

Abstract
Identification of mutations that cause rare familial forms of Parkinson’s disease (PD) and subsequent studies of genetic risk factors for sporadic PD have led to an improved understanding of the pathological mechanisms that may cause nonfamilial PD. In particular, genetic and pathological studies strongly suggest that alpha-synuclein, albeit very rarely mutated in PD patients, plays a critical role in the vast majority of individuals with the sporadic form of the disease. We have extensively characterized a mouse model over-expressing full-length, human, wild-type alpha-synuclein under the Thy-1 promoter. We have also shown that this model reproduces many features of sporadic PD, including progressive changes in dopamine release and striatal content, alpha-synuclein pathology, deficits in motor and nonmotor functions that are affected in pre-manifest and manifest phases of PD, inflammation, and biochemical and molecular changes similar to those observed in PD. Preclinical studies have already demonstrated improvement with promising new drugs in this model, which provides an opportunity to test novel neuroprotective strategies during different phases of the disorder using endpoint measures with high power to detect drug effects.
Electronic supplementary material
The online version of this article (doi:10.1007/s13311-012-0104-2) contains supplementary material, which is available to authorized users.
Keywords: Parkinson’s disease, Alpha-synuclein, Mouse model, Thy1-aSyn, Progressive
Introduction
Despite decades of improved symptomatic treatment, Parkinson’s disease (PD) remains a devastating disorder that progressively leads to disability and early death because there is no treatment currently available to slow, stop, or reverse the course of the disease [1]. Neurodegeneration in PD primarily affects the nigrostriatal dopaminergic neurons [2]. However, many other neurons degenerate in the course of PD, and extensive pathology leading to neuronal dysfunction may precede the onset of the typical neurological symptoms of PD by many years [3]. Research on PD has long been dominated by the study of nigrostriatal dopaminergic neuron loss and its consequences for the function of basal ganglia motor circuits [4]. Although, presently, this disease is diagnosed based on characteristic motor symptoms that occur when a substantial number of dopaminergic neurons have died, and more subtle motor symptoms, as well as a host of nonmotor symptoms, may precede the onset of characteristic motor deficits and could lead to earlier diagnosis [5]. This prompted many investigators to focus on elucidating mechanisms of cellular dysfunction that precede cell death and can provide new therapeutic targets to stop or reverse the pathological process before neurons are irremediably lost. Early diagnosis is unnecessary and raises ethical concerns in the absence of therapeutic options to stop the course of the disease. However, the prognosis of PD may change in the coming years with the development of both neuroprotective treatments and earlier diagnosis.
An essential step in drug discovery remains the testing of new compounds in animal models of the disease. This is a challenging endeavor, as the predictability of animal models for future clinical applications is often difficult to establish. In fact, the predictive validity of a model can only be established when effective therapies exist for the particular disorder that is being modeled. In the case of PD, models that reproduce the loss of dopaminergic neurons have demonstrated predictive validity for symptomatic treatments that have changed the lives of patients [6]. However, the same models have not yet proven predictive for neuroprotective treatments [7]. Failed clinical trials of drugs that seemed highly promising when tested in these models have led many investigators to turn to other models in the hope of developing neuroprotective strategies for PD. These new models have the combined purpose of helping to understand disease process and providing preclinical evidence of efficacy for new compounds.
PD has long been exclusively considered an environmental disorder, with little or any genetic influence, except for very rare familial forms of the disease [8]. This view has now dramatically changed. Even though the role of accidental risk factors (e.g., traumatic brain injury) or environmental factors (e.g., exposure to agricultural pesticides or industrial solvents) is increasingly documented [9], it is clear that these environmental factors act in concert with genetic risk factors [10, 11]. Furthermore, the identification of several mutations that cause familial forms of PD has revealed mechanisms shared by these rare disease forms and the much more common sporadic forms. For example, many disease-causing mutations affect mitochondrial function, which also appears to be compromised in sporadic PD [12]. More directly, alpha-synuclein, when mutated, causes familial PD, which also accumulates in the brain of all patients with the sporadic form of PD [13]. Furthermore, polymorphisms in the alpha-synuclein gene influence PD risk and rate of progression, either alone or in combination with other risk factors [14–16].
Not surprisingly, these findings led to the generation of a large number of genetic mouse models based on the expression of either mutated or wild-type forms of alpha-synuclein driven by a variety of promoters [17]. These have all contributed important information to the field, but the large number of options may be confusing to investigators in search of the “perfect” model. In fact, perfection is in the eye of the beholder because no model can possibly suit every experimental purpose [18]. In this review, we present an overview of data obtained in one extensively characterized mouse model that has recently emerged as particularly suitable for preclinical drug testing. For clarity, the present report will focus on findings in this model because extensive descriptions of other alpha-synuclein over-expressing mice have been recently published by us and other groups [17–20].
General Description
Mice over-expressing human wild-type alpha-synuclein driven by the murine Thy-1 promoter (Thy1-aSyn mice) (Fig. 1) were developed in the laboratory of Eliezer Masliah at UCSD [21] and were designated “line 61” to differentiate them from a previously published line driven by the PDGF promoter (line D: [22]). The 2 lines markedly differ in the pattern of alpha-synuclein over-expression and level [21, 23]. Only the Thy1-aSyn mice will be described in this review.
Fig. 1.
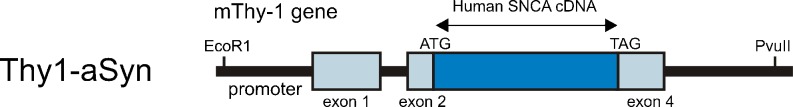
Schematic diagram of Thy1-aSyn construct: over-expression of full length human wild-type alpha-synuclein under the murine Thy-1 promoter. (See Rockenstein, et al., 2002 [21] for details)
Genetic Background and Transgene Insertion
The original line 61 was developed on a C57Bl6/DBA2 background, which may influence some aspects of their phenotype. Our laboratory has generated fully backcrossed Thy1-aSyn mice on the C57Bl6 background. Preliminary characterization of these backcrossed mice indicates that they exhibit similar deficits as the mixed lineage mice, described as follows. Importantly, the transgene is inserted in the X chromosome, which may be the reason for diminished motor deficits in females due to random inactivation of the X chromosome carrying the mutation, described as follows. For this reason, experiments in our laboratory only use male mice, although both males and females lose striatal dopamine at 14 months of age [24].
The Murine Thy-1 Promoter
Importantly, in contrast to other lines of mice using the Thy-1 promoter [25], the Thy1-aSyn mice of line 61 do not exhibit obvious motor neuron pathology [21] and do not show motor neuron deficits in behavioral tests [26]. The absence of motor neuron degeneration in Thy1-aSyn mice is critically important, as their behavioral deficits are more likely to reflect impairments that are relevant to PD and single out the Thy1-aSyn mice as particularly suitable for preclinical drug testing, described as follows. The use of the Thy-1 promoter offers distinct advantages compared to the tyrosine hydroxylase (TH) and prion protein (PrP) promoters (2 promoters often used in other models). Although models using the TH promoter were generated in the hope to reproduce the loss of catecholaminergic neurons, a major pathological aspect of PD, they fail to model the broad alpha-synuclein pathology that has been described in postmortem brains of patients with PD, as well as in incidental Lewy body disease, which may correspond to a pre-manifest phase of PD [3]. In contrast to the TH promoter, which restricts the expression of the transgene to catecholaminergic neurons, the Thy-1 promoter is broadly expressed and drives over-expression of alpha-synuclein in all brain regions (Fig. 2A; Fig. 4; Table 1). In contrast to the Thy1-aSyn, most lines using the PrP promoter do show motor neuron pathology and prominent motor deficits, which often lead to death, and these lines are related to their spinal pathology rather than to their PD-like deficits [17, 18].
Fig. 2.

Brain sections (1 hemisphere) show increased immunostaining for mouse/human aSyn (mouse anti-alpha-synuclein); clone 42, 1:500 (BD Biosciences, San Jose, CA) (A) in Thy1-aSyn mice (5 months old) compared to wild-type (WT) littermates. (B) 3.4-fold increase in aSyn in the SN pars compacta of Thy1-aSyn mice (SNc; arrows in [A]) (Image J, mean + SEM; n = 5; **p < 0.01 Student’s t test)
Fig. 4.

Alpha-synuclein protein over-expression in brain regions of Thy1-aSyn mice assessed by Western blotting using an antibody against mouse and human alpha-synuclein (mouse anti-alpha-synuclein); clone 42, 1:3000 (BD Biosciences, San Jose, CA). Left: Western blot of alpha-synuclein in the substantia nigra. Right: Alpha-synuclein expression (17 kDa) in fold change compared to wild-type (wt). Thy1-aSyn (tg) mice brain in comparison to wt. Normalized to alpha-tubulin for loading control. Substantia nigra (SN), striatum (str), thalamus (thal), cortex (cor), frontal cortex (f.cor), hippocampus (hipp), cerebellum (CB), and olfactory bulb (OB) (mean ± SEM of 3-6 mice; *p < 0.05; **p < 0.01; ***p < 0.001; 2-way repeated measures (RM) analysis of variance, main genotype effect p < 0.001, student’s t-test planned comparison for each subregion
Table 1.
Alpha-Synuclein Over-Expression in Brain Regions of Thy1-aSyn Mice Assessed with Immunohistochemistry
| Brain Regions | Wild-Type | Thy1-aSyn | ||
|---|---|---|---|---|
| Substantia nigra pars reticulata | 13,821 | ± 2466 | 20858 | ± 2621 |
| Substantia nigra pars compacta | 5175 | ± 611 | 17,536* | ± 761 |
| Thalamus | 4465 | ± 457 | 28,209* | ± 616 |
| Dentate gyrus | 19,975 | ± 1252 | 39,856* | ± 2305 |
| Hippocampus CA1 | 18,361 | ± 1540 | 43,918* | ± 1529 |
| Hippocampus CA2 | 22,786 | ± 1722 | 40,252* | ± 2027 |
| Hippocampus CA3 | 23,949 | ± 1979 | 43,739* | ± 2301 |
| anterior olfactory nucleus | 21,067 | ± 2336 | 41,296* | ± 2986 |
| Olfactory bulb, granule cell layer | 17,675 | ± 1870 | 26,421* | ± 2167 |
| Orbital/association cortex | 17,448 | ± 2215 | 43,714* | ± 2679 |
| Motor cortex | 12,472 | ± 746 | 38,101* | ± 2816 |
| Sensory cortex | 12,249 | ± 1008 | 32,126* | ± 1726 |
| Striatum | 14,285 | ± 1232 | 21,692 | ± 1510 |
Anti-alpha-synuclein antibody that recognizes both mouse and human alpha-synuclein, mouse anti-alpha-synuclein; clone 42, 1:500 (BD Biosciences, San Jose, CA); immunofluorescence intensity units, mean ± SEM; *p < 0.05
Human Wild-Type Alpha-Synuclein
The vast majority of PD patients do not show mutations in alpha-synuclein. Therefore, even though alpha-synuclein carrying mutations found in patients (i.e., A53T, A30P, E46K, or combined A53T and A30P, or specially designed mutations to increase aggregation) may cause a faster pathology than wild-type alpha-synuclein [18], the over-expression of the nonmutated protein is closer to the situation encountered in sporadic PD. The use of a model expressing the wild-type human protein to mimic sporadic PD is clearly validated by genetic studies showing that the multiplication of the gene encoding wild-type alpha-synuclein causes familial forms of PD, and polymorphisms in noncoding regions of the gene increase the risk of PD and cause an earlier onset or a faster progression of the disease [14–16].
Alpha-Synuclein Over-Expression Occurs in the First Postnatal Weeks in Thy1-aSyn Mice
Over-expression of a transgene during development can cause unwanted effects that are unrelated to pathology occurring in humans later in life. Driven by the Thy-1 promoter, alpha-synuclein over-expression does not begin until postnatal day 10, thus avoiding this potential problem. However, this limits the use of this model for cell culture experiments that require harvesting tissue during the prenatal or immediately postnatal period, unless they are kept long enough to express the transgene at the corresponding postnatal age (J. Watson and C. Ghiani, UCLA, personal communication).
Lifespan and General Health in Thy1-aSyn Mice
Up to 14 months of age, prior to dopamine loss, described as follows, Thy1-aSyn mice do not show increased mortality or morbidity, and they do not need special husbandry. Male Thy1-aSyn mice show a slightly decreased body weight gain starting at 4 months of age with a body weight difference of approximately 3 g compared to the same age wild-type littermates. Aged wild-type mice can reach body weights of 50 g or more, whereas aged Thy1-aSyn mice weigh on average approximately 31 g. Beyond 14 months of age, mortality increases in Thy1-aSyn mice, and their survival can be improved by providing moistened mashed food and avoiding stress.
Alpha-Synuclein Pathology in the Thy1-aSyn Mice
Thy1-aSyn Mice Express Human Alpha-Synuclein in Nigrostriatal Dopaminergic Neurons
As previously discussed, even though widespread over-expression of alpha-synuclein is preferable to expression restricted to catecholaminergic neurons to model PD, it is important to ensure that alpha-synuclein over-expression does occur in the nigrostriatal dopaminergic neurons, which are severely affected in the disease. In contrast, models with primary accumulation of alpha-synuclein in the forebrain more closely model dementia with Lewy bodies, in which severe cognitive deficits appear earlier than extrapyramidal motor deficits [23, 27].
Both mRNA measurements in laser-captured nigrostriatal dopaminergic neurons (Fig. 3A, B) and immunohistochemical detection of human alpha-synuclein in the substantia nigra [21] (Fig. 2B) indicate that the Thy1-aSyn mice express the transgenic protein in dopaminergic neurons of the substantia nigra pars compacta.
Fig. 3.

Human alpha-synuclein (SNCA) is expressed in nigral dopaminergic neurons of Thy1-aSyn mice. (A) Gel electrophoresis of polymerase chain reaction (PCR) products (cDNA synthesized from amplified mRNA of 500 neurons per sample) from laser-captured microdissected tyrosine hydroxylase (TH)-positive neurons of the substantia nigra from wild-type (WT) and Thy1-aSyn transgenic (TG) mice. Human WT alpha-synuclein (SNCA 68 bp) is expressed in samples of TG mice only (positive control [+Ctrl] is cDNA from total human brain RNA). Laser captured nigral neurons used for this analysis express TH (Th, ~200 bp) and the housekeeping gene hypoxanthine guanine phosphoribosyl transferase (Hprt, ~200 bp), but not glutamic acid decarboxylase 1 (Gad1, gamma-aminobutyric acid (GABA)ergic neuron marker, ~200 bp) and glial fibrillary acidic protein (Gfap, glial cell marker, ~200 bp). Positive control for Th, Hprt, Gad1, and Gfap is cDNA from total mouse brain RNA. Negative control is a nontemplate control (water, H2O). (B) Relative expression of SNCA transcript in TH-positive neurons of the substantia nigra pars compacta (quantitative real-time PCR, n = 3 per group)
Thy1-aSyn Mice Express Moderate Levels of Human Alpha-Synuclein
One concern with transgenic models is that massive over-expression of a foreign protein could lead to nonspecific disruption of cell function, unrelated to the effects of moderate protein accumulation, as seen in diseases. Several approaches can be used to estimate the level of over-expression of alpha-synulein in transgenic animals. None is completely reliable, because the protein likely takes on various forms that can react differently with antibodies used either for immunohistochemistry or Western blots. Furthermore, changes in conformation can affect the migration of the protein in the gel, with aggregated proteins remaining in the stacking gel. Therefore, it is impossible to be sure that all transgenic protein is detected. Nevertheless, these approaches provide useful estimates.
We have measured the level of expression of alpha-synuclein in several brain regions of Thy1-aSyn mice by using a novel method for quantitatively assessing immunofluorescence levels with a microarray scanner. This approach is more reliable than traditional optical density measurements of immunohistochemical detection of protein with horseradish peroxidase-linked secondary antibodies and diaminobenzidine as a substrate. Indeed, the use of fluorescent dye-conjugated antibodies eliminates the nonlinear enzymatic step, and the use of a microarray scanner to assess fluorescence levels ensures a broad linear range for the measurements. With this approach, we found that alpha-synuclein, detected with an antibody that recognizes both mouse and human proteins, was over-expressed to a moderate degree (1.5- to 3.4-fold) in most brain regions examined in Thy1-aSyn mice compared to their wild-type littermates, with a higher (6.5-fold) over-expression in the thalamus (Table 1). A limitation of this approach is that levels will be underestimated in regions with high tissue heterogeneity, such as the mature striatum, which is crossed by myelinated fiber tracks. A moderate level of over-expression was confirmed by Western blot analysis in striatum, and this approach showed a similar range of expression as the histological approach in most other brain regions, with the exception of the cerebellum, which showed a much larger increase by Western blot than by immunohistochemical detection (Fig. 4) On the Western blots of brain tissue from Thy1-aSyn mice, antibodies that recognize both mouse and human or only human alpha-synuclein detect several bands, often described as “dimers” or “oligomers” in the literature (Fig. 4). However it is not possible to ensure that these higher molecular bands correspond to structures present in the tissue of origin rather than formed during processing. Therefore, a more conservative interpretation is warranted.
Together, these data indicate that Thy1-aSyn mice over-express alpha-synuclein across many brain regions to levels that are in the range of what would be expected to occur in the brains of individuals with alpha-synuclein gene triplication, a cause of familial PD. Accordingly, the levels of alpha-synuclein over-expression in this model is unlikely to cause nonspecific toxic effects simply due to high levels of protein over-expression.
Proteinase K Resistant Alpha-Synuclein Aggregates are Present in the Brain of Thy1-aSyn Mice
Alpha-synuclein is present in insoluble protein aggregates in PD [28] (Fig. 5A, B). A moderate treatment with proteinase K can be used before the immunostaining to hydrolyze soluble proteins while maintaining the integrity of formaldehyde fixed tissue and retaining insoluble protein aggregates [29]. In postmortem human brains, proteinase K resistant alpha-synuclein aggregates correlate with Lewy body pathology, supporting the significance of these aggregates in synucleinopathies [30]. When submitted to moderate proteinase K treatment before immunohistochemical detection of alpha-synuclein, proteinase K-resistant alpha-synuclein aggregates are evident in sections of several brain regions in Thy1-aSyn mice. These aggregates range in size from <1 μm to 16 μm, and include small spherical aggregates, larger clumps, and sometimes seem to cover the entire neuronal cell body (Fig. 5C). Interestingly, in contrast to levels of alpha-synuclein detected without proteinase K treatment, the number and size of aggregates vary considerably from region to region. In the brain of 1-month-old Thy1-aSyn mice, small punctate aggregates are abundant in the dorsal nucleus of the vagus and olfactory bulb, whereas larger aggregates are found throughout the dorsal lateral geniculate nucleus, thalamus, locus coeruleus, and cerebellum. Proteinase K resistant aggregates of variable sizes are detected in the substantia nigra at 5 months. In contrast, proteinase K-resistant aggregates are not present in the cerebral cortex, despite high levels of expression of the transgenic human protein. This pattern is consistent across ages, with increases in size and number of aggregates in older mice. This indicates that the ability to form aggregates of alpha-synuclein is regionally specific and that the model reproduces features of PD rather than those observed in other synucleinopathies (e.g., diffuse Lewy body disease in which cortical pathology is severe) [21].
Fig. 5.

Alpha-synuclein pathology in human (A, B) and mouse (C) substantia nigra neurons. (A) Lewy body (black arrow) in melanin containing nigral neuron of a Parkinson’s disease (PD) patient, hematoxylin & eosin stain. (B) Alpha-synuclein accumulated in melanin containing nigral neuron of a PD patient. (C) Proteinase K resistant alpha-synuclein aggregate in substantia nigra of a 5-months-old Thy1-aSyn mouse detected with mouse anti-alpha-synuclein; clone 42, 1:250 (BD Biosciences, San Jose, CA). Scale bar = 20 μm. (Figures [A] and [B] courtesy of Spencer Tung and Harry V. Vinters)
Thy1-aSyn Mice Show High Levels of 129S-Phosphorylated Alpha-Synuclein
Levels of phosphorylation at serine 129 of alpha-synuclein are markedly increased in PD [31]. In fact, this may be the predominant form of alpha-synuclein found in Lewy bodies, the characteristic protein aggregates present throughout the brain and peripheral nervous system in PD. The Thy1-aSyn mice also reproduce this key feature of PD with high levels of alpha-synuclein phosphorylated at serine 129 in ventral mesencephalon/substantia nigra, striatum, cortex, frontal cortex, and hippocampus (Fig. 6A-C). No increases in phosphorylation of alpha-synuclein were observed in thalamus, cerebellum, and olfactory bulb. The cellular distribution of Ser129–P alpha-synuclein also changes dramatically in the transgenic mice compared to wild-type mice. In wild-type mice, phosphorylated S129 alpha-synuclein is detected in nerve terminals, but not in the cytoplasm of neurons in the cortex, hippocampus, and substantia nigra. In Thy1-aSyn mice phosphorylated S129 alpha-synuclein is nearly absent from nerve terminals, but accumulates in the cell soma and nucleus in these brain regions (Fig. 6A).
Fig. 6.

Serine 129 phosphorylated alpha-synuclein protein accumulation in brain regions of Thy1-aSyn mice (antibody is rabbit anti-phosphoser129-alpha-synuclein (Abcam, USA; at 1:500 dilution, ab59264). (A) Immunostaining in the substantia nigra of wild-type (WT) (black arrow points to dark brown stained fibers) and Thy1-aSyn mice (black arrow points to dark brown cytoplasmic and nuclear staining) at 5 months. Scale bar = 20 μm. (B) Western blot of phosphorylated alpha-synuclein in the substantia nigra. (C) Serine 129 phosphorylated alpha-synuclein protein as fold change compared to wild-type (wt). Thy1-aSyn (tg) mice brain in comparison to wt. Normalized to alpha-tubulin for loading control. Substantia nigra (SN), striatum (str), thalamus (thal), cortex (cor), frontal cortex (f.cor), hippocampus (hipp), cerebellum (CB), and olfactory bulb (OB) (mean ± SEM of 3-6 mice; *p < 0.05; **p < 0.01; ***p < 0.001; 2-way RM analysis of variance, main genotype effect p < 0.001, student's t-test planned comparison for each subregion
Alpha-Synuclein Accumulation in Peripheral Neurons of the Thy1-aSyn Mice
The pattern of expression of human alpha-synuclein in the Thy1-aSyn mice has not been fully examined in all regions of the peripheral nervous system. In the myenteric plexus of the distal colon, abundant alpha-synuclein varicose terminals surrounded neurons expressing immunoreactivity to peripheral choline acetyl transferase, indicating that they are cholinergic. In contrast, few alpha-synuclein terminals contacted neurons immunoreactive for neuronal nitric oxide synthase. No proteinase K-resistant alpha-synuclein aggregates were observed in myenteric neurons at 8 months of age (Wang et al., submitted). Alpha-synuclein pathology is observed in the enteric nervous system in PD patients [32, 33], and abnormal accumulation of alpha-synuclein in these neurons may contribute to the digestive pathology observed both in PD patients and in the Thy1-aSyn mice, described as follows.
Progressive Nigrostriatal Pathology in Thy1-aSyn Mice
Thy1-aSyn Mice Exhibit a Progressive Loss of Striatal Dopamine
Based on the particular vulnerability of nigrostriatal dopaminergic neurons in PD, the presence of progressive alterations in these neurons is usually considered an essential requirement for a model of PD [34]. The Thy1-aSyn mice lose 40% of striatal dopamine by 14 months of age [24] (Fig. 7A). This is accompanied by a more moderate loss of TH, the rate limiting enzyme of dopamine synthesis, as detected by Western blot (−23%) and immunoreactivity (−17%). Importantly, this loss of striatal dopamine is progressive, with normal levels of dopamine (and TH) being present at 8 and even 12 months of age. These data indicate that, once initiated, the loss of dopamine progresses rapidly, suggesting that age-related factors precipitate the expression of this pathology. Accordingly, this model is experimentally useful to identify the factors that trigger the demise of nigrostriatal dopaminergic neurons, which, if better known, could lead to therapeutic strategies to delay the onset of disease in persons at risk.
Fig. 7.

Parkinsonism-like phenotype in Thy1-aSyn mice aged 14 months. (A) Thy1-aSyn mice show a 40% loss of dopamine in the striatum compared to age matched wild-type (WT) mice (mean ± SEM; n = 8; *p < 0.05; Student’s t test). (B) Thy1-aSyn mice need longer to remove a sticker from their nose compared to wild-type. This behavioral deficit can be reversed by L-dopa (mean ± SEM; n = 6-7; **p < 0.01 compared to wild-type vehicle, Mann-Whitney U test). (See Lam, et al., 2011 [24] for details)
Evidence from brain imaging studies suggests that axonal pathology precedes dopaminergic cell loss in PD [35]. However, even when examined at 22 months of age, Thy1-aSyn mice did not show any reduction in the number of TH-positive neurons, the classical marker for dopaminergic neurons, in the substantia nigra pars compacta, even though their diameter was slightly reduced. Thus, the model lacks a cardinal feature of PD, but reveals a progressive process of terminal loss. This observation is not unusual for mouse models of neurodegenerative diseases, and dopaminergic cell loss has been reported in only 2 of the many mouse models over-expressing alpha-synuclein, a line expressing a doubly mutated protein, which is not found in patients and in a line expressing a truncated alpha-synuclein [36, 37]. Similarly most other mouse models of PD based on disease-causing mutations also lack dopaminergic cell death, although a conditional model of Parkin deletion has recently been reported to show progressive loss of dopaminergic neurons, suggesting that early deletion of the gene elicits compensatory mechanisms [18, 38].
It is possible that a combination of genetic and environmental manipulations is necessary to elicit cell death in Thy1-aSyn mice, as suggested by the “multiple hits” hypothesis of PD [33, 39]. However, the pesticide Paraquat caused a 25% loss of nigrostriatal dopaminergic neurons both in wild-type and Thy1-aSyn mice, indicating that its effect on dopaminergic cell death was not increased in the transgenic mice, despite a marked increase in alpha-synuclein aggregation [29]. This is reminiscent of experiments in other lines of alpha-synuclein over-expressing mice that showed either no effect or neuroprotection, perhaps due to an increase in neuroprotective mechanisms in the transgenics [40]. As both environmental and genetic risk factors for PD become better known, it is likely that experimental manipulations will be found that cause dopamine cell loss in the Thy1-aSyn mice. In the meantime, neuroprotective strategies can be tested before and after the onset of striatal dopamine loss to determine whether they can prevent or slow down this critical aspect of PD pathology, which likely precedes cell body loss in patients as well [35].
Thy1-aSyn Mice Show Progressive PD-Like Motor Deficits
In humans, the loss of dopamine in the striatum, a brain region involved in the control of movement, leads to akinesia, rigidity, and tremor [2]. At 14 months of age, Thy1-aSyn mice show decreased locomotion in the open field, catalepsy in the block test, and a slowness in sensory motor tests, which, as in humans, is reversed by the dopamine precursor L-dopa [24, 41] (Fig. 7B). With increasing age, progressively more severe deficits are evident from the decreased ability of mice to eat standard chow, thus requiring moistened mashed food. Mice showed increased akinesia in their home cage or during handling. Reduced motor activity together with decrease in fine motor skills results in reduced grooming, which is evident by rough and dirty fur. The mice huddle in cage corners in hunched postures, which is reminiscent of the behavior of PD patients before the onset of L-dopa therapy. Pharmacological treatment with L-dopa may help prolong the life of these mice up to a point when cell loss might occur, but this is not the most critical aspect of this model; its value resides in its potential to help us understand the early features of the disease that may be the most amenable to treatment to reverse or slow the progressive demise of neurons and the extranigral pathology that occurs in PD.
Loss of Striatal Dopamine is Preceded by Increased Extracellular Striatal Dopamine in Thy1-aSyn Mice
Extracellular dopamine levels in the striatum of these mice were examined from an early age with in vivo microdialysis [24]. Tonic striatal extracellular dopamine and 3-methoxytyramine levels were elevated in Thy1-aSyn mice at 6 months, prior to any reduction in total striatal tissue content. Dopamine clearance and amphetamine-induced dopamine efflux were unchanged. These changes in dopamine release were accompanied by hyperactivity in the open field. Indeed, analysis of distance traveled (Fig. 8A), move time, number of move episodes, movement velocity (Fig. 8B), distance per move episode, move time per episode, and center entries in the open field at 4 to 5 months and at 6 to 7 months revealed a significant increase in all parameters in the Thy1-aSyn mice, except for number of move episodes, which significantly decreased in Thy1-aSyn mice, indicating a hyperactive phenotype. When placed in a mesh cylinder, Thy1-aSyn mice climb more and with a reduced latency at 4 to 5 months (Fig. 8C), a manifestation of the hyperactivity they also manifest in the open field at this age. This unexpected observation is not an isolated feature of the Thy1-aSyn mice. Indeed, other lines of mice over-expressing alpha-synuclein show a phase of hyperactivity at an early age, suggesting that they may exhibit similar increases in extracellular dopamine, which has been shown to cause hyperactivity [18]. More intriguingly, imaging studies in LRRK2 carriers also reveal alterations compatible with excess extracellular dopamine, just as we observed in the Thy1-aSyn mice [42].
Fig. 8.

Hyperactivity at 4 to 5 months of age. Thy1-aSyn mice (A) travel more distance and (B) show higher velocity in the open field and show increased numbers of climbing in a wire cage cylinder compared to wild-type (WT) mice (mean ± SEM; *p < 0.05 Student’s t test; n = 10-11 per group)
It is conceivable that dysregulation of striatal dopamine is an early feature of PD, occurring many years before the loss of dopamine that leads to the characteristic features of the disease and to its diagnosis. This early dysregulation could contribute to the later demise of the neurons, as dopamine has repeatedly shown to be toxic when it escapes normal regulatory mechanisms [43, 44]. In support of this hypothesis, polymorphisms in genes involved in dopamine regulation, such as the vesicular and cytoplasmic dopamine transporters have been associated with changes in PD risk in humans and modulation of toxicity to dopamine neurons in experimental models [45, 46], and lack of the vesicular transporter induces a loss of dopaminergic neurons in mice [47].
Thy1-aSyn Mice Reproduce Multiple Deficits Observed in Pre-Manifest PD
Although the loss of nigrostriatal dopaminergic neurons remains the most characteristic feature of PD, much attention has recently been devoted to the wide range of nonmotor symptoms that are present in these patients [5]. Some of these nonmotor symptoms are improved by dopaminergic therapy, but most are not, and their presence many years before the onset of the dopamine-responsive neurological symptoms suggests that they may be due to the dysfunction of other neuronal systems. This would explain that they are not always reproduced in models based on toxin-induced selective dopaminergic cell loss [48]. Indeed, extensive extra-nigral alpha-synuclein pathology is detected in PD brains and may be present even before the onset of motor symptoms [3]. This may cause neuronal dysfunction that is responsible for nondopaminergic deficits in patients [49].
Data in patients, however, are only correlative, and it is important to determine whether modest alpha-synuclein over-expression is sufficient to cause all deficits observed in PD. The Thy1-aSyn mice provide an ideal model to test this hypothesis because, as previously discussed, they show broad over-expression of alpha-synuclein, both in the central and peripheral nervous system, and also a delayed nigrostriatal pathology, suggesting that they may reproduce the extended pre-manifest phase of the disease.
Thy1-aSyn Mice Exhibit Early and Sustained Olfactory Deficits
Olfactory deficits are one of the most frequent nonmotor deficits observed in PD, and their presence may precede motor impairments by many years [50]. We have used 3 different tests of olfactory function in Thy1-aSyn mice 3 and 9 months old and their wild-type littermates [51]. The transgenic mice showed deficits at both ages in the woodblock test in which mice are first exposed to 4 blocks previously buried in their own bedding and then to a block previously buried into the bedding of another mouse. The increased sniffing shown by wild-type mice in response to the odor from the different mouse is significantly reduced in Thy1-aSyn mice. To rule out a decreased interest in social cues, mice were subsequently tested in a habituation/de-habituation test in which mice are exposed to cotton pellets infused with distinct odors. Both genotypes sniffed the cartridge containing the scented cotton at first presentation, although the response of the transgenic was often blunted, and then habituated to the odor by decreasing their sniffing time when presented the same odor several times. When the stimulus was switched to a novel odor, the increased sniffing observed in wild-type mice was again markedly decreased in transgenics. Finally, to eliminate any differences in sniffing abilities, mice were tested in the buried pellet test. After moderate food deprivation, mice were put in a cage with a cereal pellet buried in the bedding. The time to find the pellet was measured on 5 consecutive trial days in which the location of the pellet was changed each day. To control for differences in appetite or motor abilities, mice where then tested to find the pellet placed on the surface of the bedding. Transgenic mice took considerably more time on average to find the buried pellets but were identical to wild-type mice in finding the surface pellet. Taken together, deficits in the 3 tests strongly support a decrease in olfactory function. Both detection and discrimination were affected, without total loss of olfaction, as is usually reported in patients. The deficits were not progressive, again as described in PD, suggesting that they are established very early in the course of the disease [51]. These data indicate that alpha-synuclein pathology is sufficient to induce olfactory deficits, but the mechanism of this effect is not fully understood. Thy1-aSyn mice show aggregates of alpha-synuclein in the olfactory bulb, but could also have deficits in neurogenesis, as described in other lines of mice over-expressing alpha-synuclein [51, 52].
Thy1-aSyn Mice Exhibit Colonic Motor Alterations from a Young Age
Thy1-aSyn mice have gut dysfunction indicated by reduced basal colonic transit and increased response to stress in 12-month-old mice [53]. Younger Thy1-aSyn mice (2.5-3, 4-5, or 7-8 months old) showed marked reductions in fecal pellet output when exposed to a novelty stress compared with age-match wild-type littermates (Fig. 9) (Wang et al., submitted). By contrast, Thy1-aSyn mice aged 4 to 18 months did not show delay in basal gastric emptying of solid or semi-liquid meals. Although both deficits in gastric emptying and colonic motor function have been described in patients, epidemiological studies indicate that decreased colonic motility can occur many years before the onset of motor symptoms, with constipation being the best predictor of developing PD in a large, retrospective study [54, 55]. Furthermore, neuropathological analyses indicate that alpha-synuclein pathology can occur very early in myenteric neurons in PD, prompting the provocative hypothesis that the pathological process could start in the gut [32, 33]. Our data in Thy1-aSyn mice [53] indicated for the first time that alpha-synuclein over-expression is sufficient to induce colonic deficits, an observation later confirmed in other lines of mice [56]. However, neither study addressed if the digestive deficits are of central or peripheral origin, and this question remains unanswered.
Fig. 9.
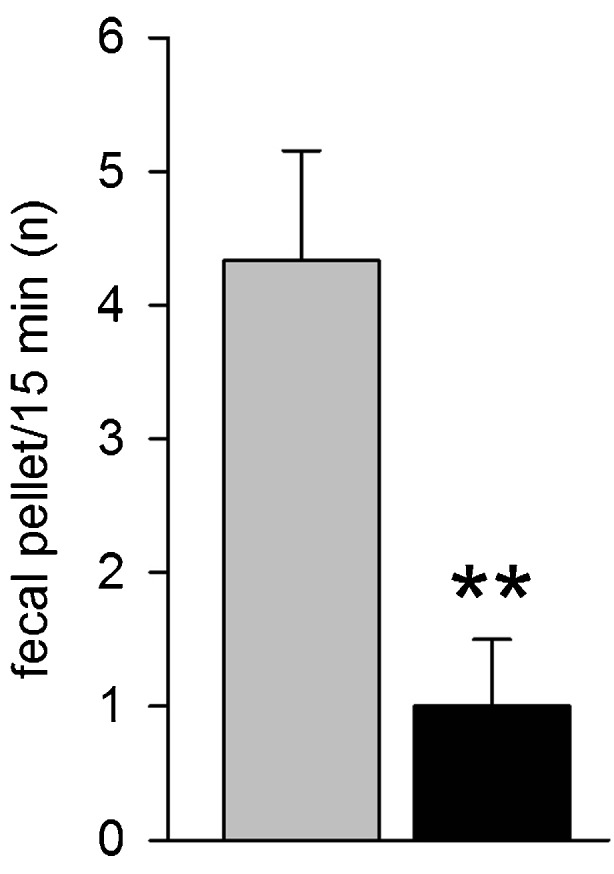
Early nonmotor deficits in Thy1-aSyn (black) versus wild-type (grey). Colonic dysfunction at 4 to 5 months (n = 8-12) (mean + SEM; **p < 0.01 Student’s t test)
Thy1-aSyn Mice Show Disrupted Circadian Rhythm
Circadian rhythm alterations are frequent in PD patients and accompany early perturbations in sleep patterns with insomnia, sleep fragmentation, and daytime sleepiness [5, 57]. The Thy1-aSyn mice also show severe disruptions in the circadian regulation of their locomotor activity from an early age (3-4 months) with reduced amplitude of rhythms and greater fragmentation in their activity/rest cycle. These circadian alterations worsened progressively up to 12 months of age [58]. Sleep patterns were also altered in Thy1-aSyn mice, but a more thorough study of sleep in this model is warranted. Interestingly, although the peak/trough of the period 2 gene was unaffected in the suprachiasmatic nucleus, the daytime firing of neurons in this region was reduced in the transgenics [58]. These data suggest that the Thy1-aSyn mice provide a useful platform to further elucidate mechanisms of circadian rhythm and possibly sleep dysregulation in PD.
Thy1-aSyn Mice Exhibit Early Deficits in Cognitive Domains that are Affected in PD
Mild cognitive dysfunction can be observed at early stages of PD, and its presence may predict later development of more severe cognitive troubles [59]. At 4 to 6 months of age, Thy1-aSyn mice made fewer spontaneous alternations in the Y-maze, and showed deficits in tests of novel object recognition (NOR), object-place recognition, and operant reversal learning, compared to age-matched wild-type littermates (Fig. 10) [60]. In the operant-reversal task, deficits were not apparent at the acquisition phase of the task, in which mice had to achieve the initial criterion before reversing the contingency, but only in the reversal phase, indicating that Thy1-aSyn mice could see and learn a rule as efficiently as their wild-type littermates, but they had difficulties switching to a new, reversed rule. This is reminiscent of the cognitive inflexibility reported in PD, as patients learn a sequential learning task as accurately as controls but tend to make more errors when switched to a new sequence [61]. These data indicate that cognitive impairments that resemble early PD manifestations are reproduced by alpha-synuclein over-expression in mice [60, 62].
Fig. 10.

Early nonmotor deficits in Thy1-aSyn (black) versus wild-type (grey). Cognitive deficits in (A) One-trial object-place recognition test at 4 to 5 months (mean + SEM; n = 10-12; *p < 0.05 compared to wild-type; Student’s t test), (B) Y-maze at 7 to 9 months (mean + SEM; **p < 0.01; n = 10-12; Student’s t test), and (C) novel object recognition test at 4 to 5 months (mean + SEM; n = 17-18; *p < 0.01 Student’s t test).
Thy1-aSyn Mice Exhibit Increased Anxiety
Most studies indicate that affective disorders are extremely frequent in PD and can include increased anxiety, depression, and apathy. When tested in classical Pavlovian fear conditioning at 4 months of age, Thy1-aSyn mice show enhanced freezing compared to wild-type mice during the tone fear test, but not during the context test [63]. The increased tone fear response was retained for 1 month in the transgenics, whereas fear extinction was no different when compared to wild-types and this was not due to increased sensitivity to pain during fear acquisition. Overall, Thy1-aSyn mice show a phenotype consistent with increased anxiety, which can be masked in some tests by their increased activity and impulsivity [64]. A more thorough exploration of depressive behaviors remains to be performed in these mice, but this is challenging because their motor deficits prevent the use of the classical Porsolt swim test, and their olfactory impairments may interfere with other tests.
Thy1-Asyn Mice Show Early Motor Deficits in Challenging Tests
Interestingly, recent studies indicate that carriers of the LRRK2 mutation, a frequent cause of familial PD, display early deficits in challenging motor tests, before the onset of parkinsonian symptoms [65]. Young Thy1-aSyn mice also show progressive deficits in challenging motor tests, such as a beam overlaid with a grid, the pole test, or when observing limb movements in a cylinder [26]. For the challenging beam test, mice are trained to run toward their home cage on a narrowing beam; on the day of the test, a mesh grid is positioned approximately 1 cm above the beam and mice are videotaped during 5 runs. Time to traverse, number of steps, and number of foot slips (or “errors” easily visible through the grid) are recorded. The number of “errors per step” proved to be the most reliable and robust measure to detect drug effects in this test, with 16 mice being sufficient to detect a 30% change with 80% power. Deficits were recorded as early as 2 months of age and increased progressively up to 8 months of age (Fig. 11A). These deficits proved to be extremely stable for many generations of mice and still continue to be reliably detected almost 10 years after we first observed them in this line of mice (Fig. 11B). Deficits increase with age and are severe in 14-months-old males (Fig. 11C), with females being less affected, as previously discussed. Although they require the use of challenging motor tests, these deficits are in no way subtle, even at the early ages, and they provide robust endpoint measures for evaluating drug effects (Table 2).
Fig. 11.

Errors per step on the challenging beam. Thy1-aSyn (also known as ASO) mice make more errors compared to wild-type (WT) as early as 2 months of age. (A) Originally published (see Fleming, et al., 2004 [26] for details) (n = 7 Thy1-aSyn and n = 17 wild-type), and (B) observed in 2010 to 2011 (n = 19 at 2 and 4 months, n = 4-8 at 6 months) (mean ± SEM, **p < 0.01 compared to wild-type; ^^p < 0.01 compared to 2 months of age; 2-way RM analysis of variance). Note the high robustness of the progressive deficit before and after 6 years of breeding (more than 12 generations of mice). (C) This deficit persists and progresses to 14 months of age, when dopamine loss occurs. Female mice show a strong trend to increased errors per step at this age (mean ± SEM; n = 4-5 males; n = 7-11 females; **p < 0.01 Student’s t test)
Table 2.
Power Analysis for Major Endpoints in Thy1-aSyn Mice
| Tests | Parameters with Impairment or Changes in Thy1-aSyn Mice (Major Endpoints)-Age | n for 80% Power to Detect % Improvement |
|---|---|---|
| Beam motor test | Errors per step (increased) 4 months | n = 16 for 30% |
| Pole motor test | Time to turn, time to descend (increased) 4 months | n = 16 for 30% |
| Open field test | Total move time (increased in young mice) 7 months | n = 2 for 30% |
| Olfaction skills | Time to find buried food reward (increased) 4.5 months | n = 16 for 40% |
| Cognitive function | Object place recognition (decreased) 4.5 months | n = 9 for 50% |
| Cognitive function | Alternations in Y-maze (decreased) 7-9 months | n = 7 for 50% |
| Adhesive removal | Time to remove adhesive sticker (increased) 14 months | n = 18 for 30% |
| Catalepsy test | Latency to move (increased) 14 months | n = 20 for 50% |
| Substantia nigra | Alpha-synuclein positive aggregates (transgenics) 5 months | n = 6 for 30% |
| Inflammation | Microglia (IBA-1) activation (increased) 5 months | n = 6 for 12% |
| Striatum HPLC | Dopamine and metabolites (decreased) 14 months | n = 18 for 50% |
“Power” is defined as the probability of correctly rejecting the null hypothesis. The Student’s t test sample size is determined using the following parameter: expected difference in means, expected standard deviation (drawn from previous experience with data), desired power, and alpha (probability to incorrectly rejecting the null hypothesis, set to 0.05). Sufficiency is considered to be 80% power. HPLC = High-performance liquid chromatography
The challenging beam assesses general motor activity, but more specifically fine motor control that is necessary to accurately grasp the grid. Another advantage of this test is that it can be repeated (although, too frequent motor testing should be avoided, because exercise and environmental enrichment simulated by frequent handling can have a protective effect in a number of mouse models of neurodegenerative diseases). Indeed, we found that the deficit is stable or increases when the test is performed several times at 2 months intervals in the same mice [26, 66]. This allows for mouse comparison of progression and eventually drug effects when the test is performed before, and at several time points during drug treatment.
Other measures of fine motor skills are altered in the Thy1-aSyn mice. Nest building is a simple test that avoids interference from the investigator and minimizes stress to the animal [26]. Mice are housed individually for the duration of the test only. Cotton is placed in the feeding bin and measured before, and after 24, 48, and 72 h; both male and female mice grasp cotton to build a nest in their cage, and the amount of cotton left in the bin reflects front paw dexterity. At 4 months of age, the Thy-1-aSyn mice show a marked decrease in their ability to pick up the cotton compared to their wild-type littermates, a deficit that greatly increases at 8 months of age. To control for decreased interest in nest building rather than impaired motor skills, cotton is placed in the cage and the remaining amount is measured. Although Thy1-aSyn mice do show a delay in using the cotton, they rapidly reach the same level of usage as the wild-type mice, in contrast with what is observed when they need to extract the cotton from the bin. Deficits in rearing are not involved as shown by results in the cylinder at the same age, described as follows.
Other evidence of early deficits in fine motor skills can be observed in the hole board, a behavioral test that does not require single housing of mice. The hole board is usually used to test memory deficits that require the animal to reach and grab a food pellet in holes. Although Thy1-aSyn mice did not demonstrate cognitive deficits in this test at 3 to 4 months of age, they did spend more time in the baited hole they correctly identified, indicating a difficulty in grasping the pellet [60].
In contrast to these tests previously described, several behavioral tests assess general motor skills, activity, and balance. The pole test is a simple way to assess motor coordination. Mice are placed head up near the top of a small wooden pole (50 cm long, 1 cm diameter) and the times they take to turn and descend the pole are measured with a stopwatch. Mice receive 2 days of training with 5 trials for each session. On the test day, their performance is recorded for 5 trials. Thy1-aSyn mice present profound deficits in this task at 2 months of age, and deficits worsen progressively with age [26]. Because some animals do not turn at all, it is necessary to determine an arbitrary cutoff time, usually 30 seconds for time to turn and 30 seconds for time to descend. This also allows 1 to determine the number of “performers” and “nonperformers” (i.e., the number of animals at the cutoff). In addition to the average time to turn and to descend, comparing the number of nonperformers with or without drug treatment can be a useful way to assess drug effects on this behavioral deficit.
Thy1-aSyn mice also show deficits in the cylinder [26]. Mice are placed in a transparent cylinder (height, 15.5 cm; diameter, 12.7 cm), and number of rears, movements of the hind limbs and forelimbs on the floor of the cylinder are counted on videotapes for accuracy. Thy1-aSyn mice show progressive deficits in hind limb movements from 2 months. Rearing and front limb movements are also reduced at 2 months of age, but are less affected than hind limb movements at later time points. Grooming is only significantly reduced at 8 months of age in Thy1-asyn mice compared to age-matched wild-type littermates [66].
These deficits are distinct from parkinsonism because they occur long before any loss of dopamine, and certainly the administration of L-dopa or dopamine agonists tend to make them worse [66], in contrast to the later deficits that are improved by the dopaminergic treatment [24]. The worsening of these early deficits with dopaminergic agonists is not surprising in view of the detrimental effect of L-dopa on motor behavior in wild-type mice [67, 68]. This is reminiscent of the detrimental effects of dopaminergic agonists on cognitive function in patients with PD, which is interpreted as resulting from the excess dopamine in regions without loss of endogenous dopamine [69]. Thus, too much dopamine may impair both cognitive and motor function. This raises the question of the mechanisms underlying these early motor deficits. In wild-type mice, lesions of the locus coeruleus, which contains the cell bodies of noradrenergic neurons that project throughout the brain, lead to deficits on the challenging beam [70] that are very similar to those we have observed in the Thy1-aSyn mice, suggesting that early deficits in noradrenaline could, if present, contribute to the observed deficits. In addition, young Thy1-aSyn mice exhibit profound alterations at the corticostriatal synapse (and perhaps other synaptic connections) that are described as follows. Finally, even when no dopamine loss is present, nigrostriatal dopaminergic neurons may be functionally abnormal. We have already indicated that extrastriatal dopamine levels are transiently elevated at 6 months of age, but are unchanged at 10 months in Thy1-aSyn mice [24]. The nigrostriatal dopaminergic neurons also display alterations in numerous cellular mechanisms, including expression of ion channel genes, described as follows. Together these data indicate that the Thy1-aSyn mice exhibit early neuronal dysfunction, which precedes the loss of dopamine and the emergence of the classical PD neurological deficits. This provides a useful platform to elucidate the earliest aspects of neuronal dysfunction that could also occur in patients long before they present with the symptoms that permit the diagnosis of PD.
Early Electrophysiological Alterations in Thy1-aSyn Mice Striatum
Unlike studies in patients, mouse models allow for a thorough exploration of mechanisms that may cause early deficits. From an early age, the Thy1-aSyn mice show profound deficits in corticostriatal transmission [71]. Corticostriatal synaptic function was examined with whole-cell patch clamp recordings from visually identified medium-size spiny neurons in striatal slices from Thy1-aSyn mice and their littermate wild-type controls. The neurons displayed significant decreases in the frequency of spontaneous excitatory postsynaptic currents (sEPSCs) in Thy1-aSyn mice at 35, 90, and 300 days of age. Subsequent studies indicated that the effect was not present at 21 days of age, indicating that the corticostriatal deficit developed progressively and was not due to developmental defects. The difference in sEPSCs persisted in the presence of tetrodotoxin, indicating that it was independent of action potentials. Stimulation thresholds for evoking EPSCs were significantly higher and responses were smaller in the Thy1-aSyn mice. These data suggest a decrease in neurotransmitter release at the corticostriatal synapse. At 90 days, the frequency of spontaneous GABAA receptor-mediated synaptic currents was decreased in medium-size spiny neurons, but it was increased in cortical pyramidal neurons. These observations indicate that high levels of expression of alpha-synuclein alter corticostriatal synaptic function early, and they provide evidence for early synaptic dysfunction in a pre-manifest model of PD. Of importance, these changes are opposite to those found in dopamine-depletion models, suggesting that before degeneration of dopaminergic neurons in the substantia nigra, synaptic adaptations occur at the corticostriatal synapse that may initiate neuronal dysfunction. These mice also show altered presynaptic plasticity in the corticostriatal pathway, possibly also reflecting a reduction in glutamate at corticostriatal synapses due to modulation of adenylyl cyclase signaling pathways [72].
As indicated earlier, young Thy1-aSyn mice show sustained increases in the basal level of extracellular striatal dopamine [24]. Whole-cell patch clamp recordings from medium-size spiny neurons in striatal slices revealed parallel alterations of dopaminergic modulation of corticostriatal synaptic function in the Thy1-aSyn mice. Amphetamine reduced sEPSC frequency in wild-types, but produced no effect in Thy1-aSyn mice. Furthermore, while the dopaminergic D2 agonist quinpirole reduced and the dopaminergic D2 antagonist sulpiride increased sEPSC frequency in wild-type mice, it produced the opposite effects in Thy1-aSyn mice [24]. These observations indicate that over-expression of alpha-synuclein alters dopamine efflux and D2 receptor modulation of corticostriatal glutamate release in these mice at a young age. Interestingly, in parallel with the lack of effects of amphetamine on sEPSC in Thy1-aSyn mice, we have shown that these mice do not increase grooming in response to amphetamine treatment, despite a normal effect of the drug on dopamine release in their striatum [66].
Thus, electrophysiological findings have demonstrated that there are major, progressive, circuit alterations in the Thy1-aSyn mice, which start long before the loss of striatal dopamine and could explain the early behavioral deficits observed in these mice (Fig. 12)
Fig. 12.

Corticostriatal synapse alterations in 6 months old Thy1-aSyn mice, prior to dopamine loss. Terminals from nigral dopaminergic neurons and from cortical glutamatergic neurons project to the striatum and form synapses onto GABAergic medium spiny neurons. Increased extrasynaptic dopamine results in alteration of dopaminergic modulation of the corticostriatal synapse. Reduced spontaneous and evoked excitatory postsynaptic currents (EPSCs) indicate decrease in neurotransmitter release at the corticostriatal synapse. Increased microglia activation and tumor necrosis factor-α (TNF-alpha) release might contribute to neuronal dysfunction in the striatum. AMPAR = xxxx; D1/2R = xxxx; D2R = xxxx; DAT = xxxx; NMDAR = xxxx; VMAT = xxxx.
Thy1-aSyn Mice Show Inflammatory Changes in the Innate and Adaptive Immune Systems
The Thy1-aSyn mice show early innate inflammatory reactions in the brain and late peripheral adaptive immune responses. Similar to what has been observed in PD patients [73–75], microglial activation and the proinflammatory cytokine tumor necrosis factor-α are increased in the striatum and substantia nigra of young Thy1-aSyn mice, and microglial activation persists in both regions up to 14 months of age [76]. Importantly, microglial activation occurs first in the striatum (1 month of age) and later in the substantia nigra (5-6 months of age), which correlates with recent imaging studies in PD patients [75]. In addition to innate immune abnormalities in PD, there are recent observations of alterations in the adaptive immune response. In PD patients, increased percentages of cytotoxic T cells have been observed in the blood [77, 78] and the brain [79, 80]. Correspondingly, there were significant increases in the percentages of cytotoxic T cells in the blood of 22-month-old Thy1-aSyn mice compared with age-matched WT mice (Watson et al., in preparation).
Thy1-aSyn Mice Show Progressive Molecular Alterations in Mechanisms Implicated in PD
Mechanisms of neurodegeneration in PD are generally deduced from observation in postmortem brains, determined years after disease progression. Although informative, these observations leave the question open as to early molecular changes that are likely to be a critical target for neuroprotection. We reasoned that analysis of the Thy1-aSyn mice at very early stages of the disease could shed new lights onto the progressive pathological mechanisms that eventually lead to the demise of dopaminergic neurons.
Morphological studies have shown increased sensitivity of mitochondria to the complex 1 inhibitor MPTP in Thy1-aSyn mice [81]. In agreement with the presence of mitochondrial dysfunction, neurons of the substantia nigra in the Thy1-aSyn mice show the presence of DNA damage, as revealed by staining for H2A.X at 5 months of age (Fig. 13C). In addition, the mice exhibit a progressive increase in iron deposits in the substantia nigra, although their level is not different from those seen in wild-type littermates (Fig. 13A, B). Thus, the model reveals that pathological features detected in postmortem brains of PD patients (iron accumulation, oxidative stress) may already be present at early stages of the disease and vice versa, and the presence of these anomalies further validates the model as it indicates the replication of pathological processes known to occur in PD.
Fig. 13.

Iron accumulation and oxidative stress. (A, B) Progressive iron accumulation in the substantia negra (SN) of Thy1-aSyn mice (A) quantification of iron stain at 5, 12, and 22 months of age shown in (B) perl stain, total iron. Scale bar = 30 μm. Optical density (OD) in the SN (Image J) shows 2.6-fold increase of iron accumulation between 5 m and 12 m, and further 3.1-fold increase between 12 m and 22 m (**p < 0.01 compared to 5 m, ##p < 0.01 compared to 12 m; one-way analysis of variance followed by Fisher’s Least Significant Difference (LSD), n = 5 each). (C) Increased staining for DNA damage marker (anti-phospho-Histone H2A.X [Ser139] Millipore, green) in nigral TH (rabbit anti-TH antibody; 1:500, Millipore, red) positive neurons of Thy1-aSyn mice, but not wild-type (WT) at 5 months of age (40 μm brain sections, 100 x /1.6-fold zoom confocal image of 1 focal plane). Scale bar = 25 μm. TH = tyrosine hydroxylase
Based on this evidence, it is likely that a more in-depth analysis of this model could provide clues to the molecular deficits occurring in nigrostriatal dopamine neurons during the early stages of PD. Indeed, transcriptome analysis of laser-captured dopamine neurons from these mice uncovered alterations in many cellular pathways thought to be implicated in PD [82]. For example, Weighted Gene Co-expression Network Analysis [83] of microarray data revealed that superoxide dismutase 2 and endogenous alpha-synuclein are highly correlated hub genes, driving gene expression alterations involved in synaptic and mitochondrial function. At 4 months of age, Thy1-aSyn mice showed altered gene expression in canonical pathways highly relevant for neuronal function (e.g., signaling in 5' adenosine monophosphate-activated protein kinase (AMPK), apoptosis, axonal guidance, integrin, RhoA, ERK/MAPK, HIF1alpha, and toll-like receptor); in addition, the Weighted Gene Co-expression Network Analysis reveals alterations in a module comprised of genes involved in Nfe2l2 (also known as Nrf2) oxidative stress response and Dnajc (heat shock protein). Alterations in this pathway worsen at 8 months, as shown by an upregulation of Nfe2l2 itself accompanied by upregulation, for example of Rras2, Mapk8, Mapk9 upstream and Cat, thioredoxin-disulfide reductase (Trxr1), and several heat shock proteins downstream from Nfe2l2. Iron-related genes show similar progression effects, whereby the transferrin receptor is a hub gene, driving gene expression alterations at 4 months of age. At 8 months, several key genes regulating iron homeostasis (e.g., iron regulatory proteins 1 [Aco1] and 2 [Ireb2], transferrin [Tf]) show altered expression in differential gene expression analysis. The worsening of transcriptome alterations in these pathways between 4 and 8 months show that the model is progressive [82]. Thus, analysis of changes in gene expression in this well-characterized model can point toward key molecular pathways affected at different stages of PD, both protective and presumably detrimental, and can identify new targets for neuroprotection.
The Thy1-aSyn Mice: A Model Suitable for Preclinical Drug Testing of Novel Neuroprotective Therapies
In addition to providing an experimental organism to uncover pathophysiology at disease stages that are difficult to study in humans, mammalian models are necessary for preclinical testing of novel drug therapies before clinical trials. This requires robust endpoint measures with high statistical power to detect drug effects with tests that are cost effective and can be optimally performed repeatedly to follow disease progression, while reducing the number of animals needed for the study. It is also important for the model to reproduce the pathological mechanism that is being targeted by the drug (e.g., it only makes sense to test an inhibitor of alpha-synuclein aggregation in a model that presents with this feature). It is not indispensable, however, for a model to reproduce behavioral features identical to those seen in humans to provide valid information. Behavioral phenotypes, even when different from clinical manifestations in humans can provide valid endpoints if they are an expression of the pathophysiological mechanism that is being targeted. With their large variety of endpoints in multiple behavioral domains, and their wide range of molecular disturbances similar to those suspected to occur in PD, the Thy1-aSyn mice provide a good model to test potential neuroprotective therapies in PD. Treatments can be started at different stages of disease progression to reproduce different clinical situations. Treatment early in the disease process are unlikely to be feasible in the clinic, but are useful as a proof of principle and less costly than treatment started after dopamine loss, which would better mimic clinical situations, and can be initiated if early experiments show positive results. Thus, this genetic mouse model of PD allows for testing the ability of drugs to protect against both early motor and nonmotor manifestations of PD, and later deficits related to dopamine loss. In addition, systemic alterations (inflammation), which may turn out to provide an accessible index of treatment effects in future clinical trials can be assessed in these mice. Indeed, several compounds have already shown behavioral and pathological or molecular improvements in the Thy1-aSyn mice. We have shown for the first time a behavioral improvement (a 38% decrease in the number of errors per step in the challenging beam traversal test) in these mice after daily administration of the microtubule-interacting peptide NAPVSIPQ (NAP; also known as davunetide or AL-108) intranasally for 2 months starting at 1 month of age, together with a reduction in proteinase K-resistant alpha-synuclein inclusions in the substantia nigra compared to vehicle-treated transgenics [84]. More recently, Lee et al. [85] have shown that treatment of Thy1-aSyn mice with eicosanoyl-5-hydroxytryptamide increased neuronal activity and dendritic arborizations while reducing glial activation and improved motor performance. These effects were attributed to an increased methylation of the phosphatase PP2A, resulting in decreased alpha-synuclein phosphorylation at Serine 129 and alpha-synucein aggregation in brain. Lovastatin, a cholesterol synthesis inhibitor, decreased alpha-synuclein aggregation and pathology in the Thy1-aSyn mice [86]. Interestingly, the cholesterol-oximes TRO19622 and TRO40303, 2 drugs that target outer mitochondrial membrane proteins and are cytoprotective in several cell and animal models involving oxidative stress, trophic factor withdrawal, and Fas (TNF receptor superfamily, member 6)-induced death pathways [87–89], normalized a number of transcriptome anomalies observed in laser-captured nigrostriatal dopaminergic neurons of young Thy1-aSyn mice after 3 months of administration in food [90]. Finally, we have investigated whether increasing the stability, trafficking, and activity of glucocerebrosidase with the pharmacological chaperone afegostat-tartrate (AT2101) in mice that over-express human wild-type alpha-synuclein (Thy1-aSyn mice) could be a therapeutic approach for synucleinopathies. AT2101 administered orally for 4 months to Thy1-aSyn mice improved motor function and reduced the number of small alpha-synuclein aggregates while increasing the number of large alpha-synuclein aggregates in the substantia nigra of Thy1-aSyn mice [91]. These data support the further investigation of pharmacological chaperones that target GCase as a therapeutic approach for PD and other synucleinopathies.
In conclusion, the Thy1-aSyn mice reproduce key features of Parkinson’s disease, including a prolonged “pre-manifest” phase preceding the loss of striatal dopamine, nonmotor deficits, pathological, biochemical, and molecular alterations that are reminiscent of those observed in the disease, and at older ages, alterations in nigrostriatal dopaminergic neurons, including loss of striatal dopamine and TH, that lead to L-dopa responsive motor deficits. These observations support the contention that pathological accumulation of alpha-synuclein is sufficient to cause many of the symptoms and anomalies observed in sporadic PD. To our knowledge, these mice do not develop a measurable loss of dopaminergic cell bodies in their lifetime, but they do provide a useful platform to elucidate the mechanism of early pathological mechanisms in sporadic PD and test new approaches for neuroprotection in this still incurable disorder.
Electronic supplementary materials
(PDF 511 kb)
Acknowledgments
Supported by PHS grant P50 NS38367 (UCLA Morris K. Udall Parkinson Disease Research Center of Excellence), and gifts to the Center for the Study of Parkinson’s Disease at UCLA.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
References
- 1.Meissner WG, Frasier M, Gasser T, et al. Priorities in Parkinson's disease research. Nat Rev Drug Discov. 2011;10:377–393. doi: 10.1038/nrd3430. [DOI] [PubMed] [Google Scholar]
- 2.DeLong MR, Juncos JL. Parkinson's disease and other extrapyramidal movement disorders. In: Fauci AS, editor. Harrison's Principles of Internal Medicine. New York: McGraw-Hill Medical; 2008. pp. 2549–2559. [Google Scholar]
- 3.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/S0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 4.Delfs JM, Ciaramitaro VM, Soghomonian JJ, Chesselet MF. Unilateral nigrostriatal lesions induce a bilateral increase in glutamate decarboxylase messenger RNA in the reticular thalamic nucleus. Neuroscience. 1996;71:383–395. doi: 10.1016/0306-4522(95)00470-X. [DOI] [PubMed] [Google Scholar]
- 5.Chaudhuri KR, Odin P. The challenge of non-motor symptoms in Parkinson's disease. Prog Brain Res. 2010;184:325–341. doi: 10.1016/S0079-6123(10)84017-8. [DOI] [PubMed] [Google Scholar]
- 6.Olanow CW, Stern MB, Sethi K. The scientific and clinical basis for the treatment of Parkinson disease. Neurology. 2009;72:S1–136. doi: 10.1212/WNL.0b013e3181a1d44c. [DOI] [PubMed] [Google Scholar]
- 7.Lohle M, Reichmann H. Clinical neuroprotection in Parkinson's disease — still waiting for the breakthrough. J Neurol Sci. 2010;289:104–114. doi: 10.1016/j.jns.2009.08.025. [DOI] [PubMed] [Google Scholar]
- 8.Langston JW. Epidemiology versus genetics in Parkinson's disease: progress in resolving an age-old debate. Ann Neurol. 1998;44:S45–52. doi: 10.1002/ana.410440239. [DOI] [PubMed] [Google Scholar]
- 9.Bronstein J, Carvey P, Chen H, et al. Meeting report: consensus statement-Parkinson's disease and the environment: collaborative on health and the environment and Parkinson's Action Network (CHE PAN) conference June 26-28, 2007. Environ Health Perspect. 2009;117:117–121. doi: 10.1289/ehp.11702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamza TH, Chen H, Hill-Burns EM, et al. Genome-wide gene-environment study identifies glutamate receptor gene GRIN2A as a Parkinson's disease modifier gene via interaction with coffee. PLoS Genet. 2011;7:e1002237. doi: 10.1371/journal.pgen.1002237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manthripragada AD, Costello S, Cockburn MG, Bronstein JM, Ritz B. Paraoxonase 1, agricultural organophosphate exposure, and Parkinson disease. Epidemiology. 2010;21:87–94. doi: 10.1097/EDE.0b013e3181c15ec6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burbulla LF, Krebiehl G, Kruger R. Balance is the challenge — the impact of mitochondrial dynamics in Parkinson's disease. Eur J Clin Invest. 2010;40:1048–1060. doi: 10.1111/j.1365-2362.2010.02354.x. [DOI] [PubMed] [Google Scholar]
- 13.Spillantini MG, Goedert M. The alpha-synucleinopathies: Parkinson's disease, dementia with Lewy bodies, and multiple system atrophy. Ann N Y Acad Sci. 2000;920:16–27. doi: 10.1111/j.1749-6632.2000.tb06900.x. [DOI] [PubMed] [Google Scholar]
- 14.Edwards TL, Scott WK, Almonte C, et al. Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet. 2010;74:97–109. doi: 10.1111/j.1469-1809.2009.00560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gatto NM, Rhodes SL, Manthripragada AD, et al. alpha-Synuclein gene may interact with environmental factors in increasing risk of Parkinson's disease. Neuroepidemiology. 2010;35:191–195. doi: 10.1159/000315157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang Y, Rowe DB, Halliday GM. Interaction between α-synuclein and tau genotypes and the progression of Parkinson's disease. J Parkinson's Dis. 2011;1:271–276. doi: 10.3233/JPD-2011-11027. [DOI] [PubMed] [Google Scholar]
- 17.Magen I, Chesselet MF. Genetic mouse models of Parkinson's disease: the state of the art. Prog Brain Res. 2010;184:53–87. doi: 10.1016/S0079-6123(10)84004-X. [DOI] [PubMed] [Google Scholar]
- 18.Chesselet MF, Richter F. Modelling of Parkinson's disease in mice. Lancet Neurol. 2011;10:1108–1118. doi: 10.1016/S1474-4422(11)70227-7. [DOI] [PubMed] [Google Scholar]
- 19.Dawson TM, Ko HS, Dawson VL. Genetic animal models of Parkinson's disease. Neuron. 2010;66:646–661. doi: 10.1016/j.neuron.2010.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harvey BK, Richie CT, Hoffer BJ, Airavaara M. Transgenic animal models of neurodegeneration based on human genetic studies. J Neural Transm. 2011;118:27–45. doi: 10.1007/s00702-010-0476-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rockenstein E, Mallory M, Hashimoto M, et al. Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters. J Neurosci Res. 2002;68:568–578. doi: 10.1002/jnr.10231. [DOI] [PubMed] [Google Scholar]
- 22.Masliah E, Rockenstein E, Veinbergs I, et al. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- 23.Hashimoto M, Rockenstein E, Masliah E. Transgenic models of alpha-synuclein pathology: past, present, and future. Ann N Y Acad Sci. 2003;991:171–188. doi: 10.1111/j.1749-6632.2003.tb07475.x. [DOI] [PubMed] [Google Scholar]
- 24.Lam HA, Wu N, Cely I, et al. Elevated tonic extracellular dopamine concentration and altered dopamine modulation of synaptic activity precede dopamine loss in the striatum of mice overexpressing human alpha-synuclein. J Neurosci Res. 2011;89:1091–1102. doi: 10.1002/jnr.22611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van der Putten H, Wiederhold KH, Probst A, et al. Neuropathology in mice expressing human alpha-synuclein. J Neurosci. 2000;20:6021–6029. doi: 10.1523/JNEUROSCI.20-16-06021.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fleming SM, Salcedo J, Fernagut PO, et al. Early and progressive sensorimotor anomalies in mice overexpressing wild-type human alpha-synuclein. J Neurosci. 2004;24:9434–9440. doi: 10.1523/JNEUROSCI.3080-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lim Y, Kehm VM, Lee EB, et al. α-Syn suppression reverses synaptic and memory defects in a mouse model of dementia with Lewy bodies. J Neurosci. 2011;31:10076–10087. doi: 10.1523/JNEUROSCI.0618-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 29.Fernagut PO, Hutson CB, Fleming SM, et al. Behavioral and histopathological consequences of paraquat intoxication in mice: effects of alpha-synuclein over-expression. Synapse. 2007;61:991–1001. doi: 10.1002/syn.20456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neumann M, Muller V, Kretzschmar HA, Haass C, Kahle PJ. Regional distribution of proteinase K-resistant alpha-synuclein correlates with Lewy body disease stage. J Neuropathol Exp Neurol. 2004;63:1225–1235. doi: 10.1093/jnen/63.12.1225. [DOI] [PubMed] [Google Scholar]
- 31.Halliday GM, Holton JL, Revesz T, Dickson DW. Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathol 2011;122(2):187-204. [DOI] [PubMed]
- 32.Braak H, de Vos RA, Bohl J, Del Tredici K. Gastric alpha-synuclein immunoreactive inclusions in Meissner's and Auerbach's plexuses in cases staged for Parkinson's disease-related brain pathology. Neurosci Lett. 2006;396:67–72. doi: 10.1016/j.neulet.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 33.Hawkes CH, Del Tredici K, Braak H. Parkinson's disease: a dual-hit hypothesis. Neuropathol Appl Neurobiol. 2007;33:599–614. doi: 10.1111/j.1365-2990.2007.00874.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beal MF. Parkinson's disease: a model dilemma. Nature. 2010;466:S8–S10. doi: 10.1038/466S8a. [DOI] [PubMed] [Google Scholar]
- 35.Cheng HC, Ulane CM, Burke RE. Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol. 2010;67:715–725. doi: 10.1002/ana.21995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Richfield EK, Thiruchelvam MJ, Cory-Slechta DA, et al. Behavioral and neurochemical effects of wild-type and mutated human alpha-synuclein in transgenic mice. Exp Neurol. 2002;175:35–48. doi: 10.1006/exnr.2002.7882. [DOI] [PubMed] [Google Scholar]
- 37.Wakamatsu M, Ishii A, Iwata S, et al. Selective loss of nigral dopamine neurons induced by overexpression of truncated human alpha-synuclein in mice. Neurobiol Aging. 2008;29:574–585. doi: 10.1016/j.neurobiolaging.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 38.Shin JH, Ko HS, Kang H, et al. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson's disease. Cell. 2011;144:689–702. doi: 10.1016/j.cell.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sulzer D. Multiple hit hypotheses for dopamine neuron loss in Parkinson's disease. Trends Neurosci. 2007;30:244–250. doi: 10.1016/j.tins.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 40.Manning-Bog AB, McCormack AL, Purisai MG, Bolin LM, Di Monte DA. Alpha-synuclein overexpression protects against paraquat-induced neurodegeneration. J Neurosci. 2003;23:3095–3099. doi: 10.1523/JNEUROSCI.23-08-03095.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hean S, Richter F, Torres ES, et al. Mice overexpressing human alpha synuclein (Thy1-aSyn) show dopamine loss, catalepsy and severe motor deficits partially rescued by L-DOPA at 14 months of age. Neurosci Abstr 2010; 750.27/H20:
- 42.Sossi V, de la Fuente-Fernandez R, Nandhagopal R, et al. Dopamine turnover increases in asymptomatic LRRK2 mutations carriers. Mov Disord. 2010;25:2717–2723. doi: 10.1002/mds.23356. [DOI] [PubMed] [Google Scholar]
- 43.Chesselet MF. Dopamine and Parkinson's disease: is the killer in the house? Mol Psychiatry. 2003;8:369–370. doi: 10.1038/sj.mp.4001289. [DOI] [PubMed] [Google Scholar]
- 44.Hattoria N, Wanga M, Taka H, et al. Toxic effects of dopamine metabolism in Parkinson's disease. Parkinsonism Relat Disord. 2009;15(suppl 1):S35–S38. doi: 10.1016/S1353-8020(09)70010-0. [DOI] [PubMed] [Google Scholar]
- 45.Lawal HO, Chang HY, Terrell AN, et al. The Drosophila vesicular monoamine transporter reduces pesticide-induced loss of dopaminergic neurons. Neurobiol Dis. 2010;40:102–112. doi: 10.1016/j.nbd.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ritz BR, Manthripragada AD, Costello S, et al. Dopamine transporter genetic variants and pesticides in Parkinson's disease. Environ Health Perspect. 2009;117:964–969. doi: 10.1289/ehp.0800277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Caudle WM, Richardson JR, Wang MZ, et al. Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J Neurosci. 2007;27:8138–8148. doi: 10.1523/JNEUROSCI.0319-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McDowell K, Chesselet MF. Animal models of the non-motor features of Parkinson’s disease. Neurobiol Dis 2012;(in press). [DOI] [PMC free article] [PubMed]
- 49.Lang AE, Obeso JA. Time to move beyond nigrostriatal dopamine deficiency in Parkinson's disease. Ann Neurol. 2004;55:761–765. doi: 10.1002/ana.20102. [DOI] [PubMed] [Google Scholar]
- 50.Ross GW, Petrovitch H, Abbott RD, et al. Association of olfactory dysfunction with risk for future Parkinson's disease. Ann Neurol. 2008;63:167–173. doi: 10.1002/ana.21291. [DOI] [PubMed] [Google Scholar]
- 51.Fleming SM, Tetreault NA, Mulligan CK, Hutson CB, Masliah E, Chesselet MF. Olfactory deficits in mice overexpressing human wildtype alpha-synuclein. Eur J Neurosci. 2008;28:247–256. doi: 10.1111/j.1460-9568.2008.06346.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marxreiter F, Nuber S, Kandasamy M, et al. Changes in adult olfactory bulb neurogenesis in mice expressing the A30P mutant form of alpha-synuclein. Eur J Neurosci. 2009;29:879–890. doi: 10.1111/j.1460-9568.2009.06641.x. [DOI] [PubMed] [Google Scholar]
- 53.Wang L, Fleming SM, Chesselet MF, Tache Y. Abnormal colonic motility in mice overexpressing human wild-type alpha-synuclein. Neuroreport. 2008;19:873–876. doi: 10.1097/WNR.0b013e3282ffda5e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abbott RD, Petrovitch H, White LR, et al. Frequency of bowel movements and the future risk of Parkinson's disease. Neurology. 2001;57:456–462. doi: 10.1212/wnl.57.3.456. [DOI] [PubMed] [Google Scholar]
- 55.Savica R, Carlin JM, Grossardt BR, et al. Medical records documentation of constipation preceding Parkinson disease: a case-control study. Neurology. 2009;73:1752–1758. doi: 10.1212/WNL.0b013e3181c34af5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kuo YM, Li Z, Jiao Y, et al. Extensive enteric nervous system abnormalities in mice transgenic for artificial chromosomes containing Parkinson disease-associated alpha-synuclein gene mutations precede central nervous system changes. Hum Mol Genet. 2010;19:1633–1650. doi: 10.1093/hmg/ddq038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abbott RD, Ross GW, White LR, et al. Excessive daytime sleepiness and subsequent development of Parkinson disease. Neurology. 2005;65:1442–1446. doi: 10.1212/01.wnl.0000183056.89590.0d. [DOI] [PubMed] [Google Scholar]
- 58.Kudo T, Loh DH, Truong D, Wu Y, Colwell CS. Circadian dysfunction in a mouse model of Parkinson's disease. Exp Neurol 2011;232(1):66-75. [DOI] [PubMed]
- 59.Janvin CC, Aarsland D, Larsen JP. Cognitive predictors of dementia in Parkinson's disease: a community-based, 4-year longitudinal study. J Geriatr Psychiatry Neurol. 2005;18:149–154. doi: 10.1177/0891988705277540. [DOI] [PubMed] [Google Scholar]
- 60.Magen I, Fleming S, Garcia E, et al. Cognitive deficits in a mouse model of pre-manifest Parkinson’s disease. Eur J Neurosci 2011 (in press). [DOI] [PMC free article] [PubMed]
- 61.Mochizuki-Kawai H, Mochizuki S, Kawamura M. A flexible sequential learning deficit in patients with Parkinson's disease: a 2 x 8 button-press task. Exp Brain Res. 2010;202:147–153. doi: 10.1007/s00221-009-2119-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Magen I, Chesselet MF. Mouse Models of Cognitive Deficits Due to Alpha-Synuclein Pathology. J Parkinsons Dis. 2011;1:217–227. doi: 10.3233/JPD-2011-11043. [DOI] [PubMed] [Google Scholar]
- 63.Torres ES, Zelikowsky M, Richter F, et al. Mice overexpressing human alpha synuclein under the Thy1-promotor show increased fear conditioning and altered responses in anxiety related behavior. Neurosci Abstr 2010;750.25/H18.
- 64.Mulligan CK, Fleming SM, Dorriz P, Masliah E, Chesselet MF. Mice overexpressing human wildtype alpha-synuclein under the Thy-1 promoter exhibit anomalies in behavioral tests of anxiety. Neurosci Abstr 2008;742.19/U28.
- 65.Mirelman A, Gurevich T, Giladi N, Bar-Shira A, Orr-Urtreger A, Hausdorff JM. Gait alterations in healthy carriers of the LRRK2 G2019S mutation. Ann Neurol. 2011;69:193–197. doi: 10.1002/ana.22165. [DOI] [PubMed] [Google Scholar]
- 66.Fleming SM, Salcedo J, Hutson CB, et al. Behavioral effects of dopaminergic agonists in transgenic mice overexpressing human wildtype alpha-synuclein. Neuroscience. 2006;142:1245–1253. doi: 10.1016/j.neuroscience.2006.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fredriksson A, Plaznik A, Sundstrom E, Jonsson G, Archer T. MPTP-induced hypoactivity in mice: reversal by L-dopa. Pharmacol Toxicol. 1990;67:295–301. doi: 10.1111/j.1600-0773.1990.tb00833.x. [DOI] [PubMed] [Google Scholar]
- 68.Oksman M, Tanila H, Yavich L. Behavioural and neurochemical response of alpha-synuclein A30P transgenic mice to the effects of L-DOPA. Neuropharmacology. 2009;56:647–652. doi: 10.1016/j.neuropharm.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 69.Cools R. Dopaminergic modulation of cognitive function-implications for L-DOPA treatment in Parkinson's disease. Neurosci Biobehav Rev. 2006;30:1–23. doi: 10.1016/j.neubiorev.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 70.Rommelfanger KS, Edwards GL, Freeman KG, Liles LC, Miller GW, Weinshenker D. Norepinephrine loss produces more profound motor deficits than MPTP treatment in mice. Proc Natl Acad Sci U S A. 2007;104:13804–13809. doi: 10.1073/pnas.0702753104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wu N, Joshi PR, Cepeda C, Masliah E, Levine MS. Alpha-synuclein overexpression in mice alters synaptic communication in the corticostriatal pathway. J Neurosci Res. 2010;88:1764–1776. doi: 10.1002/jnr.22327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Watson JB, Hatami A, David H, et al. Alterations in corticostriatal synaptic plasticity in mice overexpressing human alpha-synuclein. Neuroscience. 2009;159:501–513. doi: 10.1016/j.neuroscience.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McGeer PL, Itagaki S, Akiyama H, McGeer EG. Rate of cell death in parkinsonism indicates active neuropathological process. Ann Neurol. 1988;24:574–576. doi: 10.1002/ana.410240415. [DOI] [PubMed] [Google Scholar]
- 74.Mogi M, Nagatsu T. Neurotrophins and cytokines in Parkinson's disease. Adv Neurol. 1999;80:135–139. [PubMed] [Google Scholar]
- 75.Ouchi Y, Yagi S, Yokokura M, Sakamoto M. Neuroinflammation in the living brain of Parkinson's disease. Parkinsonism Relat Disord 2009;(15 suppl 3):S200-S204. [DOI] [PubMed]
- 76.Watson MB, Lee SK, Richter F, Masliah E, Chesselet MF. Regionally specific microglial activation precedes neuropathology and peripheral immune response in mice over-expressing wildtype alpha synuclein. Neurosci Abstr 2011;357.12/AA31.
- 77.Bas J, Calopa M, Mestre M, et al. Lymphocyte populations in Parkinson's disease and in rat models of parkinsonism. J Neuroimmunol. 2001;113:146–152. doi: 10.1016/S0165-5728(00)00422-7. [DOI] [PubMed] [Google Scholar]
- 78.Baba Y, Kuroiwa A, Uitti RJ, Wszolek ZK, Yamada T. Alterations of T-lymphocyte populations in Parkinson disease. Parkinsonism Relat Disord. 2005;11:493–498. doi: 10.1016/j.parkreldis.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 79.Brochard V, Combadiere B, Prigent A, et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest. 2009;119:182–192. doi: 10.1172/JCI36470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology. 1988;38:1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- 81.Song DD, Shults CW, Sisk A, Rockenstein E, Masliah E. Enhanced substantia nigra mitochondrial pathology in human alpha-synuclein transgenic mice after treatment with MPTP. Exp Neurol. 2004;186:158–172. doi: 10.1016/S0014-4886(03)00342-X. [DOI] [PubMed] [Google Scholar]
- 82.Richter F, Gao F, Bove N, et al. Transcriptome network and pathway analyses reveal early alterations in dopaminergic neurons of mice overexpressing human wild-type alpha-synuclein (Thy1-aSyn). Neurosci Abstr 2011;357.07/AA26.
- 83.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fleming SM, Mulligan CK, Richter F, et al. A pilot trial of the microtubule-interacting peptide (NAP) in mice overexpressing alpha-synuclein shows improvement in motor function and reduction of alpha-synuclein inclusions. Mol Cell Neurosci. 2011;46:597–606. doi: 10.1016/j.mcn.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee KW, Chen W, Junn E, et al. Enhanced phosphatase activity attenuates alpha-Synucleinopathy in a mouse model. J Neurosci. 2011;31:6963–6971. doi: 10.1523/JNEUROSCI.6513-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Koob AO, Ubhi K, Paulsson JF, et al. Lovastatin ameliorates alpha-synuclein accumulation and oxidation in transgenic mouse models of alpha-synucleinopathies. Exp Neurol. 2010;221:267–274. doi: 10.1016/j.expneurol.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bordet T, Buisson B, Michaud M, et al. Identification and characterization of cholest-4-en-3-one, oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. J Pharmacol Exp Ther. 2007;322:709–720. doi: 10.1124/jpet.107.123000. [DOI] [PubMed] [Google Scholar]
- 88.Bordet T, Pruss RM. Targeting neuroprotection as an alternative approach to preventing and treating neuropathic pain. Neurotherapeutics. 2009;6:648–662. doi: 10.1016/j.nurt.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schaller S, Paradis S, Ngoh GA, et al. TRO40303, a new cardioprotective compound, inhibits mitochondrial permeability transition. J Pharmacol Exp Ther. 2010;333:696–706. doi: 10.1124/jpet.110.167486. [DOI] [PubMed] [Google Scholar]
- 90.Richter F, Fleming SM, Michaud M, et al. The cholesterol-oximes TRO19622 and TRO40303 affect motor function, olfaction, and alpha synuclein aggregation in mice overexpressing human alpha synuclein under the Thy1 promoter. Neurosci Abstr 2010;750.28/H21.
- 91.Lemesre V, Richter F, Fleming SM, et al. The glucocerebrosidase pharmacological chaperone afegostat-tartrate (AT2101) partially improves motor and olfactory function and alters the size of alpha-synuclein inclusions in mice overexpressing alpha-synuclein. Neurosci Abstr 2010;750.29/H22.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 511 kb)