one of the most common pediatric malignancies, with prevalence increasing 2 to 3% over the last 30 years, is a primary cerebellar tumor that occurs predominantly in children between ages 5 and 10 (Louis et al., 2007). Five-year survival rates remain less than 50%, and the patients who do survive often have impaired intellectual and physical development (Zakhary et al., 1999). MB is hypothesized to arise from abnormal proliferating cerebellar granule neuron precursors (GNPs) in the external germinal layer (EGL) of the developing cerebellum (Wechsler-Reya and Scott, 2001). Our laboratory has published results suggesting that the AhR is highly expressed and transcriptionally active during the peak proliferative phase of GNP neurogenesis. Moreover, abnormal activation of the AhR by TCDD dysregulated GNP proliferation and maturation, suggesting that the AhR has a role in the proliferation of GNPs (Williamson et al., 2005; Collins et al., 2008).
Common genes are involved in medulloblastoma pathogenesis and GNP proliferation (Fogarty et al., 2005). For example, the Notch signaling pathway, which is up-regulated in MB tissue, promotes proliferation and inhibits cell cycle exit of GNPs, through the induction of the basic helix-loop-helix transcription factor Hes1 (Solecki et al., 2001; Fogarty et al., 2005). Of interest, Hes1 has been reported as an AhR target gene (Thomsen et al., 2004). In the inner EGL, several genes that act as intrinsic promoters of GNP cell cycle exit, including p27kip1 and p21cip1, which have both been suspected in MB pathogenesis, have been identified (Argenti et al., 2005). p27kip1 has also been reported as a transcriptional repression target for Hes1 in embryonic carcinoma cells (Murata et al., 2005).
This study tested the hypothesis that AhR plays a role in MB proliferation. The human MB DAOY cell line served as a model to explore whether AhR promotes MB growth. DAOY cells were shown to express a functional AhR signaling pathway. To examine the role of AhR in the absence of exogenous ligand exposure, we created a stable cell line that has reduced AhR protein levels. Our data demonstrate that reduced AhR signaling in DAOY cells attenuates proliferation, which correlated with decreases in Hes1 and increases in p27kip1 expression. Furthermore, supplementation of human AhR restored DAOY proliferation. Our findings suggest that abnormal AhR activity, in the absence of exogenous ligands, positively promotes MB cell proliferation through a signaling pathway that includes Hes1 and p27kip1.
Materials and Methods
Reagents.
The human medulloblastoma cell line (DAOY) was purchased from American Type Culture Collection (Manassas, VA). TCDD (Cambridge Isotope Laboratories, Inc., Andover, MA) was solubilized in dimethyl sulfoxide (DMSO). DMSO, phenylmethylsulfonyl fluoride, antiprotease cocktail, Triton X-100, trypsin, puromycin, and bovine serum albumin (BSA) were purchased from Sigma-Aldrich (St. Louis, MO). Dulbecco's modified Eagle's medium (DMEM), penicillin/streptomycin, fetal bovine serum, EDTA, and l-glutamine were purchased from Invitrogen (Carlsbad, CA).
DAOY Cell Culture.
The immortalized DAOY cell line was derived from biopsy tissue taken from the posterior fossa of a 4-year-old boy with MB (Jacobsen et al., 1985). DAOY cells were maintained in DMEM containing 10% FBS, 1% l-glutamine (2 nM), and 1% penicillin/streptomycin and kept in a humidified atmosphere of 5% CO2 at 37°C. This medium is referred to as a growth medium, which was replaced every 2 to 3 days.
Immunoblot Analysis.
Cells were harvested for protein analysis in ice-cold PBS supplemented with 0.1% Triton X-100, 1.0% phenylmethylsulfonyl fluoride, 1.0% EDTA, and 1.0% antiprotease cocktail. Protein concentrations were determined by using the micro-bicinchoninic acid protein assay (Thermo Fisher Scientific, Waltham, MA). Total protein (10 μg) was fractionated on 10% polyacrylamide gels and transferred to polyvinylidene difluoride membranes (Bio-Rad Laboratories, Hercules, CA). Then, membranes were blocked with 5% powdered milk containing 0.2% Tween 20 and probed for AhR (1:1000; Enzo Life Sciences, Inc., Farmingdale, NY), Cyp1A1 (1:1000; Santa Cruz Technology, Inc., Santa Cruz, CA), β-actin (1:5000; Sigma-Aldrich), p27kip1 (1:1000; Santa Cruz Technology, Inc.), cyclin D1 (1:1000; Calbiochem, San Diego, CA), p21cip1 (1:1000; Santa Cruz Technology, Inc.), Hes1 (1:1000; Millipore Bioscience Research Reagents, Temecula, CA), MYCN (1:1000; Santa Cruz Technology, Inc.), c-Myc (1:1000; Santa Cruz Technology, Inc.), or GFP (1:2000; Invitrogen) overnight at 4°C. After overnight incubation, membranes were probed with the appropriate horseradish peroxidase-conjugated secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) for 1 h at room temperature. Proteins were visualized with LumiGLO chemiluminescent substrate reagent (Kirkegaard and Perry Laboratories, Gaithersburg, MD). Semiquantitative densitometric analysis of proteins was performed with ImageJ software (http://rsbweb.nih.gov/ij/).
Plasmids.
Lentiviral shRNA transfer plasmid (pLKO.1) targeting human Aryl hydrocarbon receptor (hAhR) mRNA and empty control vector (empty shRNA) were purchased from Open Biosystems (Huntsville, AL). Addgene plasmid 12260 packaging plasmid (psPAX2) and 12259 VSV-G envelope plasmid were a kind gift from Dr. Didier Trono (TronoLab, Lusanne, Switzerland). pcDNA3.1/CT-GFP-TOPO (Invitrogen) vector was used to tag the cDNA coding sequence of hAhR with GFP, enabling our laboratory to confirm positive transfection and to identify the localization of hAhR in live cells. DRE-EGFP reporter plasmid (pGreen1.1) was a kind gift from Dr. Michael Denison (University of California, Davis).
Transient Transfections.
DAOY cells were seeded in six-well plates (BD Falcon; BD Biosciences Discovery Labware, Bedford, MA) at a density of 105 cells/well and were grown to 70% confluence. Then 1 μg of DRE-EGFP DNA plasmid was transiently transfected using 3 μl of FuGENE 6 transfection reagent (Roche Diagnostics, Indianapolis, IN). Twenty-four hours after transfection, cells were exposed to DMSO or 1 or 10 nM TCDD for 24 h. After 24 h of exposure, protein was harvested for immunoblot assay. For hAhR rescue experiments, 1.0 μg of pcDNA3.1/hAhR-CT-GFP-TOPO was transiently transfected using 3.0 μl of FuGENE 6 transfection reagent. After 48 h of transfection, cells were harvested for confocal, immunoblot, and FACS analyses.
Lentivirus Production.
Human embryonic kidney 293FT cells (Invitrogen) were grown to 60 to 80% confluence in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum in T-175 flasks. The VSVG-pseudotyped HIV vector was generated by cotransfection with 7.3 μg of envelope plasmid (pMD2G), 18.2 μg of packaging plasmid (psPAX2), and 18.2 μg of transfer vector (shRNA hAhR), using Lipofectamine LTX (Invitrogen). Medium was changed 6 h after transfection. Viral supernatants were harvested at 48, 72, and 96 h after transfection. Viral supernatants were spun down twice for 5 min at 2000g and filtered through a 0.45-μm low protein-binding filter. Viruses were concentrated 1000-fold using a Lenti-X concentrator (Clontech, Mountain View, CA). The concentrated viruses were stored at −80°C until use.
Stable shRNA Cell Line Generation.
DAOY cells were seeded in 24-well plates (BD Falcon) at a density of 5 × 104 cells/well and grown overnight. Cells were transduced with the appropriate concentrated lentivirus (AhR shRNA or empty shRNA) at a multiplicity of infection of 20. After 48 h of transduction, puromycin (2 μg/ml) was added to the growth medium to select for virally integrated cells. Once cells reached confluence, they were trypsinized and transferred to a six-well culture dish (BD Falcon) and then to a T-75 culture dish (BD Falcon). Puromycin was maintained in the medium during stable knockdown experiments to ensure maximal transgene expression.
Cell Survival Assay.
DAOY cell viability was assessed with the Live/Dead Cytotoxicity Kit (Invitgrogen). Calcein-AM (green) and ethidium homodimer (red) were used to delineate living and dead cells, respectively. DAOY cells were seeded in 24-well plates at a density at 2 × 104 cells/well and then were exposed to DMSO or 1 or 10 nM TCDD for 24 h. Viability was also assessed in the shRNA stable cell lines in the absence of chemicals. Cells were rinsed in Dulbecco's PBS and incubated with serum-free DMEM supplemented with 2 μM calcein-AM and 4 μM ethidium homodimer at 37°C for 30 min. Fluorescence was visualized using a Nikon Eclipse T100 fluorescent microscope (20× magnification). Approximately 600 cells from three randomly selected areas were counted using Image-Pro Plus software (version 6.2; Media Cybernetics, Inc., Bethesda, MD). The percentage of live cells was determined as the ratio of green cells over the total cells stained for green and red. Serum-supplemented medium was used as a negative control, and 70% methanol served as a positive control for cell death.
[3H]Thymidine Incorporation.
DAOY cells were seeded in 96-well plates (BD Falcon) at a density of 3 × 103 cells/well until they reached 60% confluence. Cells were exposed to growth medium containing DMSO, 1 nM TCDD, or 10 nM TCDD for 48 h. For shRNA stable cell line studies, growth medium was replaced after cells reached 60% confluence, and cells were grown for 48 h. Cells were labeled with 1 μCi of [methyl-3H]thymidine (PerkinElmer Life and Analytical Sciences, Waltham, MA) during the last 2 h of incubation and harvested onto filter paper using a Skatron cell harvester. The amount of incorporated radioactivity was quantified by liquid scintillation counting. The incorporated radioactivity was normalized to the amount of protein present in individual wells to account for potential differences in cell density.
Flow Cytometry for Cell Cycle Analysis and Cell Proliferation.
DAOY cells were seeded in six-well plates at a density of 1 × 105 cells/well and were grown to 70% confluence. For TCDD exposure studies, cells were exposed to DMSO or 1 or 10 nM TCDD for 48 h. After exposures, cells were washed with PBS containing 0.5% BSA (FACS buffer), fixed with 70% ethanol, and stored at −20°C until ready for analysis. For shRNA stable cell lines, cells were trypsinized and fixed according to the protocol listed above. On the day of analysis, cells were treated with 1 mg/ml RNase A (Roche Diagnostics), and DNA was stained with 50 μg/ml propidium iodide (Invitrogen). Cells were analyzed with a FACSCantoII flow cytometer using a 488-nm argon blue laser (BD Biosciences, San Diego, CA). A total of 50,000 events were collected per sample. Cell cycle analysis was performed with FlowJo software (Tree Star Inc., Ashland, OR).
DAOY cell proliferation studies were evaluated with the CellTrace CFSE Cell Proliferation Kit (Invitrogen). Cells (3 × 106) were resuspended in 1 ml of PBS containing 0.05% BSA. Then 5 μM carboxyfluorescein diacetate succinimidyl ester (CFSE) was added to the suspension and incubated for 10 min at 37°C. The staining was quenched by addition of 5 volumes of ice-cold growth medium, and stained cells were incubated on ice for 5 min. Cells were pelleted by ultracentrifugation and resuspended in 1 ml of prewarmed growth media. Cells were seeded at a density of 7.5 × 104 cells/well. After a 96-h incubation, cells were trypsinized, pelleted, and resuspended in 0.5 ml of FACS buffer. Cells were analyzed with a FACSCantoII flow cytometer using a 488-nm argon blue laser. A total 50,000 events were collected per sample. Mean fluorescence intensity was obtained with FlowJo software.
Immunocytochemistry.
Cells were seeded in 12-well culture plates (BD Falcon) containing microglass coverslips (VWR, West Chester, PA) on the bottom of each well. After 48 h in culture, cells were rinsed with Dulbecco's PBS and then fixed with 4% paraformaldehyde at room temperature for 30 min. After fixation, cells were blocked in PBS containing 10% normal goat serum and 0.3% Triton X-100 at room temperature for 30 min. Fixed cells were then incubated with an AhR antibody (1:800; Enzo Life Sciences, Inc.) overnight at 4°C. After overnight incubation, cells were washed and incubated with the appropriate Alexa Fluor-conjugated secondary antibody (Invitrogen) for 90 min at room temperature. Nuclei were stained with DAPI (1:5000; Invitrogen) for fixed cell imaging. For live cell imaging, the nuclei were stained with DRAQ5 (1:1000; Cell Signaling Technology, Danvers, MA). Fluorescence was visualized with an Olympus FV1000 laser scanning confocal microscope (University of Rochester). Images were taken with the Olympus FV1000 software program. Colocalization was quantified using the Pearson correlation coefficient by an algorithm that measures the overlap between two fluorochromes in the same specimen (Manders et al., 1993).
Quantitative Real-Time Polymerase Chain Reaction.
For TCDD exposure studies, DAOY cells were seeded in six-well plates at a density of 105 cells/well. After overnight incubation, cells were exposed to DMSO or 1 or 10 nM TCDD for 6, 12, or 24 h. For shRNA stable cell lines, cells were grown until subconfluence. In both experiments, cells were harvested, and RNA was isolated using the RNeasy Mini Kit (QIAGEN, Valencia, CA). RNA (1 μg) was reverse-transcribed into cDNA using the SuperScript III First-Strand cDNA Synthesis Kit (Invitrogen). Real-time quantitative PCR (Q-PCR) was performed using TaqMan probes and primers, which were specific to the gene of interest. PCR mix (20 μl) consisted of 10 ng of cDNA, 200 nM forward primer, 200 nM reverse primer, 10 nM probe, and 1× IQ Supermix (Bio-Rad Laboratories). An initial denaturation was accomplished at 95°C for 2 min. Amplification was achieved by denaturation at 95°C for 30 s and annealing at 60°C for 30 s for 50 cycles. Amplified products were fluorometrically detected during the end of the 60°C annealing using an iCycler analyzer (Bio-Rad Laboratories). Relative mRNA expression was quantified using a previously published algorithm (Gilliland et al., 1990). Forward, reverse, and probe sequences for Q-PCR were as follows: Hes1, forward 5′-AGGCGGACATTCTGGAAATG-3′, reverse 5′-CGGTACTTCCCCAGCACACTT-3′, and probe 5′FAM-AGTGAAGCACCTCCGGAACCTGCAG-BHQ-3′; Cyp1A1, forward 5′-CACAGACAGCCTGATTGAGCA-3′, reverse 5′-GTGACTGTGTCAAACCCAGCTCCAAAGA-3′, and probe 5′FAM-AAGCAGCTGGATGAGAACGCCAATG-BHQ-3′; p27kip1, forward 5′-CCGGTGGACCACGAAGAGT-3′, reverse 5′-GCTCGCCTCTTCCATGTCTC-3′, and probe 5′FAM-AACCCGGGACTTGGAGAAGCACTGC-BHQ-3′; and GAPDH, forward 5′-TCAAGAAGGTGGTGAAGCAG-3′, reverse 5′-CGCTGTTGAAGTCAGAGGAG-3′, and probe 5′FAM-CCTCAAGGGCATCCTGGGCTACACT-BHQ-3′. Primers and probes were purchased from Biosearch Technologies (Novato, CA).
Statistical Analyses.
Data are expressed as means ± S.E.M. from a minimum of three independent experiments. Sample sizes are indicated in the figure legends. Statistical analyses were performed by Student's t tests. p < 0.05 was considered statistically significant.
Results
AhR Is Expressed and Transcriptionally Active in DAOY Human Medulloblastoma Cells.
The primary objective of this study was to elucidate whether the AhR signaling pathway contributes to the abnormal/unbalanced proliferation of medulloblastoma cells and begin to identify the mechanisms by which this occurs. We confirmed that the AhR protein is expressed in human DAOY MB cells (Fig. 1A). Mouse hepatoma cell lysates (Hepa) served as a positive control for AhR expression (Fig. 1A). Human AhR had a predicted molecular mass of 105 kDa, whereas mouse AhR had a predicted molecular mass of 95 kDa.
Fig. 1.

AhR is expressed and transcriptionally active in DAOY human medulloblastoma cells. A, protein was harvested from DAOY cells, and protein (10 μg) was separated by SDS-polyacrylamide gel electrophoresis and then analyzed for AhR expression by immunoblot. B, DAOY cells were treated with TCDD, and RNA was harvested as designated time points. Gene expression was normalized to GAPDH. C, cells were exposed to TCDD for 24 h, and protein (50 μg) was separated by SDS-polyacrylamide gel electrophoresis and then analyzed for Cyp1A1 and actin expression by immunoblot. D, DAOY cells were transfected with the DRE-GFP reporter construct for 24 h followed by TCDD treatment for 24 h. Protein was harvested and processed for GFP and actin detection by immunoblot. Data represent the mean ± S.E.M. (n = 4; *, p < 0. 05, significantly different from DMSO; Student's t test).
To establish that AhR is responsive and transcriptionally active, DAOY cells were exposed to TCDD, a high-affinity exogenous agonist. After 1 and 10 nM TCDD exposure, DAOY cells were harvested for mRNA analysis of the known AhR target gene, Cyp1A1 (Kress and Greenlee, 1997). After 24 h of 10 nM TCDD exposure, there was an ∼15-fold increase in Cyp1A1 mRNA (Fig. 1B). There was no statistical difference between the 1 and 10 nM TCDD-mediated increase in Cyp1A1 mRNA levels, which may be due to a maximum response ceiling effect of AhR-induced transcription. Immunoblot analysis of Cyp1A1 further confirmed that activation of the AhR signaling pathway resulted in the translation of AhR target genes (Fig. 1C).
An AhR activity reporter construct was used to further confirm that the AhR signaling is being activated in DAOY cells. The pGreen1.1 construct contains several aryl hydrocarbon response elements upstream of the EGFP coding sequence, which, when in the presence of an activated AhR complex, will result in the transcription of GFP protein molecules (Han et al., 2004). DAOY cells were transiently transfected with pGreen1.1 for 24 h and then were exposed to TCDD for 24 h, and protein was harvested for immunoblot analysis of GFP (Fig. 1D). TCDD exposure induced an ∼3-fold induction of GFP compared with that for vehicle-exposed cells. Taken together, these results suggest that AhR is transcriptionally active in DAOY cells after exposure to an exogenous ligand.
AhR Is Localized in the Nucleus and Transcriptionally Active Independent of Exogenous Ligand Exposure in MB Cells.
We hypothesize that the AhR promotes abnormal growth of MB cells. Therefore, it would be expected that AhR would be localized in the nucleus and be transcriptionally active independent of an exogenous ligand. Cellular localization of AhR in DAOY cells was evaluated by immunocytochemistry. Representative images indicate that the AhR was localized both in the nucleus and cytoplasm of DAOY cells. These two distinct populations can be seen readily from the dot plot (Fig. 2A). Using the Pearson correlation coefficient (rp) to determine colocalization of two fluorophores, we found an ∼69.5% association of AhR with the nuclear stain DAPI. This association is considered to be statistically significant on the basis of previous publications using this algorithm (Manders et al., 1993).
Fig. 2.

AhR is localized in the nucleus and is transcriptionally active independent of exogenous ligand exposure. A, DAOY cells were grown on glass coverslips and fixed with 4% paraformaldehyde. A–C, representative confocal images at 20× magnification. D–F, representative confocal images at 20× with 3× zoom. Red, AhR; blue, DAPI. Colocalization is shown in the merge panel depicting two representative DAOY cells. The graph on the right indicates overlap of the two fluorophores. B, DAOY cells were transfected with the DRE-GFP reporter construct for 24 h. Protein was harvested, and immunoblot analysis was performed for the detection of GFP and β-actin. Data represent the mean ± S.E.M. (n = 3; *, p < 0.001, significantly different from mock; Student's t test).
To confirm that the AhR was transcriptionally active independent of exogenous ligand exposure, DAOY cells were transiently transfected with the pGreen1.1 reporter construct. After 24 h of transfection, DAOY cells were harvested for immunoblot analysis of GFP. As expected, the mock-transfected group was devoid of GFP expression, whereas DAOY cells transfected with the pGreen 1.1 reporter construct exhibited an ∼8-fold induction in GFP protein expression (Fig. 2B). These results indicate that the AhR is localized in the nucleus and is transcriptionally active in DAOY human medulloblastoma cells in the absence of exogenous ligand.
Knockdown of the AhR in DAOY Cells Results in G1 Cell Cycle Arrest and Reduced Proliferation.
To understand whether the AhR promotes the abnormal growth of medulloblastoma cells, an shRNA lentiviral approach served to down-regulate AhR expression in DAOY cells. Stable cell lines were generated by transducing DAOY cells (mock) with lentiviral particles that encoded either an shRNA vector targeting human AhR (AhR shRNA) or an empty shRNA vector (empty shRNA). Compared with the empty shRNA DAOY cell line, the AhR shRNA stable cell line exhibited an ∼70% reduction in AhR protein levels (Fig. 3A). This decrease in AhR protein expression persisted between several cell passages (data not shown). Moreover, AhR shRNA DAOY cell survival did not differ compared with that of mock DAOY cells (Fig. 3B).
Fig. 3.
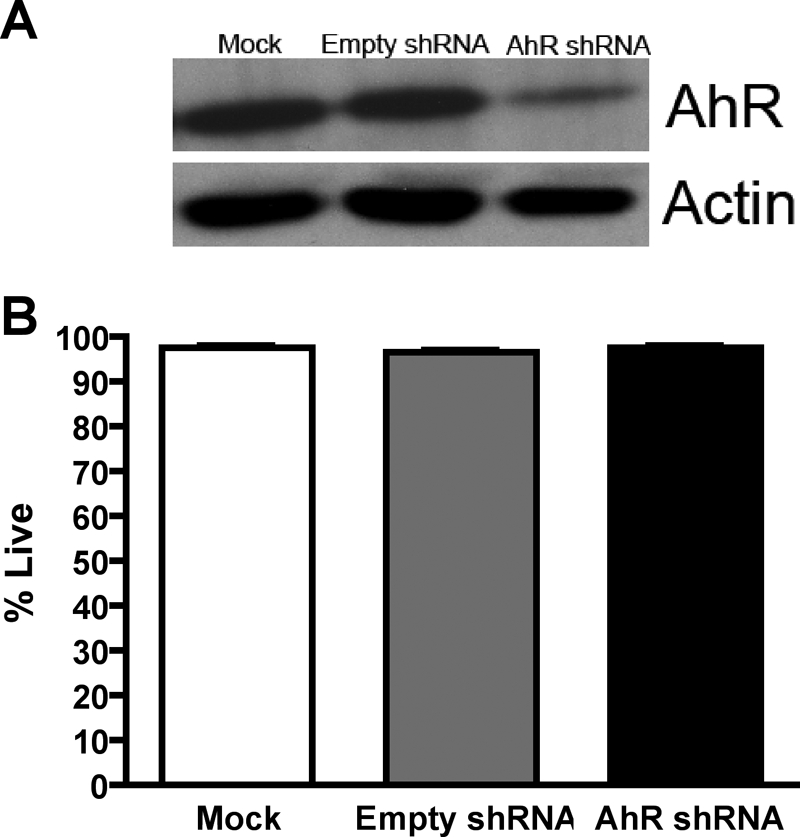
Lentiviral-mediated knockdown of AhR does not have an impact on DAOY cell survival. A, an AhR knockdown stable cell line was generated, and protein was harvested. Protein (10 μg) was separated by SDS-polyacrylamide gel electrophoresis and then was analyzed for AhR expression by immunoblot. Densitometric analysis was performed with ImageJ software. B, neuron viability was assessed with the Live/Dead Cytotoxicity kit. Data represent the mean ± S.E.M. (n = 3).
We next evaluated whether down-regulation of AhR in DAOY cells had an impact on MB cell cycle activity. Mock, AhR shRNA, and empty shRNA DAOY cells were grown in complete growth medium until 70% confluent and then were harvested and analyzed for cell cycle distribution via PI staining. Down-regulation of the AhR led to an ∼17% increase in the percentage of cells in the G0/G1 phase of the cell cycle, which was accompanied by an ∼17% decrease in the percentage of cells in the S phase of the cell cycle (Fig. 4A). To further confirm that knockdown of the AhR resulted in fewer cells entering the S phase, we investigated [3H]thymidine incorporation into AhR shRNA cells. Compared with empty shRNA DAOY cells, AhR shRNA DAOY cells incorporated ∼50% less [3H]thymidine (Fig. 4B).
Fig. 4.

Knockdown of the AhR in DAOY cells results in G1 cell cycle arrest and reduced proliferation. A, DAOY cells were harvested into a single cell suspension, fixed and stained with PI. Cells were acquired on a FACSCantoII flow cytometer, and data were analyzed with FlowJo software. Histograms are presented on the left, and graphed values are located on the right. B, cells were pulsed with 1 μCi of [methyl-3H]thymidine for 2 h. Values were normalized to the amount of protein in each well. C, CFSE-stained DAOY cells were grown for 96 h. Stained cells were analyzed with a FACSCantoII flow cytometer, and a mean CFSE intensity was calculated with FlowJo software. A graphical representation of mean CFSE staining is under the histogram. Data represent the mean ± S.E.M. (n = 3; *, p < 0.05; **, p < 0.005; ***, p < 0.001, significantly different from empty shRNA; Student's t test).
To further characterize the impact of AhR knockdown on cell cycle activity, we evaluated the number of mitotic events in AhR shRNA DAOY cells. CFSE is a membrane-permeable dye that is nontoxic and is commonly used to measure the amount of cell divisions (Hodgkin et al., 1996). The label is inherited by daughter cells after mitosis, diluting the dye in half. Therefore, increased numbers of cell divisions are inversely proportional to the levels of dye that will be present in each cell (Fig. 4C). AhR shRNA DAOY cells, mock DAOY cells, and empty shRNA DAOY cells were labeled with CFSE, grown for 96 h, and subsequently harvested for FACS analysis to quantify the intensity of the CFSE dye. AhR shRNA DAOY cells exhibited an ∼4-fold increase in the intensity of CFSE dye compared with that of empty shRNA DAOY cells (Fig. 4C). These results suggest that the AhR contributes to proliferation of MB tumor cells.
Hes1 Protein Expression Is Decreased after AhR Reduction in DAOY Cells.
Hes1 promotes proliferation of GNPs and subsequently is involved in the pathogenesis of MB (Solecki et al., 2001; Fogarty et al., 2005). In addition, Hes1 has been established as an AhR target gene (Thomsen et al., 2004). Therefore, we investigated whether Hes1 expression was altered after the knockdown of AhR in DAOY cells. Q-PCR analysis revealed a 30% reduction in Hes1 mRNA expression after knockdown of AhR in DAOY cells, but the decrease in Hes1 mRNA was not statistically significant (Fig. 5A). However, immunoblot analysis demonstrated a 50% decrease in Hes1 protein expression in AhR shRNA DAOY cells (Fig. 5B). These data suggest that AhR could regulate Hes1 expression in MB DAOY cells to promote growth.
Fig. 5.
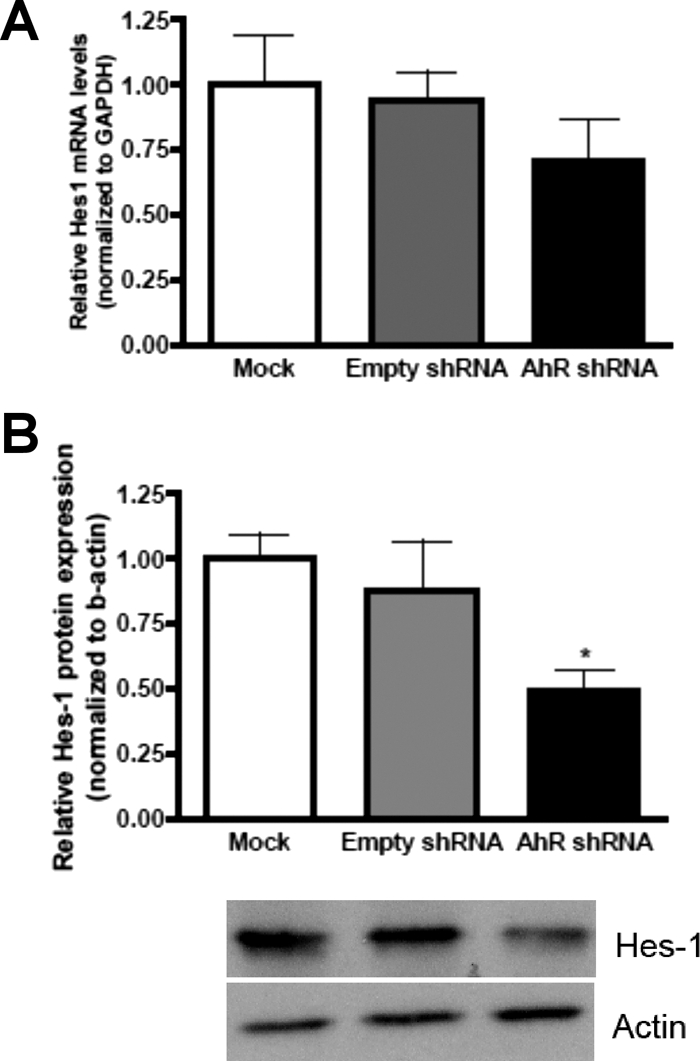
Hes1 protein expression is decreased after AhR reduction in DAOY cells. A, Hes1 mRNA was quantified by Q-PCR and normalized to GAPDH mRNA expression. B, Hes1 protein expression was quantified with ImageJ software and normalized to β-actin expression. Data represent the mean ± S.E.M. (n = 3; *, p < 0.05, significantly different from empty shRNA; Student's t test).
Loss of AhR in DAOY Cells Results in Increased p27kip1 mRNA and Protein Expression.
To further evaluate the cell cycle perturbations seen after reduction of AhR, p27kip1 was investigated because Hes1 has been shown to transcriptionally repress p27kip1 expression to promote proliferation (Murata et al., 2005). Therefore, we hypothesized that decreasing AhR levels would have a positive impact on p27kip1 expression through decreased Hes1 expression. There was an ∼2-fold increase in p27kip1 mRNA expression, which was accompanied by an ∼6-fold increase in p27kip1 protein expression (Fig. 6, A and B). In addition, we investigated the cell cycle regulatory proteins, cyclin D1, p21cip1, and c-Myc because they regulate GNP proliferation and subsequently are implicated in MB pathogenesis. We found no statistical differences in these proteins after densitometric quantification when AhR protein expression was reduced in DAOY cells (Fig. 6C). Taken together, these results demonstrate that down-regulation of AhR in DAOY MB cells is correlated with an elevation in the cell cycle arrest protein p27kip1, whereas other cell cycle proteins investigated remained unchanged.
Fig. 6.

Loss of AhR in DAOY cells results in increased p27kip1 mRNA and protein expression. A, p27kip1 mRNA was quantified by Q-PCR and normalized to GAPDH mRNA expression. B, p27kip1 protein expression was quantified with ImageJ software and normalized to β-actin expression. C, cyclin D, p21cip1, and c-Myc protein levels were assayed via immunoblot. Statistically significant differences were not observed after quantification with ImageJ software. Data represent the mean ± S.E.M. (n = 3; *, p < 0.05, significantly different from empty shRNA; Student's t test).
Supplementation of Human AhR in AhR shRNA DAOY Cells Increased Percentage of Cells in S Phase of Cell Cycle.
Supplementation experiments were performed to evaluate whether the replenishment of hAhR into AhR shRNA DAOY cells restores the proliferative capacity of MB cells. Transient transfection of shRNA AhR DAOY cells with hAhR for 48 h resulted in increased AhR expression (Fig. 7A). Furthermore, live cell confocal analysis indicated that supplemented hAhR was localized in the nucleus of AhR shRNA DAOY cells (Fig. 7B). After 48 h of transfection, AhR shRNA DAOY cells were harvested for cell cycle analysis via PI staining. Consistent with our previous observations, there was an increased population of cells in the G0/G1 phase when AhR protein expression is knocked down in DAOY cells (Fig. 7C). Compared with AhR shRNA DAOY cells, AhR shRNA DAOY cells supplemented with hAhR displayed an increased percentage of cells in the S phase (Fig. 6C). These results suggest that AhR contributed to the proliferation of MB cells by controlling the G0/G1 to S cell cycle transition.
Fig. 7.

Supplementation of human AhR in AhR shRNA DAOY cells increased the percentage of cells in the S phase of the cell cycle. A, after 48 h of transfection of hAhR, protein was harvested, and protein (10 μg) was separated by SDS-polyacrylamide gel electrophoresis and then analyzed for AhR and β-actin expression by Western blot. B, A–C, representative live cell confocal images (20× magnification) of AhR shRNA DAOY cells transfected with hAhR-GFP; D–F, 20× with 3× zoom image of one cell showing colocalization of hAhR within the nucleus. Green, hAhR-GFP; red, DRAQ5. C, DAOY AhR shRNA cells were harvested into a single cell suspension, fixed, and stained with PI. Cells were acquired on a FACSCantoII flow cytometer, and data were analyzed using FlowJo software. Data represent the mean ± S.E.M. (n = 3; *, p < 0.05; **, p < 0.001, significantly different from empty shRNA; ⋀, p < 0.05; ⋀⋀, p < 0.01; ⋀⋀⋀, p < 0.001, significantly different from AhR shRNA; Student's t test).
Increased AhR Expression in AhR shRNA DAOY Cells Correlates with Increased Hes1 and Decreased p27kip1 Levels.
Because proliferative activity was restored after the supplementation of hAhR in AhR shRNA DAOY cells, we evaluated whether Hes1 and p27kip1 expression patterns would correlate with the increased number of cells in the S phase in these cells. Densitometric quantification revealed an ∼75% increase in Hes1 protein expression in AhR shRNA DAOY cells supplemented with hAhR compared with AhR shRNA (Fig. 8B). Furthermore, an ∼72% decrease in p27kip1 expression was seen in AhR shRNA DAOY cells supplemented with hAhR (Fig. 8C). These results suggest that AhR had a positive impact on Hes1 and negatively influenced p27kip1 expression.
Fig. 8.

Increased AhR expression in AhR shRNA DAOY cells correlates with an increase in Hes1 expression and a decrease in p27kip1 expression. A, representative Western blot showing AhR, Hes1, p27kip1, and β-actin expression. B, Hes1 protein expression was quantified with ImageJ software and normalized to β-actin expression. C, p27kip1 protein expression was quantified with ImageJ software and normalized to β-actin expression. Data represent the mean ± S.E.M. (n = 3; *, p < 0.05, significantly different from empty shRNA; ⋀, p < 0.05, significantly different from AhR shRNA; Student's t test).
Discussion
Our laboratory previously established that the AhR is robustly expressed and transcriptionally active during the expansion phase of GNPs in the developing cerebellum (Williamson et al., 2005). Moreover, abnormal activation of the AhR by dioxin interfered with GNP proliferation and differentiation, suggesting that the AhR has some involvement in the maturation of these progenitors (Williamson et al., 2005; Collins et al., 2008). Of interest, MB tumors are thought to arise from GNPs that lose growth control (Wechsler-Reya and Scott, 2001). Therefore, we tested the hypothesis that AhR activity influences abnormal growth of DAOY cells, an immortalized cell culture model that was derived from a patient with MB (Jacobsen et al., 1985). We determined that the AhR is highly expressed in the absence of exogenous ligand exposure in DAOY cells. The AhR was also transcriptionally active in the absence and presence of exogenous ligand exposure. Moreover, the reduction of AhR protein expression in DAOY cells attenuated the proliferative capacity and DNA synthesis, possibly through a signaling mechanism involving Hes1 and/or p27kip1. These observations suggest that a signaling pathway involving AhR, Hes1, and p27kip1 plays an important, if not major, role in promoting growth of MB cancer cells.
Several studies have indicated that the AhR functions endogenously to regulate normal cell growth independent of exogenous ligand treatment (Puga et al., 2002). AhR was shown to regulate cell cycle progression in human breast cancer cells via a functional interaction with cyclin-dependent kinase 4, but treatment with TCDD disrupted this complex, resulting in cell cycle arrest (Barhoover et al., 2010). However, there have been conflicting reports regarding the function of AhR as a cell cycle regulator, which may be due to differential responses in various cell types. For example, AhR overexpression in neuroblastoma cells resulted in a neuronal phenotype as a result of augmented differentiation (Akahoshi et al., 2006). AhR has been reported to control the expression of genes that influence growth and differentiation, which are dysregulated in cancer, suggesting that AhR may be involved in tumor development and progression (Thomsen et al., 2004; Yang et al., 2005).
The AhR has been reported to be overexpressed in several cancers, including human lung and gastric carcinomas (Lin et al., 2003; Peng et al., 2009). In addition, expression of a constitutively active AhR variant in transgenic mice causes pro-proliferative effects, such as induction of stomach tumors and the promotion of hepatocarcinogenesis (Andersson et al., 2002; Moennikes et al., 2004). We determined that the AhR is highly expressed in human MB cells (Fig. 1). Furthermore, AhR was consistently localized in the nucleus and was transcriptionally active either in the presence or absence of exposure to an exogenous ligand (Fig. 2). In addition, several investigators have reported that the AhR is expressed at higher levels and localized predominantly in the nucleus of transformed cells compared with nontransformed cells (Yang et al., 2005). We also detected AhR in the cytoplasm of DAOY cells (Fig. 2). However, this would be expected because before activation, AhR normally resides in the cytoplasm (Hankinson, 1995). Upon activation by endogenous pathways, the AhR subsequently translocates to the nucleus and regulates transcription. These observations strongly suggest that AhR activation by endogenous pathways may play a role in promoting MB growth.
After AhR knockdown, DAOY cells exhibited decreased proliferation, diminished DNA synthesis, and a higher accumulation in the G0/G1 phase of the cell cycle (Fig. 4). However, proliferation was not completely abrogated, and there was a modest yet statistically significant percentage change in the G0/G1 and S phases of the cell cycle. A few possibilities could explain our observations in cell proliferation after reduction of AhR in DAOY cells. First, the AhR is unlikely to be a master regulator of proliferation in MB cells but may function in concert with other signaling pathways to promote abnormal growth of these cells. There may also be compensatory proliferative signals that result from the permanent decrease in AhR expression. In addition, we did not observe complete knockdown (70%) of the AhR after shRNA lentiviral-mediated stable cell line generation. Nevertheless, we observed an ∼4-fold reduction in DAOY cell proliferation over a 96-h time frame (Fig. 4), which could have implications for MB growth.
As mentioned above, MB is thought to arise from abnormally dividing GNPs residing in the EGL. Therefore, common genes such as those regulated by the Notch pathway are involved in medulloblastoma pathogenesis and GNP proliferation (Wechsler-Reya and Scott, 2001; Fogarty et al., 2005). The Notch signaling cascade initiates transcription of target genes; most characterized are the Hairy and Enhancer of Split homologs Hes1 and Hes5 (Solecki et al., 2001). Of interest, Hes1 is also an AhR target gene (Thomsen et al., 2004). In our study, Hes1 protein expression was decreased when AhR expression was knocked down in DAOY cells (Fig. 5). Hes1 functions to transcriptionally activate pro-proliferative genes, such as cyclin D1 and D2 (Fogarty et al., 2005). On the basis of our proliferation experiments, we would have expected cyclin D1 to decrease after AhR down-regulation. However, we did not observe differences in cyclin D1 protein expression after down-regulation of AhR in DAOY cells (Fig. 6). Cyclin proteins are mitogen-activated, and their activation triggers entry into the S phase of the cell cycle. Therefore, perhaps the activities of the cyclins were diminished, rather than protein expression. However, Hes1 also functions to repress cell cycle inhibitory molecules such as the cyclin-dependent kinase inhibitor p27kip1 to maintain cell proliferation (Solecki et al., 2001). We identified AhR-dependent (directly or indirectly) expression of p27kip1 in DAOY cells (Fig. 6).
Low-level p27kip1 expression has been associated with poor prognosis in a variety of neuronal tumors, including astrocytoma, glioblastoma, and neuroblastoma (Chu et al., 2008). Although MB tumors have not been investigated for p27kip1 expression, a recent report indicated that an MB mouse model expressing a mutant p27kip1 allele developed an accelerated and more severe form of MB (Ayrault et al., 2009). Therefore, we evaluated whether AhR affected p27kip1 levels, which could ultimately modulate MB growth. Our results suggest that AhR is associated with reduced p27kip1 expression, which would be consistent with promoting MB growth, possibly through Hes1 induction (Fig. 6, A and B). However, we cannot conclude that the observed differences in p27kip1 expression are directly related to Hes1 or AhR-mediated gene transcription. AhR could be destabilizing p27kip1 mRNA and/or protein. As an alternative, AhR could be repressing p27kip1 promoter activity or positively regulating a transcription factor that negatively regulates p27kip1, such as Hes1. Because cyclin-dependent kinase 2 activity is required for entry into the S phase, its inhibition by p27kip1 could ultimately result in G0/G1 cell cycle arrest (Kolluri et al., 1999). Regardless of the mechanisms involved, our experiments provide compelling reasons to further investigate the role of AhR in the regulation of expression of Hes1, p27kip1, and the MB cell cycle.
We investigated the GNP cell cycle regulatory molecules p21cip1 and c-Myc because they have been shown to regulate GNP proliferation and differentiation and are also suspected to be involved in MB pathogenesis (Yang et al., 2005; Wechsler-Reya and Scott, 2001). Furthermore, p21cip1 and c-Myc are suspected to be AhR target genes (Gasiewicz et al., 2008). However, immunoblot quantification revealed no significant changes in the expression of c-Myc and p21cip1 after AhR reduction in DAOY cells (Fig. 7C). These data further support the contention that the AhR and Notch signaling pathways may converge to promote MB proliferation, possibly through regulation of p27kip1.
Recent studies have demonstrated a relationship between AhR activity/expression and tumorigenesis. AhR gene polymorphisms have been associated with an increased risk of lung, breast, and pancreatic tumors in humans (Kim et al., 2007; Long et al., 2006). Of interest, substantial evidence regarding AhR activity and tumor growth has resulted in an increased effort for targeting this transcription factor for cancer therapy (Koliopanos et al., 2002). We propose that the AhR has a role in promoting the proliferation of MB cells, specifically through a mechanism that involves Hes1 and p27kip1 expression. Our observations suggest that the AhR has the potential of acting as a proto-oncogene in MB cells and warrants future mechanistic studies concerning the relationship between AhR and MB progression. Therefore, the AhR represents another potential therapeutic target to consider for MB treatment.
Acknowledgments
We thank Drs. Thomas Gasiewicz, Sarah Latchney, and Nina Schor for helpful comments during the preparation of the manuscript. We also acknowledge Bryan Thompson for assistance in the laboratory.
This research was supported by the National Institutes of Health National Institute of Environmental Health Sciences [Grants R01-ES016357, P30-ES01247, T32-ES07026] and The Bristol-Myers Squibb Pharmaceutical Research Institute.
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
- AhR
- aryl hydrocarbon receptor
- bHLH
- basic helix-loop-helix
- TCDD
- 2,3,7,8-tetrachlorodibenzo-p-dioxin
- MB
- medulloblastoma
- GNP
- cerebellar granule neuron precursors
- EGL
- external germinal layer
- DMSO
- dimethyl sulfoxide
- BSA
- bovine serum albumin
- DMEM
- Dulbecco's modified Eagle's medium
- PBS
- phosphate-buffered saline
- GFP
- green fluorescent protein
- shRNA
- short hairpin RNA
- hAhR
- human aryl hydrocarbon receptor
- EGFP
- enhanced green fluorescent protein
- FACS
- fluorescence-activated cell sorting
- CFSE
- carboxyfluorescein diacetate succinimidyl ester
- DAPI
- 4,6-diamidino-2-phenylindole
- PCR
- polymerase chain reaction
- Q-PCR
- quantitative real-time PCR
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- PI
- propidium iodide.
Authorship Contributions
Participated in research design: Dever and Opanashuk.
Conducted experiments: Dever.
Contributed new reagents or analytic tools: Dever and Opanashuk.
Performed data analysis: Dever and Opanashuk.
Wrote or contributed to the writing of the manuscript: Dever and Opanashuk.
References
- Abdelrahim M, Smith R, 3rd, Safe S. (2003) Aryl hydrocarbon receptor gene silencing with small inhibitory RNA differentially modulates Ah-responsiveness in MCF-7 and HepG2 cancer cells. Mol Pharmacol 63:1373–1381 [DOI] [PubMed] [Google Scholar]
- Akahoshi E, Yoshimura S, Ishihara-Sugano M. (2006) Over-expression of AhR (aryl hydrocarbon receptor) induces neural differentiation of Neuro2a cells: neurotoxicology study. Environ Health 5:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson P, McGuire J, Rubio C, Gradin K, Whitelaw ML, Pettersson S, Hanberg A, Poellinger L. (2002) A constitutively active dioxin/aryl hydrocarbon receptor induces stomach tumors. Proc Natl Acad Sci USA 99:9990–9995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argenti B, Gallo R, Di Marcotullio L, Ferretti E, Napolitano M, Canterini S, De Smaele E, Greco A, Fiorenza MT, Maroder M, et al. (2005) Hedgehog antagonist RENKCTD11 regulates proliferation and apoptosis of developing granule cell progenitors. J Neurosci 25:8338–8346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayrault O, Zindy F, Rehg J, Sherr CJ, Roussel MF. (2009) Two tumor suppressors, p27Kip1 and patched-1, collaborate to prevent medulloblastoma. Mol Cancer Res 7:33–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barhoover MA, Hall JM, Greenlee WF, Thomas RS. (2010) Aryl hydrocarbon receptor regulates cell cycle progression in human breast cancer cells via a functional interaction with cyclin-dependent kinase 4. Mol Pharmacol 77:195–201 [DOI] [PubMed] [Google Scholar]
- Brooks J, Eltom SE. (2011) Malignant transformation of mammary epithelial cells by ectopic overexpression of the aryl hydrocarbon receptor. Curr Cancer Drug Targets 11:654–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu IM, Hengst L, Slingerland JM. (2008) The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer 8:253–267 [DOI] [PubMed] [Google Scholar]
- Collins LL, Williamson MA, Thompson BD, Dever DP, Gasiewicz TA, Opanashuk LA. (2008) 2,3,7,8-Tetracholorodibenzo-p-dioxin exposure disrupts granule neuron precursor maturation in the developing mouse cerebellum. Toxicol Sci 103:125–136 [DOI] [PubMed] [Google Scholar]
- Elizondo G, Fernandez-Salguero P, Sheikh MS, Kim GY, Fornace AJ, Lee KS, Gonzalez FJ. (2000) Altered cell cycle control at the G2/M phases in aryl hydrocarbon receptor-null embryo fibroblast. Mol Pharmacol 57:1056–1063 [PubMed] [Google Scholar]
- Fogarty MP, Kessler JD, Wechsler-Reya RJ. (2005) Morphing into cancer: the role of developmental signaling pathways in brain tumor formation. J Neurobiol 64:458–475 [DOI] [PubMed] [Google Scholar]
- Gasiewicz TA, Henry EC, Collins LL. (2008) Expression and activity of aryl hydrocarbon receptors in development and cancer. Crit Rev Eukaryot Gene Expr 18:279–321 [DOI] [PubMed] [Google Scholar]
- Gilliland G, Perrin S, Blanchard K, Bunn HF. (1990) Analysis of cytokine mRNA and DNA: detection and quantitation by competitive polymerase chain reaction. Proc Natl Acad Sci USA 87:2725–2729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramatzki D, Pantazis G, Schittenhelm J, Tabatabai G, Köhle C, Wick W, Schwarz M, Weller M, Tritschler I. (2009) Aryl hydrocarbon receptor inhibition downregulates the TGF-β/Smad pathway in human glioblastoma cells. Oncogene 28:2593–2605 [DOI] [PubMed] [Google Scholar]
- Han D, Nagy SR, Denison MS. (2004) Comparison of recombinant cell bioassays for the detection of Ah receptor agonists. Biofactors 20:11–22 [DOI] [PubMed] [Google Scholar]
- Hankinson O. (1995) The aryl hydrocarbon receptor complex. Annu Rev Pharmacol Toxicol 35:307–340 [DOI] [PubMed] [Google Scholar]
- Hayashibara T, Yamada Y, Mori N, Harasawa H, Sugahara K, Miyanishi T, Kamihira S, Tomonaga M. (2003) Possible involvement of aryl hydrocarbon receptor (AhR) in adult T-cell leukemia (ATL) leukemogenesis: constitutive activation of AhR in ATL. Biochem Biophys Res Commun 300:128–134 [DOI] [PubMed] [Google Scholar]
- Hodgkin PD, Lee JH, Lyons AB. (1996) B cell differentiation and isotype switching is related to division cycle number. J Exp Med 184:277–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen PF, Jenkyn DJ, Papadimitriou JM. (1985) Establishment of a human medulloblastoma cell line and its heterotransplantation into nude mice. J Neuropathol Exp Neurol 44:472–485 [DOI] [PubMed] [Google Scholar]
- Kalmes M, Hennen J, Clemens J, Blömeke B. (2011) Impact of aryl hydrocarbon receptor (AhR) knockdown on cell cycle progression in human HaCaT keratinocytes. Biol Chem 392:643–651 [DOI] [PubMed] [Google Scholar]
- Kim JH, Kim H, Lee KY, Kang JW, Lee KH, Park SY, Yoon HI, Jheon SH, Sung SW, Hong YC. (2007) Aryl hydrocarbon receptor gene polymorphisms affect lung cancer risk. Lung Cancer 56:9–15 [DOI] [PubMed] [Google Scholar]
- Koliopanos A, Kleeff J, Xiao Y, Safe S, Zimmermann A, Büchler MW, Friess H. (2002) Increased arylhydrocarbon receptor expression offers a potential therapeutic target for pancreatic cancer. Oncogene 21:6059–6070 [DOI] [PubMed] [Google Scholar]
- Kolluri SK, Weiss C, Koff A, Göttlicher M. (1999) p27Kip1 induction and inhibition of proliferation by the intracellular Ah receptor in developing thymus and hepatoma cells. Genes Dev 13:1742–1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress S, Greenlee WF. (1997) Cell-specific regulation of human CYP1A1 and CYP1B1 genes. Cancer Res 57:1264–1269 [PubMed] [Google Scholar]
- Lin P, Chang H, Tsai WT, Wu MH, Liao YS, Chen JT, Su JM. (2003) Overexpression of aryl hydrocarbon receptor in human lung carcinomas. Toxicol Pathol 31:22–30 [DOI] [PubMed] [Google Scholar]
- Long JR, Egan KM, Dunning L, Shu XO, Cai Q, Cai H, Dai Q, Holtzman J, Gao YT, Zheng W. (2006) Population-based case-control study of AhR (aryl hydrocarbon receptor) and CYP1A2 polymorphisms and breast cancer risk. Pharmacogenet Genomics 16:237–243 [DOI] [PubMed] [Google Scholar]
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114:97–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manders EM, Verbeek FJ, Aten AJ. (1993) Measurement of co-localization of objects in dual-colour confocal images. J Microsc 169: 375–382 [DOI] [PubMed] [Google Scholar]
- Moennikes O, Loeppen S, Buchmann A, Andersson P, Ittrich C, Poellinger L, Schwarz M. (2004) A constitutively active dioxin/aryl hydrocarbon receptor promotes hepatocarcinogenesis in mice. Cancer Res 64:4707–4710 [DOI] [PubMed] [Google Scholar]
- Murata K, Hattori M, Hirai N, Shinozuka Y, Hirata H, Kageyama R, Sakai T, Minato N. (2005) Hes1 directly controls cell proliferation through the transcriptional repression of p27Kip1. Mol Cell Biol 25:4262–4271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng TL, Chen J, Mao W, Liu X, Tao Y, Chen LZ, Chen MH. (2009) Potential therapeutic significance of increased expression of aryl hydrocarbon receptor in human gastric cancer. World J Gastroenterol 15:1719–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puga A, Tomlinson CR, Xia Y. (2005) Ah receptor signals cross-talk with multiple developmental pathways. Biochem Pharmacol 69:199–207 [DOI] [PubMed] [Google Scholar]
- Puga A, Xia Y, Elferink C. (2002) Role of the aryl hydrocarbon receptor in cell cycle regulation. Chem Biol Interact 141:117–130 [DOI] [PubMed] [Google Scholar]
- Solecki DJ, Liu XL, Tomoda T, Fang Y, Hatten ME. (2001) Activated Notch2 signaling inhibits differentiation of cerebellar granule neuron precursors by maintaining proliferation. Neuron 31:557–568 [DOI] [PubMed] [Google Scholar]
- Thomsen JS, Kietz S, Ström A, Gustafsson JA. (2004) HES-1, a novel target gene for the aryl hydrocarbon receptor. Mol Pharmacol 65:165–171 [DOI] [PubMed] [Google Scholar]
- Vaziri C, Schneider A, Sherr DH, Faller DV. (1996) Expression of the aryl hydrocarbon receptor is regulated by serum and mitogenic growth factors in murine 3T3 fibroblasts. J Biol Chem 271:25921–25927 [DOI] [PubMed] [Google Scholar]
- Wechsler-Reya R, Scott MP. (2001) The developmental biology of brain tumors. Annu Rev Neurosci 24:385–428 [DOI] [PubMed] [Google Scholar]
- Williamson MA, Gasiewicz TA, Opanashuk LA. (2005) Aryl hydrocarbon receptor expression and activity in cerebellar granule neuroblasts: implications for development and dioxin neurotoxicity. Toxicol Sci 83:340–348 [DOI] [PubMed] [Google Scholar]
- Yang X, Liu D, Murray TJ, Mitchell GC, Hesterman EV, Karchner SI, Merson RR, Hahn ME, Sherr DH. (2005) The aryl hydrocarbon receptor constitutively represses c-myc transcription in human mammary tumor cells. Oncogene 24:7869–7881 [DOI] [PubMed] [Google Scholar]
- Zakhary R, Keles GE, Berger MS. (1999) Intraoperative imaging techniques in the treatment of brain tumors. Curr Opin Oncol 11:152–156 [DOI] [PubMed] [Google Scholar]