

Abstract
Diseases caused by the apicomplexan protozoans Toxoplasma gondii and Cryptosporidium parvum are a major health concern. The life cycle of these parasites is regulated by a family of calcium-dependent protein kinases (CDPKs) that have no direct homologs in the human host. Fortuitously, CDPK1 from both parasites contains a rare glycine gatekeeper residue adjacent to the ATP-binding pocket. This has allowed creation of a series of C3-substituted pyrazolopyrimidine compounds that are potent inhibitors selective for CDPK1 over a panel of human kinases. Here we demonstrate that selectivity is further enhanced by modification of the scaffold at the C1 position. The explanation for this unexpected result is provided by crystal structures of the inhibitors bound to CDPK1 and the human kinase c-SRC. Furthermore, the insight gained from these studies was applied to transform an alternative ATP-competitive scaffold lacking potency and selectivity for CDPK1 into a low nanomolar inhibitor of this enzyme with no activity against SRC.
Introduction
Like most apicomplexan parasites, Toxoplasma gondii and Cryptosporidium parvum are obligate intracellular pathogens that must invade a host cell in order to grow and replicate. Several of the processes that facilitate host cell invasion, including gliding motility and microneme secretion, are tightly regulated by oscillations in calcium ion concentrations within the parasite cell.1–5 Transmission of calcium signals to these inter-related pathways is under the control of a family of serine/threonine protein kinases called calcium-dependent protein kinases (CDPKs), which themselves are regulated by calcium and appear to have essential physiological roles. This kinase family is found in apicomplexa, plants, and ciliates, but is absent in vertebrates and is thus an intriguing family to target for anti-apicomplexan chemotherapy. T. gondii CDPK1 (TgCDPK1) has been shown genetically to be critical in cell invasion and egress, processes necessary for parasite reproduction.6 The homologous CDPK1 of C. parvum (CpCDPK1) is also likely to be critical for infection.
This family of enzymes retains the conserved features of classical kinase catalytic domains, but exhibits evolutionary differences sufficient to cause differential drug sensitivity and thus allow the design of parasite-specific inhibitors.7 Specifically, Tg/CpCDPK1 contains an abnormally small amino acid, glycine, at a conserved position (the “gatekeeper residue”) in the ATP-binding pocket adjacent to the site of adenine binding. The gatekeeper residues of kinases are typically large, and mammalian kinases that contain alanine or glycine at this position are exteremely rare.8 This rarity motivated the development of an orthogonal set of ATP-competitive inhibitors (bumped kinases inhibitors) that are able to specifically target mammalian kinases that have been engineered to contain alanine or glycine residues at the gatekeeper position without affecting wild type members of this enzyme family.9 These selective inhibitors contain a pyrazolopyrimidine (PP) scaffold that makes the same hydrophobic interactions and hydrogen bonds as the adenine ring of ATP, and a bulky aromatic group substituent at the C3-position (the R1 group) that projects into a hydrophobic pocket adjacent to the gatekeeper position (the gatekeeper pocket). The steric bulk of the substituent at the C3-position of the PP core confers selectivity for kinases that contain small gatekeeper residues.9 Therefore it is notable that TgCDPK1 and CpCDPK1 contain a glycine at the gatekeeper position, rendering them especially sensitive to pyrazolopyrimidine-based inhibitors that contain large groups at the 3-position.7,10,11 Indeed, we have shown structurally that it is possible to target the gatekeeper pocket of TgCDPK1 and CpCDPK1 with these inhibitors and that potent pharmacological inhibition of this kinase blocks parasite proliferation in vivo.7,10 In T. gondii, we have confirmed that CDPK1 is the primary cellular target of the inhibitors that we have developed by showing that a parasite cell line engineered to exogenously express a CDPK1 gene mutated to contain a methionine gatekeeper is rendered largely insensitive to inhibitor treatment.7
Drug development based on compounds targeting a class of enzymes such as kinases that contains a highly conserved active site must address concerns of toxicity to the host caused by cross-reactivity with host enzymes. As threonine, which is considerably larger than glycine, is among the smallest gatekeeper residues found in mammalian kinases, we anticipated that kinases that contain this amino acid at this position would be the most likely off-targets of inhibitors developed for CDPK1. Extensive inhibitor selectivity screens have demonstrated that PP-based inhibitors with smaller substituents (substituted phenyl groups) most potently inhibit kinsases that contain threonine residues.12,13 Therefore, a small panel of kinases that contain this residue at this position have been used as a selectivity screen for inhibitors developed against TgCDPK1 and CpCDPK1. Specifically, all of the inhibitors generated were tested against the tyrosine kinases SRC and ABL, which have been demonstrated to be among the most sensitive kinase to PP-based inhibitors.12
We now report a comparative analysis of inhibitor interactions with Tg/CpCDPK1 and with the human kinase SRC based on crystal structures of kinase:inhibitor complexes. The selectivity observed for our series of PP scaffold inhibitors was originally postulated to be due to a steric clash between the gatekeeper sidechain and the substituent displayed from the 3-position of the scaffold. This is the fundamental structural rationale for the selectivity of bumped kinase inhibitors. However the current analysis shows that selectivity of these compounds for Tg/CpCDPK1 cannot be explained entirely on this basis. We observe that substituents displayed from the 1-position of this scaffold (the R2 group) have a much greater contribution to selectivity over Src-family kinases than anticipated. In particular, we find that inhibitors with a 4-piperidinemethyl group as the R2 substituent are highly selective relative to otherwise identical compounds containing instead an isopropyl R2 substituent. Analysis of CDPK1:PP and SRC:PP crystal structures suggests that the structural basis for this additional selectivity is a difference in the relative orientation of the gatekeeper pocket and the ribose pocket within the active site of the two structures. We infer that the ribose pocket constitutes an additional selectivity-determining region despite being a conserved feature of the kinase active site.
The analysis allows us to rationalize the observed dependence of selectivity on both R1 and R2 substituents, and to structurally guide optimization of inhibitors that are highly selective for Tg/CpCDPK1 over mammalian kinases and, thus, have a relatively low probability of producing toxic side effects. Furthermore, we have been able to exploit this structural insight to modify an alternative inhibitor scaffold, initially of low potency and poor specificity, into one that is highly potent for CDPK1 and is highly selective for CDPK1 over Src-family kinases. This work provides a basis for further rational design of more potent TgCDPK1 and CpCDPK1 specific inhibitors as lead compounds for the treatment of toxoplasmosis and cryptosporidiosis.
Results and discussion
PP inhibitors with 4-piperidinemethyl at the 1-position are selective for CDPK1
Several inhibitors based on the pyrazolopyrimidine (PP) scaffold have been used as exquisitely specific chemical probes of mutant kinases (engineered to contain glycine or alanine at the gatekeeper position) in mammalian systems.9,14,15 However, we noted in the course of optimizing PP-based inhibitors with a naphthylmethyl group at the 3-position against Tg/CpCDPK1 that the substituent at the 1-position contributed to additional potency and selectivity.10 Of the substituents that were explored at the 1-position, inhibitors that contained a 4-piperidinemethyl R2 were found to be the most potent against Tg/CpCDPK1. To further explore this observation, we generated a panel of PP-based inhibitors that contain various substituents at the 3-position and either an iso-propyl or 4-piperidinemethyl group at the 1-position. Table 1 highlights the gain in potency toward Tg/CpCDPK1 afforded by changing from an isopropyl group to a 4-piperidinemethyl group at R2.
Table 1.
In vitro activity of PP scaffold compounds against CDPK1 and against two threonine-gatekeeper human kinases.
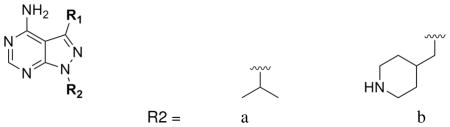 | ||||||
|---|---|---|---|---|---|---|
| R1 | R2 | cmpd # |
TgCDPK1 Ki(μM) |
CpCDPK1 Ki(μM) |
SRC Ki(μM) |
ABL Ki(μM) |
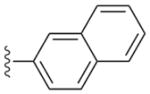 |
a | 1a | 0.0025 | 0.0047 | 0.065 | 0.069 |
| b | 1b | 0.0013 | 0.0004 | >10 | ND | |
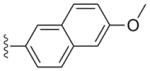 |
a | 2a | 0.003 | 0.0024 | 0.77 | 0.75 |
| b | 2b | 0.0025 | 0.0011 | >20 | >15 | |
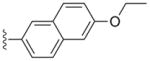 |
a | 3a | 0.0025 | 0.0057 | 0.2 | 1.6 |
| b | 3b | 0.0013 | 0.0003 | >20 | >15 | |
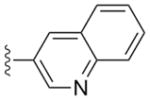 |
a | 4a | 0.012 | 0.0095 | 0.2 | 1.6 |
| b | 4b | 0.009 | 0.0024 | >20 | >15 | |
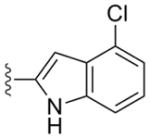 |
a | 5a | 0.0046 | 0.0076 | 1.24 | 0.28 |
| b | 5b | 0.009 | 0.030 | >10 | ND | |
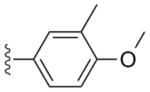 |
a | 6a | 0.0025 | 0.008 | 0.65 | ND |
| b | 6b | 0.0011 | 0.0009 | >10 | 5.5 | |
| b | 7b | >2.5 | >2.5 | >10 | >10 | |
To investigate the selectivity of these inhibitors, we assayed them in vitro for activity against Tg/CpCDPK1 and against two mammalian kinases that contain threonine at the gatekeeper position (SRC and ABL). Large scale selectivity screens have demonstrated that SRC and ABL are two of the human kinases most sensitive to PP-based inhibitors. While none of the inhibitors tested displayed greater activity against the human kinases SRC and ABL compared to activity against Tg/CpCDPK1, some, particularly those with an isopropyl group in the R2 position, did have sub-micromolar activity against these enzymes (Table 1). Substitution with a 4-piperidinemethyl group at R2, however, largely eliminated detectable activity against SRC and ABL, greatly enhancing the selectivity for the parasite kinases. Thus, the identity of the substituent at the 1-position of the PP scaffold contributes both to potency against Tg/CpCDPK1 and to selectivity over human kinases.
Structural basis of R2 contribution to selectivity
We had anticipated that the principal contribution to CDPK1-selectivity would be the R1 group itself, but the results in Table 1 illustrate that some inhibitors with a sizable substituent at this position are still able to effectively inhibit SRC and related kinases, albeit to a much lesser extent than either TgCDPK1 or CpCDPK1. Interestingly, and very encouraging from a drug design perspective, enzyme assays showed that inhibitors containing a 4-piperidinemethyl group at the R2 position are low nanomolar inhibitors of CDPK1 but do not inhibit SRC and other family members at concentrations of ≥ 5 μM (Table 1). We sought to rationalize this observation from a structural perspective by determining crystal structures of both TgCDPK1 and SRC in complex with relevant inhibitors.
Compounds 2a and 3a, both with a large (naphthyl ether) R1 substituent and an isopropyl group in the R2 position, have single-digit nanomolar IC50s for CDPK1 (Table 1). These compounds differ only by an additional methylene group in the R1 substituent and show nearly identical binding modes in TgCDPK1 (Figures 1a, 1b). Compounds 2b and 3b, containing the same R1 substituent but having a 4-piperidinemethyl group at the R2 position rather than an isopropyl group, are slightly better CDPK1 inhibitors and, surprisingly, bind to CDPK1 in an essentially identical pose (Figures 1a, 1b). Furthermore, this pose is adopted by inhibitors displaying a variety of substituents at both R1 and R2. In all cases the pyrazolopyrimidine exocyclic nitrogen hydrogen bonds to the backbone carbonyl oxygen of Glu 129 and the scaffold interacts directly with both the backbone amide nitrogen and carbonyl oxygen of Tyr 131 (Figure 1d). These structures show that the orientation of the PP scaffold with respect to the hinge region of the ATP-binding site is unaffected by the identity of the substituent at the R2 position. Notably, the binding poses of inhibitors 2b and 3b are unaltered despite the bulky R2 piperidine substituent that projects into the ribose pocket and forms a hydrogen bond with the side chain of Glu135.
Figure 1.

(a) Overlay of TgCDPK1 complexes with 2a (green) and 2b (purple). (b) Overlay of TgCDPK1 complexes with 3a (green) and 3b (purple). In each case the presence of the bulkier 4-piperidinemethyl substituent at R2 does not alter the orientation of the remainder of the PP scaffold with respect to the gatekeeper and distal portions of the binding site. (c) Overlay of TgCDPK1 complexes with 3a and 3b as in (b), and SRC in complex with 3a (yellow). The pose of the PP scaffold when bound to SRC is shifted with respect to the hinge region compared to the pose of the same compound bound to TgCDPK1. (d) Superposed binding poses of six PP-scaffold inhibitors displaying a variety of R1 and R2 substituents: 3b, 4a, 5a (Table 1); 11, 12, 13 (Table S3). Dotted lines indicate conserved inhibitor-protein distances <3.2Å. The view at left is approximately normal to the plane of the scaffold pyrazolopyrimidine; the view at right is rotated so that the view is in the plane of the scaffold.
In contrast to the consistent pose observed for PP scaffold compounds bound to CDPK1, the PP scaffold pose when binding to SRC varies with the nature of both the R1 and R2 substituents. Compounds 2a and 3a have high nanomolar to low micromolar activity (IC50s 200–1700 nM) against threonine gatekeeper-containing kinases SRC and ABL (Table 1). The crystal structure of SRC in complex with 3a shows a similar binding mode to that seen for TgCDPK1 in that the PP scaffold associates with the protein backbone in the hinge region and the R1 substituent occupies the gatekeeper pocket. However, the orientation of the PP scaffold in relation to the hinge is shifted in SRC as compared to TgCDPK1 (Figures 1c, 2b). This is presumably due to steric clash between the threonine gatekeeper sidechain and the R1 substituent, which forces the scaffold to pivot in order to maintain favorable interactions with the hinge residues.
Figure 2.

(a) Superposed crystal structures of a compound with a large R1 substituent (2b light green) and a compound with no R1 substituent (7b dark green) bound to TgCDPK1. Both compounds contain a 4-piperidinemethyl group at R2. In this case the presence or absence of an R1 group does not affect the scaffold pose. (b) Superposition of the TgCDPK1 complexes shown in panel (a) onto the crystal structures of SRC complexes with 3a (large R1, small R2, gold) and 7b (no R1, large R2, dark brown). In contrast to CDPK1, the SRC active site can accommodate either a large R2 group or a large R1 group, but not both. The SRC complexes show that the scaffold pose pivots in opposite directions to accommodate the large group at R1 or at R2. (c) The position of the piperidine group of compound 7b in the ribose pocket of TgCDPK1 (green) and SRC (orange). The protein molecular surface is shown for TgCDPK1. The protein backbone of SRC residues 342–346 is positioned closer to the binding site of the core PP scaffold, restricting the space available to accommodate an R2 group. Note that SRC residue Gly 344 can be seen extending through the superimposed molecular surface of TgCDPK1.
Based on analysis of crystal structures of CDPK1 and SRC in complex with several different inhibitors, we hypothesized that the difference in the binding angle of the scaffold in relation to the hinge is responsible for a small degree of selectivity towards CDPK1 afforded by the 4-piperidinemethyl R2 group itself. However, it is the combined presence of large substituents at both the R1 and R2 positions that dramatically hinders binding to SRC and ABL. This hypothesis led to the prediction that a compound comprised of the PP scaffold with a 4-piperidinemethyl group at the R2 position but lacking an R1 substituent would be free of the pressure exerted on the inhibitor by the gatekeeper sidechain and should be able to adopt a permissive binding mode in SRC. Compound 7b was therefore generated for crystallographic analysis to shed light on the structural basis of the R2 contribution to selectivity. This compound is not active against CDPK1 or SRC at concentrations less than 5 μM (Table 1) but we pursued structural characterization in order to gain insight into the binding pose of the scaffold.
Whereas we had been unsuccessful in growing cocrystals of SRC in complex with inhibitors that contain a 4-piperidinemethyl group at the R2 position of PP-scaffold inhibitors containing a R1 substituent, consistent with their poor IC50s, we were able to obtain cocrystals of SRC in complex with compound 7b, which containins a 4-piperidinemethyl R2 group but no R1 substituent. We also cocrystallized the same compound in complex with TgCDPK1. As expected, for both kinases this compound occupies the site of adenine binding and makes hydrogen bonds to the backbone of the hinge region. The absence of an R1 substituent does not affect the binding mode of the scaffold or the position of the piperidine moeity when bound to TgCDPK1 (Figure 2a). However, in the SRC complex the hinge contacts are conserved but there is a shift of approximately 10°–15° in the binding angle of the scaffold relative to the hinge compared to that observed for inhibitor 3a, which contains a large R1 substituent and a small (isopropyl) R2 substituent (Figure 2b). This skewed binding mode is apparently not available to compounds containing a large R1 substituent in addition to the 4-piperidinemethyl group at R2, and explains the lack of inhibition of SRC and ABL by these compounds.
Model for a secondary selectivity determining region
The large difference in the binding angle of inhibitor 7b relative to the hinge regions of CDPK1 and SRC suggested that there are structural differences outside of the gatekeeper pocket in the active sites of these kinases that are responsible for the CDPK1 selectivity afforded by the 4-piperidinemethyl group at the R2 position. In both complexes the piperidine extends into the ribose pocket in the kinase active site. In CDPK1 it makes a hydrogen bond with Glu 135.10 The homologous residue in SRC is Ser 345, but in the crystal structure of the SRC:7b complex the spatially equivalent sidechain is that of Asp 348 (Fig 2c). The conformation of the SRC protein backbone in this region differs from that of CDPK1 in such a way that the space available to an R2 substituent is restricted relative to the equivalent ribose pocket in Tg/CpCDPK1. In particular the backbone of SRC residues 344–346 impinges into the area of the binding pocket as seen in TgCDPK1, which necessitates a corresponding displacement of the piperidine binding pose (Fig 2c). Thus these crystal structures provide a structural basis for the observed R2-dependent selectivity of inhibitors for the parasite CDPK1 versus the threonine gatekeeper human kinases SRC and ABL. Taken together these observations strongly suggest that the vicinity of the ribose pocket serves as an additional selectivity-determining region. A conceptual model is depicted in Figure 3.
Figure 3.

Both the R1 and R2 substituents contribute to the specificity of PP-scaffold inhibitors for Tg/CpCDPK1 (panel a) relative to kinases containing a Thr gatekeeper (panels b,c,d). (a) The parasite enzymes can accommodate a large group at both the R1 and R2 positions. (b) SRC can accommodate some compounds with a large R1 group if the corresponding R2 group is small enough to allow the scaffold to pivot, displacing the R1 group from a clash with the gatekeeper residue. (c) In the absence of a large R1 group SRC can accommodate a large R2 substituent, in this case the 4-piperidinemethyl group, if the scaffold can pivot in the opposite direction. (d) The presence of large groups at both R1 and R2 cannot be simultaneously accommodated. The orientation of the cartoon is approximately the same as in Figure 2b.
Exploiting the second selectivity-determining region via an alternative scaffold
We used the structural insight gained from characterization of CDPK1 selective inhibitors based on the PP scaffold to advance parallel development of an alternative molecular scaffold, acylbenzimidazole, first identified through a high throughput fluorescence-based thermal shift screen. The initial lead compound based on this scaffold series (8) was not very potent against TgCDPK or CpCDPK, and less than 10-fold selective with respect to the human kinase ABL (Table 2). Structures of both TgCDPK1 and SRC in complex with 8 showed a binding mode reminiscent of the PP scaffold, in which the scaffold benzamidazole and the carbamate nitrogen associate with the backbone of the hinge region residues, while the phenone moiety lies adjacent to the gatekeeper residue (not shown). Based on these structures, we hypothesized that derivatization of the benzamidazole ring at the 1-position would be sterically equivalent to substitutions at position R2 of the original PP scaffold. Following this strategy, we found that removal of the methoxycarbonyl moiety at C2 (yielding compound 9) increases ligand efficiency, while addition of 4-piperidinemethyl at N1 (compound 10) significantly improves potency (Table 2). As intended in the design, the piperidine moiety extends into the secondary specificity determining region to form a favorable interaction with Glu 135 (Figure 4). Compound 10 exhibits low nanomolar potency against Tg/CpCDPK1 (Table 2) and good selectivity against SRC and ABL.
Table 2.
In vitro activity of acylbenzimidazole scaffold compounds
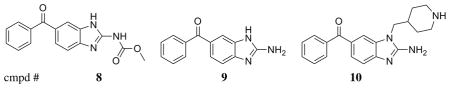 | ||||
|---|---|---|---|---|
| cmpd # | TgCDPK1 Ki(μM) |
CpCDPK1 Ki(μM) |
SRC Ki(μM) |
ABL Ki(μM) |
| 8 | 0.34 | 0.44 | >10 | 4.6 |
| 9 | 0.12 | 0.28 | >10 | ~10 |
| 10 | 0.0075 | 0.017 | >10 | >10 |
Figure 4.

Superposition of TgCDPK1:2b (green) and TgCDPK1:10 (cyan), showing the similar spatial positioning of the 4-piperidinemethyl group and the interaction of each scaffold with the hinge region backbone. The molecular surface is shown for the protein in the TgCDPK1:2b complex. In both structures the piperidine N forms a hydrogen bond to the sidechain of Glu 135 (immediately behind the surface at the right of the figure).
Discussion
T. gondii and C. parvum are highly successful protozoan parasites of major health concern for pregnant women, children, immunocompromised and immunocompetent hosts, as well as being a significant concern in veterinary medical care. The incidence of Toxoplasma infection is high and available toxoplasmosis drugs can cause rash, nephrotoxicity, and complications during pregnancy.16 There is no standard treatment for cryptosporidiosis despite its recent identification as being responsible for 15–20% of childhood diarrheal disease in the developing world.17,18 Paromomycin, nitozoxanide and new macrolides are sometimes used for cryptosporidiosis treatment with frequent risk of recurrences because elimination of the parasite is extremely difficult to achieve with these drugs.19 These challenges motivate a search for improved treatment options based on detailed target analysis of biochemical and functional pathways essential for survival.
Structure determination of T. gondii and C. parvum CDPK1 in complex with biochemically-characterized inhibitors has allowed us to understand the specific molecular interactions that influence enzyme inhibition. Paired structure determination of inhibitors bound to the target parasite CDPK1 and to potential off-target human kinases has further allowed us to better understand the molecular interactions leading to selective inhibition. This analysis provides a structural understanding for the observed dependence of selectivity on both R1 and R2 substituents, and can be used to guide optimization of the PP-scaffold series against Tg/CpCDPK1 while maintaining a relatively low probability of eliciting toxic side effects arising from inhibition of mammalian kinases. Furthermore, the identification of a secondary specificity-determining region that distinguishes Tg/CpCDPK1 from Src-family kinases allowed us to introduce specificity and nanomolar potency into compounds based on an alternative scaffold. This series of acylbenzamidazole compounds has high ligand efficiency compared to the original pyrazolopyrimidine scaffold series, affording good opportunity for optimization of solubility, pharmacokinetic, and metabolic properties. Further work is in progress to identify compounds from both series that are suitable for evaluation in the treatment of toxoplasmosis and cryptosporidiosis.
Experimental
Cloning and protein production
Full-length or truncated (residues 30–507) TgCDPK1 (GI:12484153, ToxoDB ID 162.m00001) was cloned into pAVA0421 containing a cleavable N-terminal hex-ahistidine tag and expressed in E. coli BL21*(DE3). CpCDPK1 (CryptoDB accession number cgd3_920) was expressed using a pET15-MHL-based construct coding for residues 70–538 kindly provided by Dr. Raymond Hui.20 Expressed protein was purified by Ni-NTA affinity and size-exclusion chromatography as previously described.7,10 SRC and ABL were produced using published protocols.21
Structure Analysis
Diffraction-quality crystals for inhibitor complexes of TgCDPK1 and SRC were grown by co-crystallization from sitting drops (0.9 μL protein solution + 0.9 μL crystallization buffer) set up by a Phoenix crystallization robot (Art Robbins Instruments). Drops were equilibrated by vapor diffusion against a reservoir of the crystallization buffer. Crystal growth conditions were in each case optimized by expanding around previously established growth conditions for the apo protein. For TgCDPK1 the starting point for expansion was 0.25 M ammonium citrate (pH 6.5–7.5), 25% polyethylene glycol (PEG) 3350, 5 mM dithiothreitol, and 2–2.5 mM inhibitor. For SRC it was 100 mM MES pH 6.5, 6% PEG 20000, 5 mM dithiothreitol, 2 mM inhibitor.
Diffraction data were collected at beamline 9–2 of the Stanford Synchrotron Radiation Light-source. The crystal structures were refined iteratively using refmac and manual adjustment in coot.22,23 Crystallographic statistics are given as Supplementary material. Model quality was validated using the molprobity and parvati servers prior to deposition with the PDB.24,25 Structural superpositions shown in Figures 1 and 2 were calculated using the protein backbone for residues in the hinge region (TgCDPK1 residues 125–138). The crystal structures shown in Figures 1, 2, and 4 have been deposited in the PDB as entries 3t3v, 3sxf, 3t3u, 3sx9, 3upz, 3upx, 3uqf, 3uqg, 3v51, 3v5p, and 3v5t. Figures were drawn using Pymol and Raster3D.26,27
Activity assays
In vitro inhibition of recombinant TgCDPK1 and CpCDPK1 activity was determined using a non-radioactive Kinaseglo luciferase assay (Promega, Madison, USA); inhibition of SRC and ABL was determined using a radiometric assay. Details of both assays were described previously.7,10 IC50 values were converted to Ki values using the Cheng-Prusoff equation.28 The experimentally determined KM;ATP was used for each kinase (TgCDPK1 = 10 μM; CpCDPK1 = 9.0 μM; SRC = 80 μM; Abl = 33 μM).
Chemistry
PP scaffold compounds were synthesized as described separately.10,29 Synthesis of compounds based on the acylbenzimidazole scaffold (8, 9, 10) is described in the Experimental section of the supporting information.
Supplementary Material
Acknowledgments
This work was funded by National Institute of Allergy and Infectious Diseases Grants R01AI080625 (W.C.V.V.), R01AI067921 (D.J.M., C.L.M.J.V., E.F., W.C.V.V., and E.A.M.), and R01AI089441 (W.C.V.V and E.A.M.), National Institute of General Medical Sciences Grant R01GM086858 (D.J.M.), and a generous contribution from G. and K. Pigotti. Portions of this research were carried out at the Stanford Synchrotron Radiation Light Source, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences.
We thank Marilyn Parsons, Amy DeRocher, Jennifer Geiger, Suzanne Scheele, Clinton White, Alejandro Castellanos-Gonzalez, Cassie Bryan, Lynn Barrett, Natascha Mueller, Rob Gillespie, Fred Buckner, and Frank Zucker for valuable discussion and technical assistance.
Abbreviations
- CDPK
Calcium Dependent Protein Kinase
- PP
pyrazolopyrimidine
Footnotes
Supporting Information Available
Crystallographic data collection and refinement statistics for all structures shown are provided in Tables S1 and S2. Additional information on the compounds shown in Figure 1d is provided in Table S3. Difference electron density supporting the crystallographics model for the pose of each inhibitor is shown in Figures S1 and S2.
This material is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Kieschnick H, Wakefield T, Narducci CA, Beckers C. Toxoplasma gondii attachment to host cells is regulated by a calmodulin-like domain protein kinase. J Biol Chem. 2001;276:12369–12377. doi: 10.1074/jbc.M011045200. [DOI] [PubMed] [Google Scholar]
- 2.Lovett JL, Sibley LD. Intracellular calcium stores in Toxoplasma gondii govern invasion of host cells. J Cell Sci. 2003;116:3009–3016. doi: 10.1242/jcs.00596. [DOI] [PubMed] [Google Scholar]
- 3.Chen XM, O’Hara SP, Huang BQ, Nelson JB, Lin JJ, Zhu G, Ward HD, LaRusso NF. Apical organelle discharge by Cryptosporidium parvum is temperature, cy-toskeleton, and intracellular calcium dependent and required for host cell invasion. Infect Immun. 2004;72:6806–6816. doi: 10.1128/IAI.72.12.6806-6816.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagamune K, Sibley LD. Comparative genomic and phylogenetic analyses of calcium ATPases and calcium-regulated proteins in the apicomplexa. Mol Biol Evol. 2006;23:1613–1627. doi: 10.1093/molbev/msl026. [DOI] [PubMed] [Google Scholar]
- 5.Billker O, Lourido S, Sibley LD. Calcium-dependent signaling and kinases in apicomplexan parasites. Cell Host Microbe. 2009;5:612–622. doi: 10.1016/j.chom.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lourido S, Shuman J, Zhang C, Shokat KM, Hui R, Sibley LD. Calcium-dependent protein kinase 1 is an essential regulator of exocytosis in Toxoplasma. Nature. 2010;465:359–362. doi: 10.1038/nature09022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ojo KK, et al. Toxoplasma gondii calcium-dependent protein kinase 1 is a target for selective kinase inhibitors. Nat Struct Mol Biol. 2010;17:602–607. doi: 10.1038/nsmb.1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang C, Kenski DM, Paulson JL, Bonshtien A, Sessa G, Cross JV, Templeton DJ, Shokat KM. A second-site suppressor strategy for chemical genetic analysis of diverse protein kinases. Nat Methods. 2005;2:435–441. doi: 10.1038/nmeth764. [DOI] [PubMed] [Google Scholar]
- 9.Bishop AC, Kung C-y, Shah K, Witucki L, Shokat KM, Liu Y. Generation of Monospecific Nanomolar Tyrosine Kinase Inhibitors via a Chemical Genetic Approach. J Am Chem Soc. 1999;121:627–631. [Google Scholar]
- 10.Murphy RC, Ojo KK, Larson ET, Castellanos-Gonzalez A, Perera BG, Keyloun KR, Kim JE, Bhandari JG, Muller NR, Verlinde CL, White AC, Merritt EA, Van Voorhis WC, Maly DJ. Discovery of Potent and Selective Inhibitors of Calcium-Dependent Protein Kinase 1 (CDPK1) from C. parvum and T. gondii. ACS Med Chem Lett. 2010;1:331–335. doi: 10.1021/ml100096t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sugi T, Kato K, Kobayashi K, Watanabe S, Kurokawa H, Gong H, Pandey K, Takemae H, Akashi H. Use of the kinase inhibitor analog 1NM-PP1 reveals a role for Toxoplasma gondii CDPK1 in the invasion step. Eukaryotic Cell. 2010;9:667–670. doi: 10.1128/EC.00351-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Apsel B, Blair JA, Gonzalez B, Nazif TM, Feldman ME, Aizenstein B, Hoffman R, Williams RL, Shokat KM, Knight ZA. Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat Chem Biol. 2008;4:691–699. doi: 10.1038/nchembio.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD, Wood JL, Morgan DO, Shokat KM. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature. 2000;407:395–401. doi: 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- 15.Bishop AC, Buzko O, Shokat KM. Magic bullets for protein kinases. Trends Cell Biol. 2001;11:167–172. doi: 10.1016/s0962-8924(01)01928-6. [DOI] [PubMed] [Google Scholar]
- 16.Jacobson JM, Davidian M, Rainey PM, Hafner R, Raasch RH, Luft BJ. Pyrimethamine pharmacokinetics in human immunodeficiency virus-positive patients seropositive for Toxoplasma gondii. Antimicrob Agents Chemother. 1996;40:1360–1365. doi: 10.1128/aac.40.6.1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Samie A, Bessong PO, Obi CL, Sevilleja JE, Stroup S, Houpt E, Guerrant RL. Cryptosporidium species: preliminary descriptions of the prevalence and genotype distribution among school children and hospital patients in the Venda region, Limpopo Province, South Africa. Exp Parasitol. 2006;114:314–322. doi: 10.1016/j.exppara.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 18.Ajjampur SS, Rajendran P, Ramani S, Banerjee I, Monica B, Sankaran P, Rosario V, Arumugam R, Sarkar R, Ward H, Kang G. Closing the diarrhoea diagnostic gap in Indian children by the application of molecular techniques. J Med Microbiol. 2008;57:1364–1368. doi: 10.1099/jmm.0.2008/003319-0. [DOI] [PubMed] [Google Scholar]
- 19.Pandak N, Zeljka K, Cvitkovic A. A family outbreak of cryptosporidiosis: Probable noso-comial infection and person-to-person transmission. Wien Klin Wochenschr. 2006;118:485–487. doi: 10.1007/s00508-006-0637-7. [DOI] [PubMed] [Google Scholar]
- 20.Wernimont AK, Artz JD, Finerty P, Lin YH, Amani M, Allali-Hassani A, Senisterra G, Vedadi M, Tempel W, Mackenzie F, Chau I, Lourido S, Sibley LD, Hui R. Nat Struct Mol Biol. 2010;17:596–601. doi: 10.1038/nsmb.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seeliger MA, Young M, Henderson MN, Pellicena P, King DS, Falick AM, Kuriyan J. High yield bacterial expression of active c-Abl and c-Src tyrosine kinases. Protein Sci. 2005;14:3135–3139. doi: 10.1110/ps.051750905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta Cryst. 2011;D67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 24.Lovell S, Davis I, Arendall WB, III, de Bakker P, Word J, Prisant M, Richardson J, Richardson D. Structure Validation by Cα Geometry: φ, ψand Cβ Deviation. Proteins: Structure, Function, and Genetics. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 25.Zucker F, Champ PC, Merritt EA. Validation of crystallographic models containing TLS or other descriptions of anisotropy. Acta Cryst. 2010;D66:889–900. doi: 10.1107/S0907444910020421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DeLano W. The PyMOL Molecular Graphics System. 2002 http://www.pymol.org.
- 27.Merritt EA, Bacon DJ. Raster3D - photorealistic molecular graphics. Meth Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- 28.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 29.Johnson SM, et al. Development of Toxoplasma gondii Calcium-Dependent Protein Kinase 1 Inhibitors with Potent Anti-Toxoplasma Activity. J Med Chem. 2012 doi: 10.1021/jm201713h. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.