

Abstract
The underlying cause of aging remains one of the central mysteries of biology. Recent studies in several different systems suggest that not only may the rate of aging be modified by environmental and genetic factors, but also that the aging clock can be reversed, restoring characteristics of youthfulness to aged cells and tissues. This Review focuses on the emerging biology of rejuvenation through the lens of epigenetic reprogramming. By defining youthfulness and senescence as epigenetic states, a framework for asking new questions about the aging process emerges.
Introduction
The inexorable tolls of aging are evident in almost all living beings. From the onset of reproductive maturity, organismal aging is generally characterized by a decline in fecundity, an increased susceptibility to disease and tissue dysfunction, and increased risk of mortality (Kirkwood, 2005; Hayflick, 2007; Kirkwood and Shanley, 2010). Aging is associated with a gradual loss of homeostatic mechanisms that maintain the structure and function of adult tissues. A major challenge of aging research has been to distinguish the causes of cell and tissue aging from the myriad of changes that accompany it. One of the hallmarks of cellular aging is an accumulation of damaged macromolecules such as DNA, proteins, and lipids. These become chemically modified by reactive molecules, such as free radicals, that are generated during normal cellular metabolism and whose production increases with age (Haigis and Yankner, 2010). DNA damage may lead to cellular dysfunction directly by altering the expression of specific genes or indirectly as result of cellular responses to damage that can alter gene expression more globally (Seviour and Lin, 2010; Campisi and Vijg, 2009). Damage to proteins may independently contribute to cellular aging if mis-folded or damaged proteins are replaced more slowly than they are generated, especially when they form stable aggregates that are not degraded by the cell (Koga et al., 2011). Such “proteotoxicity” has been postulated to underlie many age-related diseases and may also be an important part of normal cellular aging (Douglas and Dillin, 2010).
The consequences of age-related changes to the macromolecular components of a cell, particularly for long-lived postmitotic cells like neurons and myofibers, lead to gradual loss of normal structure and function—so-called “chronological aging,” marked simply by the passage of time. For continuously dividing cells, like those of the epithelia of the skin or gut, there is the added challenge of “replicative aging,” referring to the accumulation of cellular damage, such as telomere shortening and replication-associated DNA mutations, that occurs during the process of cell division (Rando, 2006; Liu and Rando, 2011). This is particularly relevant for adult stem cells because they divide throughout the life of the individual and therefore experience both chronological and replicative aging (Charville and Rando, 2011). As the burden of mutations increases with age, the likelihood that a cell will undergo apoptosis, malignant transformation, or senescence, which for diving cells means irreversible cell-cycle arrest (Kuilman et al., 2010), also increases. Although cellular function invariably declines with age, it may be that some of the changes, for example senescence and apoptosis, are actually adaptive in order to prevent cellular transformations such as metaplasia or neoplasia that may result from age-related genomic instability.
Despite the fact that aging appears to be inexorable, with the ultimate result being the death of the organism, it is incontrovertible that life span itself can be experimentally manipulated. An unlimited number of genetic defects and environmental challenges that may have no relation to the normal drivers of aging can shorten life span, but both genetic and environmental interventions have been shown to extend the life span of model organisms such as the nematode worm Caenorhabditis elegans (C. elegans), the fruit fly Drosophila melanogaster, and laboratory mice (Kenyon, 2010; Fontana et al., 2010). For example, mutations in individual genes in the insulin/insulin-like growth factor signaling pathway, in the pathways of protein translation involving the enzyme mammalian target of rapamycin (mTOR), and in energy-sensing pathways involving AMPK have all been shown to extend life span in model organisms (Kimura et al., 1997; Kapahi et al., 2004; Apfeld et al., 2004). In terms of environmental influences that can extend life span, none is better studied or more broadly effective, from C. elegans to mammals, than dietary restriction (Fontana et al., 2010). However, extending life span is not equivalent to delaying aging. Interventions may prevent common causes of death (for example, improved safety features to prevent automobile accidents as a sociological intervention or treatment of acute infectious illnesses as a medical intervention), without changing the fundamental rate of organismal aging. Nevertheless, it does seem that many so-called “longevity genes,” as well as dietary restriction, appear to extend not only life span, but also “health span” (Kauffman et al., 2010; Luo et al., 2010). In that regard, it does appear that it is possible to experimentally slow the rate of aging. Still, in each case, aging does continue on as if there is some clock that is driving individual aging, ticking relentlessly toward old age and death.
Though these examples support the notion that the process of aging can be slowed, there are also clear examples in nature when the aging clock appears to be temporarily arrested. The average life expectancy of C. elegans is about 2 weeks. However, under specific conditions such as food scarcity, the developing larvae can adopt an alternate fate, called a dauer stage, that does not feed and is metabolically relatively inactive (Fielenbach and Antebi, 2008). Dauer larva can survive for months, effectively prolonging the life span of the worm by an order of magnitude. Such diapauses are common in nature and reflect evolutionarily conserved responses to periods of adverse environmental conditions when survival is at stake. Even more dramatic examples of prolonged periods of survival in an arrested state come from the study of seeds and spores. Viable seeds capable of germination and growth have been obtained from excavations and dated as being 2,000 years old (Sallon et al., 2008). Viable bacterial spores have been found preserved in amber for 25–30 million years (Cano and Borucki, 1995) and preserved in salt crystals for 250 million years (Vreeland et al., 2000). Such examples reflect the remarkable survival ability of life forms under extreme conditions. These forms of life exist in suspended states that appear to arrest fundamental biological processes, including any aging that may accompany those processes, uncoupling the process of biological aging from chronological aging measured by the passage of time. However, is there reason to hypothesize that it is possible not only to arrest, but to reverse the aging clock?
Resetting of the Aging Clock
Despite the apparently unidirectional and inexorable process of aging of individual metazoans, the ability of the aging clock to be not only halted but reversed, or “reset to zero,” is so deeply embedded in the nature of life itself that it should not be surprising. Yet the process appears so mysterious that it is difficult to reconcile with concepts of individual aging. We are referring here to the resetting of the aging clock that comes with every fertilization event, giving rise to a zygote that will ultimately mature into an adult member of the species. For humans, this involves the fusion of two cells, a sperm and an egg, each of whose chronological age is measured in decades, to form a single cell that somehow erases any trace of the age of the parental cells. This resetting, or “reprogramming,” of the zygotic nucleus, rewinding the aging clock to begin anew, is mediated by factors in the oocyte cytoplasm that are at the heart of this mystery of rejuvenation. Granted, though the “biological age” of germ cells may differ from the biological age of other cells in the soma, there is no evidence that germ cells exist in any kind of diapause or metabolic arrest, completely resistant to the myriad of age-related changes that occur in cells over time. Therefore, the erasure of any manifestations of germ cell aging is central to the survival of the species. Were it not for this resetting of the aging clock, species would age with each generation and ultimately fail to propagate as the germline increasingly bore the burden of the effects of aging that occur during maturation to reproductive maturity.
The reprogramming process that is so central to fertilization, even though poorly understood, was exploited in the very earliest cloning experiments using somatic cell nuclear transfer (SCNT), in which the nucleus of a mature somatic cell is transferred to an enucleated oocyte (Briggs and King, 1952). The pioneering work of Dr. John Gurdon in this area showed for the first time that differentiated nuclei from tadpole intestinal or muscle cells could be transferred into enucleated Xenopus eggs and give rise to mature and fertile male and female frogs (Gurdon, 1962). Like the fused nucleus of the sperm and egg during fertilization, the somatic nucleus is reprogrammed by the oocyte cytoplasm to allow the development of a new member of the species, resetting any hallmarks of aging that the somatic nucleus bore upon transplantation. Importantly, these studies challenged the dogma at the time that the process of aging and differentiation from a single fertilized egg to a mature adult involved the loss of genetic material, which would in essence be an irreversible process rendering the resulting nuclei incapable of recapitulating the embryological developmental program. This early work demonstrated unequivocally that the full battery of genetic material present at fertilization and necessary to give rise to an adult organism is maintained in cells through development and maturation to adulthood. They also showed that the nuclei of adult somatic cells, just like that of the genetic material in the adult sperm and egg, can be apparently rejuvenated and can have pluripotency restored in the context of the oocyte cytoplasm.
The fact that the nuclei were capable of giving rise to viable embryos that were themselves capable of developing into fertile adults and did not exhibit premature aging is evidence that the chronological age of the donor nuclei had been reset. Thus, just like the example of fertilization, SCNT appears to be capable of resetting the aging clock for propagating a species. Whether there are any age-related alterations of the transplanted nucleus, for example in the genome, that were not erased by the process of SCNT cannot be ruled out and raises the fundamental question of how to define a young or old nucleus on a molecular level (this issue is considered in a later section). SCNT is, of course, also the process that gave rise to the first cloned mammal, Dolly the sheep (Campbell et al., 1996), which led to an explosion of research in cloning. In those studies, nuclei from different developmental and adult tissues were used and yielded viable lambs. Dolly actually died young (at 6 years of age as opposed to the typical life span of about 12 years for her breed). But the cause of death was a viral illness, not a premature aging syndrome. Dolly was fertile and gave birth to numerous offspring. Still, whether animals that are cloned from adult nuclei by SCNT are normal in terms of health and longevity remains to be fully determined. The majority of cloned animals often die in early development or display growth defects postnatally, possibly due to incomplete epigenetic reprogramming (Rideout et al., 2001) but also possibly due to genomic changes in the donor nucleus or technical limitations associated with the SCNT process. Still, SCNT demonstrates the remarkable ability of the oocyte cytoplasm to reprogram the donor nucleus, not only erasing manifestations of differentiation, but also resetting the chronological age.
Recent advances in stem cell biology have begun to unlock the molecular secrets behind these reprogramming events that occur during fertilization and SCNT. Specifically, these advances refer to the discovery of the process to create induced pluripotent stem cells (iPSCs) (Takahashi and Yamanaka, 2006), by which a variety of terminally differentiated adult cells, initially from mouse and human and more recently from other species, can be converted to pluripotent stem cells by the introduction of a small number of transcription factors such as Oct4, Sox2, and Klf4 (Stadtfeld and Hochedlinger, 2010). Pluripotency is a cellular property defined as the ability to give rise to differentiated cells in all three germ layers of the embryo: the ectoderm, the endoderm, and the mesoderm. In that regard, iPSCs resemble embryonic stem cells (ESCs), pluripotent stem cells derived from the inner cell mass of an early embryo and able to both give rise to any cell type in the body and support complete fetal development (Rossant, 2001). Likewise, iPSCs can generate an entire mouse embryo, including the germline (Stadtfeld and Hochedlinger, 2010). Indeed, the ability of a limited number of transcription factors to effect iPSC reprogramming stems from their key roles in the ESC gene expression program (Young, 2011). Furthermore, detailed analyses of multiple iPSC lines have shown that their global gene expression programs and chromatin states are remarkably similar to those of ESCs, though not equivalent (Loh and Lim, 2010; Ouyang et al., 2010).
One possible rejuvenating consequence of iPSC reprogramming is reactivation of the expression of telomerase (Marión and Blasco, 2010), the enzyme responsible for maintaining telomere length and long-term self-renewal potential in ESCs (Zeng, 2007). Genetic defects in components of the telomerase complex may prevent the restoration of telomere length and full telomerase activity during iPSC reprogramming in some conditions but not others (Agarwal et al., 2010; Batista et al., 2011). Nevertheless, the fact that differentiated adult cells can be directly reprogrammed to iPSCs by known factors rather than by the complex oocyte cytoplasm has generated yet another paradigm whereby the aging clock is reset. However, just like SCNT, the process of generating iPSCs is inefficient, and the vast majority of cells fail to attain the status of a pluripotent stem cell capable of giving rise to a new, fertile organism (Stadtfeld and Hochedlinger, 2010). There is clearly an important process of selection of those cells that are most fit and amenable to reprogramming, although the molecular bases of this fitness test are only now beginning to be revealed.
Although we describe each of these processes in the context of resetting of the aging clock, the major emphasis of research in the field of reprogramming is, in fact, reversal of the differentiation program and the attainment of a pluripotent state (Hanna et al., 2010), not the reversal of aging. In all of the examples presented, rewinding of the aging clock is coupled to the reversal of the differentiation program. The cell, whether a sperm or egg in the case of fertilization or an adult somatic cell (or its nucleus) in the case of SCNT or iPSC generation, not only rejuvenates, but also completely loses its differentiated characteristics. Indeed, this “dedifferentiation” is at the essence of the process of fertilization, as well as the ability of either SCNT or iPSC generation to create ESC-like cells. In each case, the cell (or its nucleus) ceases to maintain its identity as a particular differentiated cell type and, instead, adopts a pluripotent state coupled to the adoption of a more youthful state. However, to restore youthful properties to aged tissues for therapeutic purposes, for example to improve wound healing in aged skin or to improve cardiac function in the aged heart, the ideal would be to reset the aging clock but to leave the differentiation program untouched. Converting aged cardiomyocytes to pluripotent stem cells might yield no beneficial effect and might, in fact, have profound detrimental consequences. One of the major limitations of ESCs and iPSCs therapeutically is the fact that, upon transplantation, they have the propensity to form teratomas, tumors that have the features of all three germ layers (Takahashi and Yamanaka, 2006). By contrast, converting aged cardiomyocytes into young cardiomyocytes, without going through a pluripotent state, could improve cardiac function directly. This raises the fundamental question: Is it possible to uncouple the resetting of the aging clock from the resetting of the differentiation program?
Rejuvenation without Dedifferentiation
Recent studies have begun to test the potential of different interventions to restore youthfulness to aged cells or tissues. Although not specifically designed to address the question posed above as to whether cell or tissue rejuvenation can be achieved without dedifferentiation, evidence suggests that it may be possible to uncouple the processes that “maintain” the aged state (a concept that we will return to) from those that maintain the differentiated state. The following are three examples of apparently rejuvenating interventions–one environmental, one genetic, and one pharmacologic–that result in apparently more youthful states of aged cells that retain their differentiated states.
To test whether cells and tissues from an old animal can be restored to a more youthful state by environmental exposure, experimental approaches have included heterochronic (i.e., young-to-old or old-to-young) transplantations and heterochronic parabiosis, whereby the systemic circulations of two animals are joined together (Bunster and Meyer, 1933). Historically, this has been used to explore age-related physiological and pathological changes (Finerty, 1952; Tauchi and Hasegawa, 1977) and even longevity (McCay et al., 1957). Recently, heterochronic parabiosis (Figure 1) has been used to test whether tissue-specific stem cells (i.e., stem cells that are not pluripotent but are already committed to a specific germ layer or even a specific tissue) from old mice could be rejuvenated by exposure to a young environment (Conboy et al., 2005). Not only did the aged cells in muscle and liver adopt a more youthful functional phenotype, but the molecular signatures of aging were restored to a more youthful state (Conboy et al., 2005). Likewise, the young stem cells adopt a more aged molecular and functional state in these heterochronic parabiotic pairings (Brack et al., 2007; Villeda et al., 2011), demonstrating that the systemic environment is a powerful determinant of the “age” of cells in an organism (determined functionally, not chronologically). It may be that systemic influences are mediated by different signaling pathways in different tissues, but studies in muscle suggest that the manifestations of age are due to changes in the Notch-, Wnt-, and TGF-β-signaling pathways (Conboy et al., 2005; Brack et al., 2007; Carlson et al., 2008) and that the rejuvenating effects of heterochronic parabiosis may be due to restoration of a more youthful systemic milieu in terms of protein components, particularly chemokines and cytokines, in the blood and tissues (Villeda et al., 2011). In addition to illuminating the influence of the systemic environment on cellular function, such heterochronic studies emphasize the potential role of environmental factors in rejuvenating aged cells.
Figure 1. Rejuvenation without Dedifferentiation.
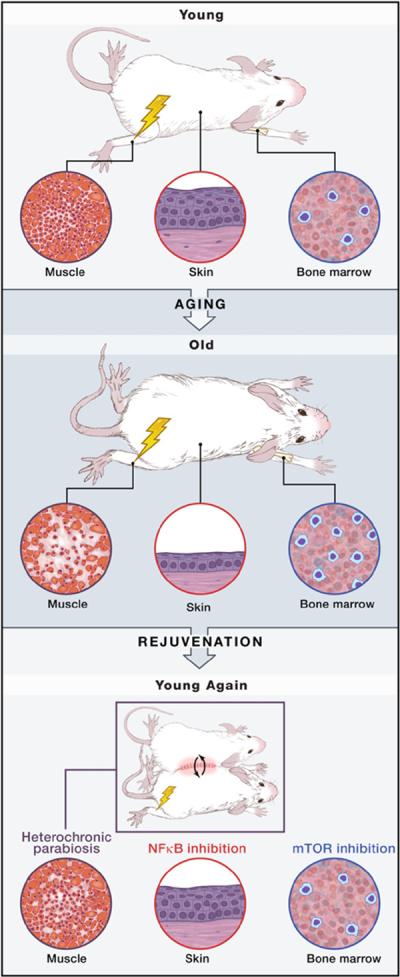
With age, distinct changes are evident in any mammalian tissue examined, such as impaired regenerative responses in skeletal muscle, thinning of the skin epithelium, and hypercellularity of the bone marrow. Different interventions have been shown to restore youthful function in each of these tissues in aged mice. Heterochronic parabiosis (or inhibition of Wnt or TGF-β signaling) enhances aged muscle regeneration, increasing the formation of new muscle and reducing fibrosis (left). Inhibition of NF-κB signaling restores the youthful skin phenotype, expanding the thickness of the epithelium (middle). Inhibition of mTOR signaling with rapamycin restores the youthful state of the hematopoietic system, reducing the hypercellularity that characterizes the aged tissue (right). In each case, not only are the youthful cell and tissue phenotypes restored, but the molecular signatures of youthfulness are also induced in the aged cells during the period of treatment.
Molecular signatures of aging have been directly tested as determinants of the aged state by genetically manipulating specific biochemical pathways. A recent example demonstrates the power of transcriptional profiling and bioinformatic analysis to reveal an aging signature that can be genetically engineered to reflect a more youthful state (Adler et al., 2007). In a comparison of old and young tissues from mice and humans, old tissues were found to express at significantly higher levels a set of genes that contained sequences in their 5′ regulatory regions, indicative of regulation by the NF-κB-signaling pathway (Adler et al., 2007). This led to the hypothesis that an increase in NF-κB activity was necessary to maintain the aged phenotype, which introduced the novel concept that the aged state, much like the differentiated state, might require active maintenance. The direct test of this involved the generation of a transgenic mouse strain in which NF-κB could be conditionally inhibited in the skin. After these mice were allowed to age normally, expression of the NF-κB inhibitor resulted in the skin rapidly reverting to a more youthful state and manifesting the molecular signature of youthfulness (Adler et al., 2007). Markers of cell senescence, such as the cell-cycle inhibitor protein p16, disappeared, and skin progenitor cells regained their proliferative activity, rebuilding atrophic skin to a youthful depth while maintaining the rigors of stratification and differentiation (Figure 1).
A third example illustrates that pharmacological targeting of pathways that have been implicated in promoting aging may also restore youthfulness at cellular and biochemical levels. Among the key regulators associated with interventions that extend life span is the enzyme mTOR, which senses cellular nutrient levels and in turn regulates rates of protein synthesis and energy utilization. Notably, administration of rapamycin, an mTOR inhibitor, starting at midlife can extend the life span of mice, suggesting that aging can be delayed or reversed in multiple cell types (Harrison et al., 2009). In the hematopoietic system, aging is associated with an increase in mTOR activation in stem cells and progenitors (Chen et al., 2009). Administration of rapamycin to old mice to inhibit mTOR not only limited the normal age-related increases in hematopoietic stem cells and biomarkers of aging in those cells, but also enhanced the performance of the stem cells to become as effective as young stem cells in heterochronic transplantation experiments (Chen et al., 2009) (Figure 1).
Together, these kinds of studies indicate that it may be possible to uncouple the aging clock from the differentiation program. In each case, the old cells are induced to adopt a more youthful phenotype without losing their differentiated characteristics; the muscle stem cells, the epithelial cells, and the hematopoietic stem cells all maintained their individual identities but functioned as if there had been a partial rewinding of the aging clock. Intriguingly, the reactivation of telomerase in adult tissue stem cells with experimentally shortened telomeres is sufficient to reverse degenerative pathologies—some typical of aged tissues—in multiple organs (Jaskelioff et al., 2011). As with the rejuvenating interventions described above, there does not appear to be any loss of differentiated phenotypes. In what ways are all of these processes and their resulting cellular states different from reprogramming that occurs with fertilization, SCNT, and iPSC generation, in which there is both dedifferentiation and rejuvenation? To address that question, it is first necessary to understand the molecular mechanisms that underlie reprogramming to the pluripotent state.
Epigenetics and Epigenetic Reprogramming
The mechanisms that underlie reprogramming of nuclei and erasure of the differentiation program during fertilization, SCNT, and iPSC generation are at the core of the field of epigenetics. Virtually all cells in a multicellular organism have the same DNA but can turn on different genes as a result of epigenetic mechanisms, thus allowing genetically identical cells to adopt divergent fates such as hepatocytes in the liver, neurons in the brain, or macrophages in the blood. Epigenetic regulation can occur by the direct methylation and demethylation of DNA bases, so called “cis-epigenetics” (Bonasio et al., 2010). The extent of cytosine methylation of the 5° regulatory region is an important determinant of the expression of a gene, with high levels of methylation associated with repression. Regulation of chromatin adds another level of epigenetic complexity. Histones come in several variants and can be altered by a number of modifications, including methylation and acetylation. Specific histone modifications are associated with expressed genes and others with repressed genes (Rando and Chang, 2009). For example, genes that are enriched for histone 3 trimethylated at lysine 4 (H3K4me3) tend to be expressed. By contrast, genes that are enriched for histone 3 trimethylated at lysine 27 (H3K27me3) tend to be repressed. Histone acetylation is generally associated with gene expression (Wang et al., 2008a). Finally, regulation of gene expression can also occur by “trans-epigenetics” (Bonasio et al., 2010), in which proteins and RNAs influence gene expression and repression. Stable transcription factor networks are an example of trans-epigenetics (Young, 2011). Clearly, enzymes that modify DNA and histones (methyltransferases, demethylases, acetyltransferases, deacetylases) are central epigenetic regulatory mechanisms (Rando and Chang, 2009).
The essence of epigenetics is not only the establishment, but also the maintenance, of a biological state, as cells need to maintain their identity over time through rounds of cell division and in response to a myriad of environmental influences. The robustness of an epigenetic state is referred to as “canalization” (Rando and Verstrepen, 2007), the concept of a particular state being buffered against change (by being “canalized”); the more canalized an epigenetic state is, the more stable it is over time, during cell division, and in the face of environmental changes. Clearly, the differentiated state of cells in the adult organism is highly canalized.
Although epigenetic mechanisms confer stability to cell states, early SCNT experiments clearly demonstrated that epigenetic states are plastic and reversible. Epigenetic reprogramming, like epigenetics itself, refers to changes in the stable transcriptional profile of a cell and changes in the very nature of that cell without changes in DNA sequences. Chromatin states can be erased if the modifications on DNA and histones that define them are altered. This can occur in a passive manner through successive cell divisions if the enzymes that modify DNA and histones fail to reinforce the modifications on newly synthesized chromatin during and after DNA replication (Rando and Chang, 2009). The passive erasure of chromatin states can be gradual and may require multiple rounds of cell division to “dilute out” the original pattern (Dodd et al., 2007). The phenomenon of position effect variegation (PEV) provides a prime example of this principle (Girton and Johansen, 2008). PEV can occur when a reporter gene is inserted near transcriptionally silent heterochromatin; the silent state can spread over to the reporter gene and be passed on to progeny cells. The strength of epigenetic silencing can be read out by the degree of variegation among cell clones. Strong silencing leads to uniform, large clones, whereas weak silencing leads to a salt-and-pepper intermingling of small clones as evidence of transcriptional reactivation after a few cell divisions.
This passive mechanism of epigenetic reprogramming can be viewed as a natural time-keeping strategy; a new biological state will emerge over time after a certain number of cell divisions via stochastic loss or degradation of epigenetic information (Rando and Verstrepen, 2007). The requirement for multiple rounds of cell division makes this mechanism particularly relevant to tissue-specific stem cells, which in many adult tissues are capable of multiple rounds of cell division throughout the life span of the organism. Passive epigenetic reprogramming may also be naturally coupled to environmental conditions. For instance, if tissue injury leads to compensatory cell divisions, then the process of regeneration creates a situation in which passive epigenetic reprogramming can be revealed. Indeed, Drosophila imaginal disc cells can change their positional identity, changing from a leg disc cell to a wing disc cell, for example, upon tissue fragmentation and regeneration. This process involves weakening of epigenetic silencing by passive mechanisms (Lee et al., 2005). Alternatively, chromatin states can be reprogrammed actively by regulated relocalization of one or more regulatory enzymes. Many chromatin modification enzymes do not have intrinsic DNA binding specificity and need to be recruited to their target genes on chromatin. These recruitment mechanisms include sequence-specific transcription factors, pre-existing chromatin marks that serve as docking sites, and both long and short noncoding RNAs (Ruthenburg et al., 2007; Bonasio et al., 2010; Hung and Chang, 2010). Thus, a dynamic steady state between both passive and active mechanisms underlies the apparent stability, as well as potential for reprogramming, of the epigenetic landscape.
The processes described above leading to dedifferentiation are prime examples of active epigenetic reprogramming. During early development, two major waves of epigenetic reprogramming occur (Feng et al., 2010). After fertilization, the zygotic genome undergoes dramatic epigenetic reprogramming. DNA methylation is erased wholesale and later re-established (Feng et al., 2010; Meissner, 2010), and histone modifications are also extensively reorganized. A second wave of DNA demethylation and resetting of imprinting marks occurs in the gonad of the zygote, when the primordial germ cells are produced. Many of the epigenetic changes that occur in the former setting—in the early zygote—are mimicked by iPS reprogramming (Mikkelsen et al., 2008). The embryonic genome is believed to be transcriptionally inactive until maternal zygotic transition. This transition also corresponds to a period of enrichment of H3K4me3 on specific gene loci and the formation of so-called bivalent domains, enriched for both H3K27me3 (repressive) and H3K4me3 (active) marks and therefore poised for future activation (Vastenhouw et al., 2010). This choreography of chromatin marks also occurs on enhancers, which are typically marked by histone H3 monomethylated on lysine 4 (H3K4me1). The transition of enhancers from an inactive to an active state, for example during ESC differentiation, is marked by the change of histone H3 lysine 27 from trimethylation (H3K27me3) to acetylation (H3K27ac) (Rada-Iglesias et al., 2011; Creyghton et al., 2010).
The potency and efficacy of reprogramming conditions are revealed by SCNT and cell fusion (including fertilization) studies, in which a “donor” nucleus is suddenly exposed to a different complement of trans-acting regulatory factors. The expression pattern of some genes of transferred or fused nuclei may be reprogrammed to resemble that of the host cell, but some genes retain the pattern of expression of the donor nucleus. DNA methylation is strongly associated with the loci that resist reprogramming (Lee et al., 2009a, 2009b). In addition, an apparent hierarchy of cell fate dominance can be derived by pair-wise fusion of different cell types (Terranova et al., 2006; Piccolo et al., 2011). The basis of the hierarchy of cell fate dominance is not known, but ESCs are at the top of this hierarchy because they can reprogram most fusion partners to transcribe an ESC-like gene expression program (Cowan et al., 2005; Tada et al., 2001). One potential explanation for the success of iPS reprogramming is that the ESC state is stabilized by a positive feedback loop. The core pluripotency transcription factors co-occupy their own and each other's enhancer elements, providing a strong positive feedback for maintenance of the ESC state (trans-epigenetics). Moreover, these factors directly bind to and activate genes important for ESC pluripotency and can also repress genes by controlling their epigenetic state. In ESCs, for example, Oct4, Sox2, and Nanog repress the long noncoding RNA Xist, the expression of which is necessary and sufficient to mark one of two × chromosomes in female cells for transcriptional silencing. Downregulation of the core transcription factors allows derepression of Xist and subsequent × chromosome inactivation in conjunction with ESC differentiation (Donohoe et al., 2009; Navarro et al., 2008).
One characteristic of reprogramming associated with resetting of the aging clock is low efficiency. Typical iPSC reprogramming yields < 1% conversion rate, and even among iPSC colonies, there is considerable heterogeneity (Stadtfeld and Hochedlinger, 2010). Thus, an important feature of iPS reprogramming is a selection among the starting cell population, allowing for only the most capable cells to emerge. The molecular determinants of fitness remain unclear; however, p16 and p53 proteins, both of which are associated with senescent or damaged cells, are both strong barriers to iPSC formation (Krizhanovsky and Lowe, 2009). This feature of iPSC formation, effectively a form of Darwinian selection, is likely also to account for the low efficiency of successful SCNT and may be an important determinant of successful fertilizations, in each case providing quality control and allowing only the most robust cells of highest integrity to progress.
Aging and Epigenetics
Is it reasonable to consider aging to be comparable to differentiation in terms of epigenetic determination? Although many parallels exist, an important difference is that differentiation occurs without any specific change in the genome, whereas aging is associated with (and may be due at least in part to) the accumulation of nuclear and mitochondrial DNA mutations (Garinis et al., 2008). The irreversibility of somatic mutations that accompany aging of replicative and postmitotic cells means that “rejuvenated” cells may not necessarily be identical to young cells, as mentioned with regard to SCNT. However, although the accumulation of nuclear and mitochondrial DNA mutations has clearly been correlated with aging (Vijg et al., 2005; Herbst et al., 2007) and increasing the burden of mitochondrial DNA mutations can shorten life span (Trifunovic et al., 2004), there is no direct evidence that DNA mutations are the proximal cause of cellular aging. Specifically, no experiment has demonstrated that a reduction in DNA mutations leads to an extension of life span. As such, there is currently much interest in the role of epigenetic processes as mediators of the aging process (Oberdoerffer and Sinclair, 2007; Campisi and Vijg, 2009). The observations that the aging clock can be halted in the C. elegans dauer state, reset at fertilization, and potentially rewound by the environmental influences described above suggest strongly that the manifestations, and possibly the causes, of aging may be largely epigenetic. The hypothesis is further supported by the recent finding of transgenerational epigenetic inheritance of extended life span in C. elegans (Greer et al., 2011).
Beyond the genome and the epigenome, cellular aging is characterized by the accumulation of damaged macromolecules, including proteins and lipids, and highly stable aggregates of those molecules (Campisi and Vijg, 2009). These, too, are manifestations of aging that are also potentially “reversible,” either by dilution in dividing cells or by disaggregation and degradation followed by replacement with new undamaged macromolecules. Only mutated and deleted DNA sequences, for which no template, code, or cellular machinery exists to guide their correction, are inaccessible to the rejuvenation process. The notion that aging is at least in part, if not largely, a manifestation of epigenetic changes, including those that may be secondary to genomic mutations, offers a theoretical construct for understanding the mechanisms of rejuvenation. If so, it should be possible to characterize “young” and “old” cells by specific transcriptional and epigenetic profiles and states. Furthermore, the processes that underlie aging and rejuvenation should be identifiable in terms of regulators of epigenetic states. Although biomarkers of age have remained elusive, studies have begun to reveal key epigenetic features of aging cells. We present below some specific examples of the epigenetic changes identified in aged cells and the relationship between epigenetic regulators and life span.
Aged cells show several distinctive features on their chromatin (Figure 2). The CDKN2A (encoding cyclin-dependent kinase inhibitor p16) locus becomes progressively expressed with age, eventually leading to cellular senescence, a state of irreversible cell-cycle arrest (Krishnamurthy et al., 2004). This mechanism may be particularly pronounced in adult tissue stem cells, which need to undergo long-term self-renewal, especially in the setting of tissue injury. The CDKN2A locus is under epigenetic control by the Polycomb group proteins, a gene-silencing complex. Polycomb-repressive complex 2 (PRC2, with EZH2 being the catalytic subunit) trimethylates lysine 27 of histone H3 (H3K27me3), which then recruits PRC1 to further modify the chromatin to enforce gene silencing (Margueron and Reinberg, 2011). PRC2 occupies the CDKN2A locus in young cells and prevents p16 expression. As pancreatic β cells age, EZH2 mRNA and protein levels decline, and the level of H3K27me3 at the CDKN2A locus wanes, now permitting p16 expression and cell senescence (Dhawan et al., 2009; Dhawan et al., 2009). Age-related derepression of genes is not entirely passive. For example, the H3K27me3 demethylase JMJD3 can compete with EZH2 for occupancy of CDKN2A, erase H3K27me3, and promote p16 expression. JMJD3 expression is induced by replicative exhaustion and also by the stress-responsive transcription factor NF-κB or by oncogenic stress (Agger et al., 2009; Barradas et al., 2009; De Santa et al., 2007). Interestingly, iPS experiments suggest that epigenetic features associated with aging can be reversed. In successfully reprogrammed iPSCs, the chromatin state of CDKN2A locus associated with aging is erased and restored to that of youthful cells (Meissner, 2010).
Figure 2. Epigenetic States in Young and Old Cells.

Aging is associated with specific changes in chromatin, some of which are illustrated here. In young cells, Polycomb group proteins (PcG) and sirtuins (SIRTs) silence aging genes by maintaining histone H3 lysine 27 methylation and by deacetylating multiple residues, respectively. Old cells are characterized by the appearance of DNA damage, which can titrate away sirtuins, as well as stress-inducible transcription factors like NF-κB. An exchange of PcG for Trithorax group proteins (Trx) and H3K27 demethylase JMJD3 allows accumulation of active chromatin marks such as H3K4me3 and histone acetylation, removal of H3K27me3, and increased expression of proaging genes such as the cell-cycle inhibitor p16, which drives cell senescence. Additional consequences of epigenetic dys-regulation include increased transcriptional noise and decreased coordination of gene expression that contributes to organismal aging.
The requirement for proper epigenetic gene silencing for longevity has been observed in multiple model organisms, suggesting an evolutionarily conserved process (Lin et al., 2000; Chen et al., 2005; Greer et al., 2010). The function of Polycomb group proteins is counteracted by the trithorax group proteins, which encode complexes that trimethylate histone H3 at lysine 4 (H3K4me3), a histone mark associated with gene activation. In C. elegans, inactivation of several H3K4 methylase subunits extends life span, whereas inactivation of a H3K4 demethylase shortens life span (Greer et al., 2010). The specific target genes of H3K4me3 in worms, presumably causing aging, are not yet clear, but life span extension by trithorax inactivation requires a functioning germline. Patterns of DNA methylation associated with gene repression also change with age (Maegawa et al., 2010; Murgatroyd et al., 2010). Moreover, DNMT1, the enzyme responsible for maintenance of cytosine methylation, is critical for the self-renewal of progenitor cells in the blood and skin (Trowbridge et al., 2009; Sen et al., 2010). Depletion of DNMT1 or enforced expression of Gadd45a, an enzyme involved in a repair-based mechanism of DNA demethylation, causes epidermal stem cell depletion and ectopic differentiation in a manner analogous to that of Polycomb depletion. However, the exact genes affected by Polycomb and DNMT1 are more complementary than overlapping (Sen et al., 2010), indicating that multiple silencing mechanisms may share common pathways to prevent ectopic transcription in the aging genome. The observation of age-associated increase in stochastic gene expression on a cell-by-cell level (so called transcriptional noise) in some aging tissues is consistent with this concept (Bahar et al., 2006).
The sirtuin family of NAD+-dependent lysine deacetylases has long been associated with the control of longevity, although the precise mechanisms remain controversial (Guarente, 2007; Vaquero and Reinberg, 2009). As histone acetylation is strongly associated with gene activation (Wang et al., 2008b), sirtuins are in general silencers of gene expression by the deacetylation of histones. In yeast cells, expression of the sirtuin Sir2, which is required to maintain the silent chromatin state of the ribosomal RNA genes and telomeres, decreases with replicative age (Sinclair and Guarente, 1997; Dang et al., 2009). Ectopic transcription and recombination of rRNA genes cause toxicity and limit replicative life span (Sinclair and Guarente, 1997). Although initial reports suggested that the overexpression of Sir2 extends replicative life span in yeast (Kaeberlein et al., 1999) and that overexpression of sirtuins extends life span of Drosophila and C. elegans (Tissenbaum and Guarente, 2001; Rogina and Helfand, 2004), recent evidence suggests that the effects in Drosophila and C. elegans are minimal and that initial reports may have been confounded by influences of genetic backgrounds of the strains (Burnett et al., 2011).
In mammals, seven Sir2 homologs exist and are named Sirt1–Sirt7. Sirt1 localization is mobile and stress responsive, relocalizing to sites of DNA damage where it participates in DNA repair. But this movement comes at a price: the corresponding departure of Sirt1 from basal target genes during damage allows these loci to become derepressed, promoting expression of a number of genes whose expression is known to increase with age (Oberdoerffer et al., 2008). This series of events provides one explanation for how environmental stress can indirectly trigger changes in chromatin state over time. The chromatin-associated sirtuin Sirt6 is also required for longevity; inactivation of Sirt6 results in a constellation of defects with features of premature aging (Mostoslavsky et al., 2006). Sirt6 maintains telomeric chromatin to promote replicative capacity (Michishita et al., 2008). Moreover, Sirt6 is recruited by transcription factors to target chromatin. In particular, Sirt6 directly interacts with NF-κB subunit RelA and is recruited by RelA to promoters to deacetylate H3K9Ac, a key event that promotes RelA eviction and terminates NF-κB signaling (Kawahara et al., 2009). Thus, Sirt6 may provide a mechanistic link between aging, rejuvenation, and epigenetics (Tennen and Chua, 2011). The connections between aging and epigenetics are also exemplified by the life span-extending effects of dietary restriction, which exerts multiple impacts on chromatin through sirtuins, TOR, and other factors (Vaquero and Reinberg, 2009).
Clearly, epigenetic changes are both responsive to and effectors of the aging process. With DNA damage and environmental stresses like inflammation leading to changes in chromatin, the epigenome clearly adapts to age-related changes in the genome and the local milieu. Perhaps the epigenome is a general sensor of cellular dysfunction, sensing metabolic and proteomic changes that accompany aging as well. However, the epigenome is also an effector of the aging process, enforcing different patterns of gene expression in old cells and young cells and, in many cases, resulting in cellular phenotypes associated with aging such as senescence and metaplasia (Martin, 2009). In that sense, the epigenome is rather like a lens through which genomic information is filtered (Figure 3), a lens that deteriorates with age because of both loss of integrity of genomic information and direct environmental stresses within and outside of the cell. Within the “epigenome as lens” metaphor, the process of rejuvenation is the restoration of a youthful state by actions on the epigenomic lens (Figure 3). The loss of integrity of the genomic information remains, but the rejuvenating interventions are sufficient to overcome and possibly reverse at least some of the age-related epigenetic changes. Similarly, an altered epigenome and gene expression programs may also be able to reverse or compensate for some age-dependent biochemical changes, such as protein aggregation, macromolecular oxidation, and glycation, to maintain cellular functions (Douglas and Dillin, 2010).
Figure 3. Aging and Rejuvenation through the “Epigenetic Lens”.

The process of light passing through a film, focused by a lens, and projected to create a complex image is drawn as a metaphor for genetic information being processed and interpreted by epigenetic mechanisms to create complex cellular phenotypes. In this analogy, the film represents the genome (shown as a DNA double helix) containing the fundamental information, the lens represents the epigenome (represented as a string of nucleosomes) that allows that genetic information to be translated, and the Cell cover represents the resulting complex phenotype (shown as a mature neuron).
In the vertical axis, the processes first of aging and then of rejuvenation are illustrated. With age, there is clearly a deterioration of the cellular phenotype, reflected by a blurring of the image. This may be due to intrinsic changes to DNA (depicted as double-strand breaks) and also to the epigenome (depicted as less well-organized nucleosomes), the latter resulting both from genomic changes and also from environmental influences. Together, these changes distort the genomic information of youth to create imperfect products, blurred as an image and structurally and functionally disrupted as a cell. Based on the ideas put forth in this Review, we postulate that most, if not all, of the rejuvenating effects, such as those that result from processes (e.g., fertilization, SCNT, iPS cell generation) or interventions (e.g., heterochronic parabiosis, NF-κB inhibition, mTOR inhibition) described, act by restoring the epigenomic lens back toward a more youthful state. The resulting image/cell may not be precisely “young” but has youthfulness restored by these processes and interventions that act by reprogramming the epigenome. It is thus the epigenetic lens that is critical for establishing the aged phenotype and that is the target for rejuvenating interventions and reprogramming that are responsible for the apparent rewinding of the aging clock.
Rejuvenation: Is It Epigenetic Reprogramming?
By analogy to the attainment of a pluripotent state by epigenetic reprogramming of a differentiated cell, is cellular rejuvenation by heterochronic parabiosis, NF-κB inhibition, or inhibition of mTOR signaling (Figure 1) a form of epigenetic reprogramming from an aged state to a youthful state? If so, then these would be examples of an uncoupling of the differentiation program from the aging clock, with cells in each case manifesting an apparent rewinding of the aging clock without loss of differentiation. Formal demonstration will require clear epigenetic signatures of young and old cells and evidence that the aged cells have regained a youthful signature. It should be noted that reprogramming of the epigenome to a youthful state in an aged cell has inherent risks and uncertainties. For example, the increase in proliferative activity of aged stem cells and progenitors by heterochronic parabiosis may increase the risk of developing malignancies among cells that have acquired genomic mutations during normal aging but then acquire increased proliferative potential by this rejuvenating intervention. Clearly, any therapeutic goal of cell or tissue rejuvenation would aim to restore a “young adult” state from an elderly state, not rewinding the aging clock back to embryonic or even postnatal developmental stages when growth and morphogenesis are paramount and the systemic milieu is very different from that in the adult. The challenge would be to reset the aging clock back to the appropriate adult stage. Another challenge is the coordination of reprogramming among different cell types in multicellular organisms. As such, the most feasible near-term applications of any type of rejuvenating intervention for therapeutic purposes would be those that could be administered in a temporally and spatially controlled manner (e.g., to a specific site of wound repair or tissue injury for a limited time).
Studies of cell rejuvenation without dedifferentiation, uncoupling the aging clock from the differentiation program, raise several critical experimental questions that will relate to the epigenetic mechanisms at play. First, what is the perdurance of the rejuvenated phenotype once the inducing conditions are removed? When the cells are free of the rejuvenating influence, do they immediately reacquire an aged phenotype, or does the youthful phenotype persist? In successful iPSC reprogramming, the endogenous embryonic stem cell transcription factors are stably induced, and the exogenously introduced factors are not required to sustain iPSCs (Okita et al., 2007). Is the rejuvenated state also stable after an “induction” period? Second, is it possible to rejuvenate differentiated cells with a limited number of transcription factors? As noted above, induction of the pluripotent state is possible with a very limited number of transcription factors (Takahashi and Yamanaka, 2006). Similar results have also been obtained for transdifferentiation of fibroblasts into cardiomyocytes by defined transcription factors, confirming that stable epigenetic reprogramming into multiple cell fates is possible with defined factors (Ieda et al., 2010). Clearly, inhibiting single signaling pathways (NF-κB and mTOR) is sufficient to restore some features of youthful cells, but the number of transcriptional regulators that need to be modulated to result in full rejuvenation is unknown. Third, is the youthful state or the aged state dominant? It would be interesting to determine which epigenetic and transcriptional profile is more robust in experiments of fusion of young and old cells.
Concluding Remarks
Several of the fundamental questions discussed above will be answered only with more detailed information on the genetic and epigenetic profiles associated with aging. Clearly, the establishment of transcriptional networks and epigenetic profiles for cells from different ages and across species will reveal the features that are generalizable characteristics of aging and allow for direct tests of whether it is possible, as with the relationship between pluripotent and differentiated states, to directly program a cell to be either young or old even if only transiently and incompletely. It is interesting that many of the rejuvenating interventions act on the stem cell compartments, perhaps reflecting shared genetic and biochemical pathways controlling stem cell function and longevity (Rando, 2006; Sharpless and DePinho, 2007; Jones and Rando, 2011). Many of the secrets to organismal longevity might, in fact, be linked to the biology of stem cell quiescence and self-renewal.
Although genetic and environmental interventions have clearly proven to be effective in prolonging life span, we postulate that those interventions, as well as the rejuvenating interventions described above, are, in fact, acting primarily to modify the epigenome. Consistent with this, genetic interventions directly targeting the epigenome can extend life span (Greer et al., 2010). Studying aging and rejuvenation through the lens of epigenetics and reprogramming therefore offers a fresh view of the mysteries of the aging process itself.
ACKNOWLEDGMENTS
This work was supported by grants from the Glenn Foundation for Medical Research and from the NIH (P01 AG036695, R37 AG23806, and an NIH Director's Pioneer Award) to T.A.R. and by a grant to H.Y.C. from the Ellison Medical Foundation. H.Y.C. is an Early Career Scientist of the Howard Hughes Medical Institute. The authors would like to thank Dr. Ami Okada and Jamie Brett for useful comments on the manuscript.
REFERENCES
- Adler AS, Sinha S, Kawahara TL, Zhang JY, Segal E, Chang HY. Motif module map reveals enforcement of aging by continual NF-kappaB activity. Genes Dev. 2007;21:3244–3257. doi: 10.1101/gad.1588507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal S, Loh YH, McLoughlin EM, Huang J, Park IH, Miller JD, Huo H, Okuka M, Dos Reis RM, Loewer S, et al. Telomere elongation in induced pluripotent stem cells from dyskeratosis congenita patients. Nature. 2010;464:292–296. doi: 10.1038/nature08792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agger K, Cloos PA, Rudkjaer L, Williams K, Andersen G, Christensen J, Helin K. The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A-ARF locus in response to oncogene- and stress-induced senescence. Genes Dev. 2009;23:1171–1176. doi: 10.1101/gad.510809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apfeld J, O'Connor G, McDonagh T, DiStefano PS, Curtis R. The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes Dev. 2004;18:3004–3009. doi: 10.1101/gad.1255404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahar R, Hartmann CH, Rodriguez KA, Denny AD, Busuttil RA, Dollé ME, Calder RB, Chisholm GB, Pollock BH, Klein CA, Vijg J. Increased cell-to-cell variation in gene expression in ageing mouse heart. Nature. 2006;441:1011–1014. doi: 10.1038/nature04844. [DOI] [PubMed] [Google Scholar]
- Barradas M, Anderton E, Acosta JC, Li S, Banito A, Rodriguez-Niedenführ M, Maertens G, Banck M, Zhou MM, Walsh MJ, et al. Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes Dev. 2009;23:1177–1182. doi: 10.1101/gad.511109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista LF, Pech MF, Zhong FL, Nguyen HN, Xie KT, Zaug AJ, Crary SM, Choi J, Sebastiano V, Cherry A, et al. Telomere shortening and loss of self-renewal in dyskeratosis congenita induced pluripotent stem cells. Nature. 2011;474:399–402. doi: 10.1038/nature10084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonasio R, Tu S, Reinberg D. Molecular signals of epigenetic states. Science. 2010;330:612–616. doi: 10.1126/science.1191078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brack AS, Conboy MJ, Roy S, Lee M, Kuo CJ, Keller C, Rando TA. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science. 2007;317:807–810. doi: 10.1126/science.1144090. [DOI] [PubMed] [Google Scholar]
- Briggs R, King TJ. Transplantation of Living Nuclei From Blastula Cells into Enucleated Frogs' Eggs. Proc. Natl. Acad. Sci. USA. 1952;38:455–463. doi: 10.1073/pnas.38.5.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunster E, Meyer RK. An improved method of parabiosis. Anat. Rec. 1933;57:339–343. [Google Scholar]
- Burnett C, Valentini S, Cabreiro F, Goss M, Somogyvári M, Piper MD, Hoddinott M, Sutphin GL, Leko V, McElwee JJ, et al. Absence of effects of Sir2 overexpression on lifespan in C. elegans and Drosophila. Nature. 2011;477:482–485. doi: 10.1038/nature10296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell KH, McWhir J, Ritchie WA, Wilmut I. Sheep cloned by nuclear transfer from a cultured cell line. Nature. 1996;380:64–66. doi: 10.1038/380064a0. [DOI] [PubMed] [Google Scholar]
- Campisi J, Vijg J. Does damage to DNA and other macromolecules play a role in aging? If so, how? J. Gerontol. A Biol. Sci. Med. Sci. 2009;64:175–178. doi: 10.1093/gerona/gln065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano RJ, Borucki MK. Revival and identification of bacterial spores in 25- to 40-million-year-old Dominican amber. Science. 1995;268:1060–1064. doi: 10.1126/science.7538699. [DOI] [PubMed] [Google Scholar]
- Carlson ME, Hsu M, Conboy IM. Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature. 2008;454:528–532. doi: 10.1038/nature07034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charville GW, Rando TA. Stem cell ageing and non-random chromosome segregation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011;366:85–93. doi: 10.1098/rstb.2010.0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Steele AD, Lindquist S, Guarente L. Increase in activity during calorie restriction requires Sirt1. Science. 2005;310:1641. doi: 10.1126/science.1118357. [DOI] [PubMed] [Google Scholar]
- Chen C, Liu Y, Liu Y, Zheng P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci. Signal. 2009;2:ra75. doi: 10.1126/scisignal.2000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 2005;433:760–764. doi: 10.1038/nature03260. [DOI] [PubMed] [Google Scholar]
- Cowan CA, Atienza J, Melton DA, Eggan K. Nuclear reprogramming of somatic cells after fusion with human embryonic stem cells. Science. 2005;309:1369–1373. doi: 10.1126/science.1116447. [DOI] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang W, Steffen KK, Perry R, Dorsey JA, Johnson FB, Shilatifard A, Kaeberlein M, Kennedy BK, Berger SL. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature. 2009;459:802–807. doi: 10.1038/nature08085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santa F, Totaro MG, Prosperini E, Notarbartolo S, Testa G, Natoli G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell. 2007;130:1083–1094. doi: 10.1016/j.cell.2007.08.019. [DOI] [PubMed] [Google Scholar]
- Dhawan S, Tschen SI, Bhushan A. Bmi-1 regulates the Ink4a/Arf locus to control pancreatic beta-cell proliferation. Genes Dev. 2009;23:906–911. doi: 10.1101/gad.1742609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd IB, Micheelsen MA, Sneppen K, Thon G. Theoretical analysis of epigenetic cell memory by nucleosome modification. Cell. 2007;129:813–822. doi: 10.1016/j.cell.2007.02.053. [DOI] [PubMed] [Google Scholar]
- Donohoe ME, Silva SS, Pinter SF, Xu N, Lee JT. The pluripotency factor Oct4 interacts with Ctcf and also controls X-chromosome pairing and counting. Nature. 2009;460:128–132. doi: 10.1038/nature08098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas PM, Dillin A. Protein homeostasis and aging in neuro-degeneration. J. Cell Biol. 2010;190:719–729. doi: 10.1083/jcb.201005144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Jacobsen SE, Reik W. Epigenetic reprogramming in plant and animal development. Science. 2010;330:622–627. doi: 10.1126/science.1190614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielenbach N, Antebi A. C. elegans dauer formation and the molecular basis of plasticity. Genes Dev. 2008;22:2149–2165. doi: 10.1101/gad.1701508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finerty JC. Parabiosis in physiological studies. Physiol. Rev. 1952;32:277–302. doi: 10.1152/physrev.1952.32.3.277. [DOI] [PubMed] [Google Scholar]
- Fontana L, Partridge L, Longo VD. Extending healthy life span—from yeast to humans. Science. 2010;328:321–326. doi: 10.1126/science.1172539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garinis GA, van der Horst GT, Vijg J, Hoeijmakers JH. DNA damage and ageing: new-age ideas for an age-old problem. Nat. Cell Biol. 2008;10:1241–1247. doi: 10.1038/ncb1108-1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girton JR, Johansen KM. Chromatin structure and the regulation of gene expression: the lessons of PEV in Drosophila. Adv. Genet. 2008;61:1–43. doi: 10.1016/S0065-2660(07)00001-6. [DOI] [PubMed] [Google Scholar]
- Greer EL, Maures TJ, Hauswirth AG, Green EM, Leeman DS, Maro GS, Han S, Banko MR, Gozani O, Brunet A. Members of the H3K4 trimethylation complex regulate lifespan in a germline-dependent manner in C. elegans. Nature. 2010;466:383–387. doi: 10.1038/nature09195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Maures TJ, Ucar D, Hauswirth AG, Mancini E, Lim JP, Benayoun BA, Shi Y, Brunet A. Transgenerational epigenetic inheritance of longevity in Caenorhabditis elegans. Nature. 2011;479:365–371. doi: 10.1038/nature10572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarente L. Sirtuins in aging and disease. Cold Spring Harb. Symp. Quant. Biol. 2007;72:483–488. doi: 10.1101/sqb.2007.72.024. [DOI] [PubMed] [Google Scholar]
- Gurdon JB. Adult frogs derived from the nuclei of single somatic cells. Dev. Biol. 1962;4:256–273. doi: 10.1016/0012-1606(62)90043-x. [DOI] [PubMed] [Google Scholar]
- Haigis MC, Yankner BA. The aging stress response. Mol. Cell. 2010;40:333–344. doi: 10.1016/j.molcel.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna JH, Saha K, Jaenisch R. Pluripotency and cellular reprogramming: facts, hypotheses, unresolved issues. Cell. 2010;143:508–525. doi: 10.1016/j.cell.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayflick L. Biological aging is no longer an unsolved problem. Ann. N Y Acad. Sci. 2007;1100:1–13. doi: 10.1196/annals.1395.001. [DOI] [PubMed] [Google Scholar]
- Herbst A, Pak JW, McKenzie D, Bua E, Bassiouni M, Aiken JM. Accumulation of mitochondrial DNA deletion mutations in aged muscle fibers: evidence for a causal role in muscle fiber loss. J. Gerontol. A Biol. Sci. Med. Sci. 2007;62:235–245. doi: 10.1093/gerona/62.3.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung T, Chang HY. Long noncoding RNA in genome regulation: prospects and mechanisms. RNA Biol. 2010;7:582–585. doi: 10.4161/rna.7.5.13216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ieda M, Fu JD, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, Srivastava D. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell. 2010;142:375–386. doi: 10.1016/j.cell.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaskelioff M, Muller FL, Paik JH, Thomas E, Jiang S, Adams AC, Sahin E, Kost-Alimova M, Protopopov A, Cadiñanos J, et al. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature. 2011;469:102–106. doi: 10.1038/nature09603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DL, Rando TA. Emerging models and paradigms for stem cell ageing. Nat. Cell Biol. 2011;13:506–512. doi: 10.1038/ncb0511-506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–2580. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr. Biol. 2004;14:885–890. doi: 10.1016/j.cub.2004.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman AL, Ashraf JM, Corces-Zimmerman MR, Landis JN, Murphy CT. Insulin signaling and dietary restriction differentially influence the decline of learning and memory with age. PLoS Biol. 2010;8:e1000372. doi: 10.1371/journal.pbio.1000372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, Lin M, McCord RA, Ongaigui KC, Boxer LD, Chang HY, Chua KF. SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell. 2009;136:62–74. doi: 10.1016/j.cell.2008.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon CJ. The genetics of ageing. Nature. 2010;464:504–512. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- Kimura KD, Tissenbaum HA, Liu Y, Ruvkun G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science. 1997;277:942–946. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- Kirkwood TB. Understanding the odd science of aging. Cell. 2005;120:437–447. doi: 10.1016/j.cell.2005.01.027. [DOI] [PubMed] [Google Scholar]
- Kirkwood TB, Shanley DP. The connections between general and reproductive senescence and the evolutionary basis of menopause. Ann. N Y Acad. Sci. 2010;1204:21–29. doi: 10.1111/j.1749-6632.2010.05520.x. [DOI] [PubMed] [Google Scholar]
- Koga H, Kaushik S, Cuervo AM. Protein homeostasis and aging: The importance of exquisite quality control. Ageing Res. Rev. 2011;10:205–215. doi: 10.1016/j.arr.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, Sharpless NE. Ink4a/Arf expression is a biomarker of aging. J. Clin. Invest. 2004;114:1299–1307. doi: 10.1172/JCI22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krizhanovsky V, Lowe SW. Stem cells: The promises and perils of p53. Nature. 2009;460:1085–1086. doi: 10.1038/4601085a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24:2463–2479. doi: 10.1101/gad.1971610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee N, Maurange C, Ringrose L, Paro R. Suppression of Polycomb group proteins by JNK signalling induces transdetermination in Drosophila imaginal discs. Nature. 2005;438:234–237. doi: 10.1038/nature04120. [DOI] [PubMed] [Google Scholar]
- Lee JH, Bugarija B, Millan EJ, Walton NM, Gaetz J, Fernandes CJ, Yu WH, Mekel-Bobrov N, Vallender TW, Snyder GE, et al. Systematic identification of cis-silenced genes by trans complementation. Hum. Mol. Genet. 2009a;18:835–846. doi: 10.1093/hmg/ddn409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Gaetz J, Bugarija B, Fernandes CJ, Snyder GE, Bush EC, Lahn BT. Chromatin analysis of occluded genes. Hum. Mol. Genet. 2009b;18:2567–2574. doi: 10.1093/hmg/ddp188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SJ, Defossez PA, Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000;289:2126–2128. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- Liu L, Rando TA. Manifestations and mechanisms of stem cell aging. J. Cell Biol. 2011;193:257–266. doi: 10.1083/jcb.201010131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh KM, Lim B. Recreating pluripotency? Cell Stem Cell. 2010;7:137–139. doi: 10.1016/j.stem.2010.07.005. [DOI] [PubMed] [Google Scholar]
- Luo S, Kleemann GA, Ashraf JM, Shaw WM, Murphy CT. TGF-β and insulin signaling regulate reproductive aging via oocyte and germline quality maintenance. Cell. 2010;143:299–312. doi: 10.1016/j.cell.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maegawa S, Hinkal G, Kim HS, Shen L, Zhang L, Zhang J, Zhang N, Liang S, Donehower LA, Issa JP. Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res. 2010;20:332–340. doi: 10.1101/gr.096826.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marión RM, Blasco MA. Telomere rejuvenation during nuclear reprogramming. Curr. Opin. Genet. Dev. 2010;20:190–196. doi: 10.1016/j.gde.2010.01.005. [DOI] [PubMed] [Google Scholar]
- Martin GM. Epigenetic gambling and epigenetic drift as an antagonistic pleiotropic mechanism of aging. Aging Cell. 2009;8:761–764. doi: 10.1111/j.1474-9726.2009.00515.x. [DOI] [PubMed] [Google Scholar]
- McCay CM, Pope F, Lunsford W, Sperling G, Sambhavaphol P. Parabiosis between old and young rats. Gerontologia. 1957;1:7–17. doi: 10.1159/000210677. [DOI] [PubMed] [Google Scholar]
- Meissner A. Epigenetic modifications in pluripotent and differentiated cells. Nat. Biotechnol. 2010;28:1079–1088. doi: 10.1038/nbt.1684. [DOI] [PubMed] [Google Scholar]
- Michishita E, McCord RA, Berber E, Kioi M, Padilla-Nash H, Damian M, Cheung P, Kusumoto R, Kawahara TL, Barrett JC, et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. 2008;452:492–496. doi: 10.1038/nature06736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Hanna J, Zhang X, Ku M, Wernig M, Schorderet P, Bernstein BE, Jaenisch R, Lander ES, Meissner A. Dissecting direct reprogramming through integrative genomic analysis. Nature. 2008;454:49–55. doi: 10.1038/nature07056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124:315–329. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- Murgatroyd C, Wu Y, Bockmühl Y, Spengler D. The Janus face of DNA methylation in aging. Aging (Albany NY) 2010;2:107–110. doi: 10.18632/aging.100124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro P, Chambers I, Karwacki-Neisius V, Chureau C, Morey C, Rougeulle C, Avner P. Molecular coupling of Xist regulation and pluripotency. Science. 2008;321:1693–1695. doi: 10.1126/science.1160952. [DOI] [PubMed] [Google Scholar]
- Oberdoerffer P, Sinclair DA. The role of nuclear architecture in genomic instability and ageing. Nat. Rev. Mol. Cell Biol. 2007;8:692–702. doi: 10.1038/nrm2238. [DOI] [PubMed] [Google Scholar]
- Oberdoerffer P, Michan S, McVay M, Mostoslavsky R, Vann J, Park SK, Hartlerode A, Stegmuller J, Hafner A, Loerch P, et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell. 2008;135:907–918. doi: 10.1016/j.cell.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–317. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- Ouyang Z, Zheng GX, Chang HY. Noncoding RNA landmarks of pluripotency and reprogramming. Cell Stem Cell. 2010;7:649–650. doi: 10.1016/j.stem.2010.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccolo FM, Pereira CF, Cantone I, Brown K, Tsubouchi T, Soza-Ried J, Merkenschlager M, Fisher AG. Using heterokaryons to understand pluripotency and reprogramming. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011;366:2260–2265. doi: 10.1098/rstb.2011.0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando TA. Stem cells, ageing and the quest for immortality. Nature. 2006;441:1080–1086. doi: 10.1038/nature04958. [DOI] [PubMed] [Google Scholar]
- Rando OJ, Chang HY. Genome-wide views of chromatin structure. Annu. Rev. Biochem. 2009;78:245–271. doi: 10.1146/annurev.biochem.78.071107.134639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando OJ, Verstrepen KJ. Timescales of genetic and epigenetic inheritance. Cell. 2007;128:655–668. doi: 10.1016/j.cell.2007.01.023. [DOI] [PubMed] [Google Scholar]
- Rideout WM, III, Eggan K, Jaenisch R. Nuclear cloning and epigenetic reprogramming of the genome. Science. 2001;293:1093–1098. doi: 10.1126/science.1063206. [DOI] [PubMed] [Google Scholar]
- Rogina B, Helfand SL. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc. Natl. Acad. Sci. USA. 2004;101:15998–16003. doi: 10.1073/pnas.0404184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossant J. Stem cells from the Mammalian blastocyst. Stem Cells. 2001;19:477–482. doi: 10.1634/stemcells.19-6-477. [DOI] [PubMed] [Google Scholar]
- Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol. Cell. 2007;25:15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- Sallon S, Solowey E, Cohen Y, Korchinsky R, Egli M, Woodhatch I, Simchoni O, Kislev M. Germination, genetics, and growth of an ancient date seed. Science. 2008;320:1464. doi: 10.1126/science.1153600. [DOI] [PubMed] [Google Scholar]
- Sen GL, Reuter JA, Webster DE, Zhu L, Khavari PA. DNMT1 maintains progenitor function in self-renewing somatic tissue. Nature. 2010;463:563–567. doi: 10.1038/nature08683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seviour EG, Lin SY. The DNA damage response: Balancing the scale between cancer and ageing. Aging (Albany NY) 2010;2:900–907. doi: 10.18632/aging.100248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpless NE, DePinho RA. How stem cells age and why this makes us grow old. Nat. Rev. Mol. Cell Biol. 2007;8:703–713. doi: 10.1038/nrm2241. [DOI] [PubMed] [Google Scholar]
- Sinclair DA, Guarente L. Extrachromosomal rDNA circles—a cause of aging in yeast. Cell. 1997;91:1033–1042. doi: 10.1016/s0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- Stadtfeld M, Hochedlinger K. Induced pluripotency: history, mechanisms, and applications. Genes Dev. 2010;24:2239–2263. doi: 10.1101/gad.1963910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada M, Takahama Y, Abe K, Nakatsuji N, Tada T. Nuclear reprogramming of somatic cells by in vitro hybridization with ES cells. Curr. Biol. 2001;11:1553–1558. doi: 10.1016/s0960-9822(01)00459-6. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Tauchi H, Hasegawa K. Change of the hepatic cells in parabiosis between old and young rats. Mech. Ageing Dev. 1977;6:333–339. doi: 10.1016/0047-6374(77)90034-3. [DOI] [PubMed] [Google Scholar]
- Tennen RI, Chua KF. Chromatin regulation and genome maintenance by mammalian SIRT6. Trends Biochem. Sci. 2011;36:39–46. doi: 10.1016/j.tibs.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terranova R, Pereira CF, Du Roure C, Merkenschlager M, Fisher AG. Acquisition and extinction of gene expression programs are separable events in heterokaryon reprogramming. J. Cell Sci. 2006;119:2065–2072. doi: 10.1242/jcs.02945. [DOI] [PubMed] [Google Scholar]
- Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–230. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly-Y M, Gidlöf S, Oldfors A, Wibom R, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- Trowbridge JJ, Snow JW, Kim J, Orkin SH. DNA methyl-transferase 1 is essential for and uniquely regulates hematopoietic stem and progenitor cells. Cell Stem Cell. 2009;5:442–449. doi: 10.1016/j.stem.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaquero A, Reinberg D. Calorie restriction and the exercise of chromatin. Genes Dev. 2009;23:1849–1869. doi: 10.1101/gad.1807009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vastenhouw NL, Zhang Y, Woods IG, Imam F, Regev A, Liu XS, Rinn J, Schier AF. Chromatin signature of embryonic pluripotency is established during genome activation. Nature. 2010;464:922–926. doi: 10.1038/nature08866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijg J, Busuttil RA, Bahar R, Dollé ME. Aging and genome maintenance. Ann. N Y Acad. Sci. 2005;1055:35–47. doi: 10.1196/annals.1323.007. [DOI] [PubMed] [Google Scholar]
- Villeda SA, Luo J, Britschgi M, Park J-S, Ding Z, Grant JL, Couillard-Despres S, Aigner L, Rando TA, Wyss-Coray A. Age-related changes in the systemic milieu regulate adult neurogenesis. Nature. 2011;477:90–94. doi: 10.1038/nature10357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vreeland RH, Rosenzweig WD, Powers DW. Isolation of a 250 million-year-old halotolerant bacterium from a primary salt crystal. Nature. 2000;407:897–900. doi: 10.1038/35038060. [DOI] [PubMed] [Google Scholar]
- Wang MC, O'Rourke EJ, Ruvkun G. Fat metabolism links germline stem cells and longevity in C. elegans. Science. 2008a;322:957–960. doi: 10.1126/science.1162011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, Zhao K. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008b;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RA. Control of the embryonic stem cell state. Cell. 2011;144:940–954. doi: 10.1016/j.cell.2011.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X. Human embryonic stem cells: mechanisms to escape replica-tive senescence? Stem Cell Rev. 2007;3:270–279. doi: 10.1007/s12015-007-9005-x. [DOI] [PubMed] [Google Scholar]