

Abstract
Mitochondrial DNA mutations are an important cause of human disease for which there is no effective treatment. Myoclonic epilepsy with ragged-red fibers (MERRF) is a mitochondrial disease usually caused by point mutations in transfer RNA genes encoded by mitochondrial DNA. The most common mutation associated with MERRF syndrome, m.8344A > G in the gene MT-TK, which encodes transfer RNALysine, affects the translation of all mitochondrial DNA encoded proteins. This impairs the assembly of the electron transport chain complexes leading to decreased mitochondrial respiratory function. Here we report on how this mutation affects mitochondrial function in primary fibroblast cultures established from patients harboring the A8344G mutation. Coenzyme Q10 (CoQ) levels, as well as mitochondrial respiratory chain activity, and mitochondrial protein expression levels were significantly decreased in MERRF fibroblasts. Mitotracker staining and imaging analysis of individual mitochondria indicated the presence of small, rounded, depolarized mitochondria in MERRF fibroblasts. Mitochondrial dysfunction was associated with increased oxidative stress and increased degradation of impaired mitochondria by mitophagy. Transmitochondrial cybrids harboring the A8344G mutation also showed CoQ deficiency, mitochondrial dysfunction, and increased mitophagy activity. All these abnormalities in patient-derived fibroblasts and cybrids were partially restored by CoQ supplementation, indicating that these cell culture models may be suitable for screening and validation of novel drug candidates for MERRF disease.
Electronic supplementary material
The online version of this article (doi:10.1007/s13311-012-0103-3) contains supplementary material, which is available to authorized users.
Keywords: Mitophagy, Coenzyme Q10, Mitochondrial disease, MERRF
Introduction
Mutations in mitochondrial DNA (mtDNA) cause a number of rare human diseases that affect muscle and neural tissues in particular [1]. These mutations are often heteroplasmic (i.e., the affected individual harbors both mutant and wild type mtDNA), and the severity of the disease depends on the relative proportion of the mutant species. Myoclonic epilepsy with ragged red fibers (MERRF) syndrome is a rare, maternally inherited, multi-systemic mitochondrial disorder characterized by myoclonus, epilepsy, weakness, ataxia, and ragged-red fibers in the muscle [2]. Additional clinical manifestations can include short stature, hearing loss, pigmentary retinopathy, optic atrophy, dementia, peripheral neuropathy, cardiomyopathy, or renal tubular dysfunction [3, 4]. Lactate and pyruvate are commonly elevated in serum at rest and increase excessively after moderate physical activities [5]. The m.8344A > G mutation in the mtDNA gene MT-TK, which encodes mitochondrial transfer (t)RNALysine is the most common mutation associated with MERRF syndrome, although other mutations in tRNALysine or genes encoding different tRNAs have been identified in MERRF patients [6, 7]. The m.8344A > G mutation, which is localized in the T-arm of the tRNA, is one of the most studied and prevalent pathological mtDNA mutations [8]. It has been reported to cause poor aminoacylation of the mutant tRNA [9], hypomodification of its anti-codon wobble-position [10], and premature termination of translation at some lysine codons [9]. All these alterations result in decreased activities of respiratory chain complexes I and IV, low respiration rate and decreased mitochondrial membrane potential [11, 12].
High cellular levels of the m.8344A > G mutation are correlated with decreased protein synthesis, decreased oxygen consumption, and cytochrome c oxidase deficiency [13], and larger or lysine rich polypeptides are more severely affected, suggesting that the MERRF mutation directly inhibits mitochondrial protein synthesis [14]. Furthermore, oxidative stress and oxidative damage in afected tissues, elicited by the respiratory chain deficiency, plays a crucial role in the pathogenesis and progression of MERRF disease [15]. The relationship between mtDNA mutations and cellular damage, as well as the activation of compensatory mechanisms to promote cell survival, are poorly understood. Treatments for mitochondrial diseases in general are largely inadequate [16]. As with many mitochondrial diseases, there is no cure for MERRF, and treatment is primarily symptomatic. Coenzyme Q10 (CoQ) has been used in therapy with the hope of improving mitochondrial function, although there is no definitive evidence of the efficacy of this treatment.
In this work, we performed experiments in primary fibroblast cultures derived from a MERRF patient and within a transmitocondrial hybrid (cybrid) clone containing the m.8344A > G mutation in a controlled nuclear background; this was done to determine how mtDNA mutations affect mitochondria and the mechanisms activated to eliminate the dysfunctional mitochondria. In addition, we studied the effects of CoQ supplementation on the various pathophysiological changes in the various cell cultures.
Material and Methods
Reagents
Monoclonal Anti-Actin antibody, rabbit anti-VDAC1/Porin, and rabbit anti-BECLIN1 were all obtained from Sigma-Aldrich (St. Louis, MO). Monoclonal antibodies against complex III (core 1 subunit) and complex II (30 KDa subunit I), Mitosox Red, CMH2-DCFDA, 10-N-nonyl acridine orange (NAO), MitoTracker, LysoTracker, TO-PRO-3, and Hoechst 3342 were obtained from Invitrogen/Molecular Probes (Eugene, OR). Anti-cytochrome c antibody was obtained from BD Biosciences Pharmingen (San Jose, CA) and anti-GAPDH monoclonal antibody (clone 6 C5) was from Calbiochem-Merck Chemicals Ltd. (Nottingham, UK). Anti-hATG12 and anti-hATG5 were obtained from Biosensis (South Australia, Australia). Anti-MAP LC3 (N-20), anti-catalase (H-300), anti-PDI (H-160), anti-Golgi marker (AE-6), and anti-Cathepsin D were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Protease inhibitors were obtained from Boehringer Mannheim (Indianapolis, IN). All other chemicals were purchased from Sigma-Aldrich.
Fibroblasts and Cybrids Cultures
Cultured fibroblasts, harboring a heteroplasmic m.8344A > G mutation (57%) were derived from a MERRF patient (MERRF-1). We also used 2 more fibroblasts cell lines (i.e., MERRF-2 and MERRF-3), with mutation levels of 35% and 8%, respectively. Control primary fibroblasts were obtained from healthy volunteers. Samples from patients and controls were obtained according to the Helsinki Declarations of 1964, as revised in 2001. This study has been approved by the institutional ethical committee. Control and MERRF fibroblasts were cultured at 37°C in Dulbecco’s Modified Eagle Medium (DMEM) (4.5 g/L glucose), plus pyruvate supplemented with 20% fetal bovine serum (FBS), and an antibiotic/anti-mycotic solution.
Cybrid clones were prepared by fusing enucleated fibroblasts from patients or controls with 143B osteosarcoma cells, which lack mitochondrial DNA (rho 0 cells), as described by King and Attardi [17]. Cybrid cells harboring the A8344G mutation (99% of the total copy number) were cultured in Dulbecco’s Modified Eagle Medium (4.5 g/L glucose) supplemented with 5% FBS, sodium pyruvate (100 μg/ml), uridine (50 μg/ml), penicillin (100 IU/ml), and streptomycin (100 μg/ml).
CoQ Treatment of Cell Lines
Unless otherwise stated, CoQ (in FBS) was added to cultures for 72 h at a final concentration of 100 μM.
Proliferation Rate
Two hundred thousand cells were cultured with or without 100 μM CoQ for 72 h. Cell counting was performed from 10 high power fields using an inverted microscope and a 40× objective.
Mitochondrial Respiratory Chain Enzyme Activities and CoQ Levels
Activities of NADH: cytochrome c reductase (complexes I + III) and citrate synthase were determined spectrophotometrically in sonicated, permeabilized fibroblasts using previously described methods [16, 18]. Results are expressed as units/citrate synthase (mean ± SD). Protein content was determined by the Lowry procedure [19]. CoQ levels in cultured fibroblasts and cybrids were measured as previously described [20].
Adenosine-5'-Triphosphate Levels
Adenosine-5'-triphosphate (ATP) levels were determined using a bioluminescence assay (ATP determination kit, Invitrogen-Molecular Probes, Eugene, OR).
Immunofluorescence Microscopy
Immunofluorescence microscopy was performed using SD methods as previously described [20]. Cover slips were analyzed using a fluorescence microscope (Leica DMRE, Leica Microsystems GmbH, Wetzlar, Germany). Deconvolution studies and 3-dimensional projections were performed using a DeltaVision system (Applied Precision, Issaquah, WA) with an Olympus IX-71 microscope. The deconvolved images were derived from optical sections taken at 30-nm intervals using a 60× PLAPON objective with a 1.42 numerical aperture.
Measurement of Mitochondrial Reactive Oxygen Species (ROS) Generation
Mitochondrial ROS generation was assessed using the mitochondrial superoxide indicator MitoSOX Red, according to the manufacturer’s instructions. ROS levels were expressed relative to mitochondrial mass (ROS signal/NAO signal), determined by flow cytometry and fluorescence microscopy of cells stained with 10 μM NAO for 10 minutes at 37°C in the dark.
Measurement of Intracellular H2O2 Content
H2O2 levels were measured using nonfluorescent CMH2-DCFDA (5-[and-6]-chloromethyl-2’,7’-dichlorodihydrofluorescein diacetate, acetyl ester), which diffuses across membranes and is oxidized to fluorescent dichlorofluorescein (DCF). Cultured cells were rinsed in phosphate-buffered saline (PBS), incubated with CMH2-DCFDA diluted in medium at 5 μM for 30 minutes at 37°C. After that, cells were washed, trypsinized, and re-suspended in pre-warmed PBS at 37°C. Cells were then analyzed by flow cytometry.
Lipid Peroxidation
Fibroblasts were cultured on cover slips and incubated with 1 μM C11-Bodipy (BODIPY 581/591 C11, Invitrogen/Molecular Probes, Eugene, OR) for 30 minutes at 37°C. Coverslips were then rinsed with PBS and examined by fluorescence microscopy. Fluorescent intensity was measured using Image J software.
Mitochondrial Membrane Potential (ΔΨm)
Fibroblasts were grown on 1-mm width (Goldseal No. 1, Thermo Fisher Scientific Inc, Waltham, MA) glass coverslips for 24 to 48 hours, incubated with 100 nM MitoTracker Red for 30 minutes, fixed, and immunostained with anti-cytochrome c (a mitochondrial marker), and examined by fluorescence microscopy. Colocalization of both markers and quantification of MitoTracker fluorescence in individual mitochondria was assessed by the DeltaVision software (Applied Precision).
Immunoblotting
Western blotting was performed using a standard protocol [20] and the Immun Star HRP detection kit (Bio-Rad Laboratories Inc., Hercules, CA).
Quantification of ß-Galactosidase
ß-Galactosidase was stained using a previously reported protocol [21] and quantified using ImageJ software (see: http://rsb.info.nih.gov/ij/download.html).
Loading of LysoTracker Red
LysoTracker Red (100 nM) was added to cultured fibroblasts for 30 minutes each, well was washed twice with fresh DMEM, and then the cells were fixed with 2% paraformaldehyde in PBS for 10 minutes at 4°C. LysoTracker fluorescence was quantified by flow cytometry.
Real-Time Quantitative Polymerase Chain Reaction of Autophagy Genes
The expression of ATG12, MAP1LC3, and BECLIN1 was analyzed by SYBR Green quantitative polymerase chain reaction using gene specific primers (see supporting information in Table 1) in RNA isolated from fibroblasts. β-actin was used as a housekeeping control gene.
Table 1.
List of gene specific primers used in the analysis of the expression levels of autophagy genes in primary cultured fibroblasts
| Gene Product Size | Forward Primer (5’- 3’) | Reverse Primer (5’- 3’) |
|---|---|---|
| BECLIN1 152 bp | GGATGGATGTGGAGAAAGGCAAG | TGAGGACACCCAAGCAAGACC |
| ATG12 198 bp | ATTGCTGCTGGAGGGGAAGG | GGTTCGTGTTCGCTCTACTGC |
| MAP1LC3 91 bp | GCCTTCTTCCTGCTGGTGAAC | AGCCGTCCTCGTCTTTCTCC |
Statistical Analysis
All results are expressed as mean ± SD of 3 independent experiments. The measurements were statistically analyzed using the Student’s t test for comparing 2 groups and analysis of variance for more than 2 groups. The level of significance was set at p < 0.05.
Results
Effect of CoQ Supplementation on Mitochondrial Dysfunction in MERRF Fibroblasts
CoQ is a key component of the mitochondrial respiratory chain, transferring reducing equivalents from complexes I and II to complex III, and secondary CoQ deficiency was recently identified in mitochondrial diseases [22]. Therefore, we assessed whether fibroblasts harboring the m.8344A > G mutation were deficient in CoQ. The CoQ content of MERRF fibroblasts was reduced to 46% of the average control value (Fig. 1A). Furthermore, patient fibroblasts showed a significant, 46% reduction in the combined activity of complexes I + III (Fig. 1B). This activity was increased to near normal values in MERRF cells following 100 μM CoQ supplementation (Fig. 1B).
Fig. 1.
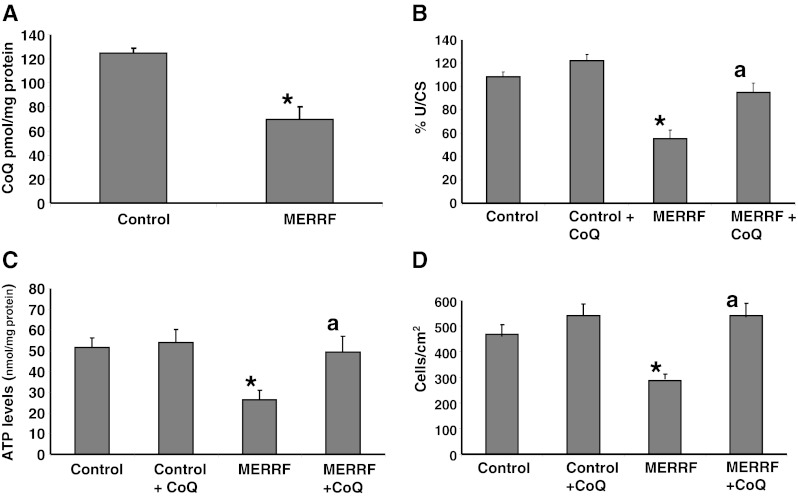
Mitochondrial function in myoclonic epilepsy with ragged-red fibers (MERRF) fibroblasts. (A) Coenzyme Q10 (CoQ) levels in control and MERRF fibroblasts. (B) Complex I + III, activity measured in cells grown in the presence or absence of CoQ (100 μM) for 72 h. Results (mean ± SD) are expressed in units per citrate synthase (U/CS). (C) Adenosine-5'-triphosphate (ATP) levels in control and MERRF fibroblasts grown in the absence or presence of CoQ (100 μM) for 72 h. (D) Cell proliferation of control and MERRF fibroblasts cultured in the absence or presence of CoQ (100 μM) for 72 h. Data represent the mean ± SD of 3 separate experiments. *p < 0.01 between control and MERRF fibroblasts. ap < 0.05 between the presence and the absence of CoQ
To determine whether the observed mitochondrial dysfunction had an effect on cellular bioenergetics, we measured intracellular ATP levels in control and MERRF fibroblasts. ATP levels were reduced to 50% of the control value in MERRF fibroblasts (Fig. 1C). CoQ supplementation (100 μM) resulted in a significant increase in cellular ATP levels in MERRF fibroblasts, but was without effect in control cells (Fig. 1C). Next we determined the effect of the m.8344A > G mutation on cellular proliferation: MERRF fibroblasts showed a reduced proliferation rate compared to control fibroblasts (Fig. 1D). The proliferation rate was increased to near normal values in MERRF cells following 100 μM CoQ supplementation (Fig. 1D).
Mitochondrial Membrane Potential (ΔΨm)
To further assess the functional consequences of reduced respiratory chain enzyme activities and CoQ levels in MERRF fibroblasts, we determined ΔΨm in fibroblast cultures using MitoTracker Red, a mitochondrion selective dye, which is concentrated by active mitochondria in a membrane potential-dependent manner. Staining and imaging analysis revealed discrete staining of mitochondria in control cells, with the mitochondria organized in a tubular network, whereas in the MERRF fibroblasts, diffuse staining was observed throughout the cytoplasm, indicating reduced mitochondrial uptake of MitoTracker Red, and the mitochondrial network was fragmented, with small, rounded mitochondria apparent throughout the cytoplasm (Fig. 2A, B). The mitotracker staining colocalized with staining for cytochrome c, a mitochondrial marker, (Fig. 2A) confirming the specificity of MitoTracker staining and verifying that mitochondrial depolarization in MERRF fibroblasts was not a result of cytochrome c release, as occurs in apoptosis [23]. Supplementation with 100 μM CoQ restored normal mitochondrial morphology and uptake of MitoTracker Red in MERRF fibroblasts, indicating restoration of the mitochondrial membrane potential (Fig. 2A, B).
Fig. 2.
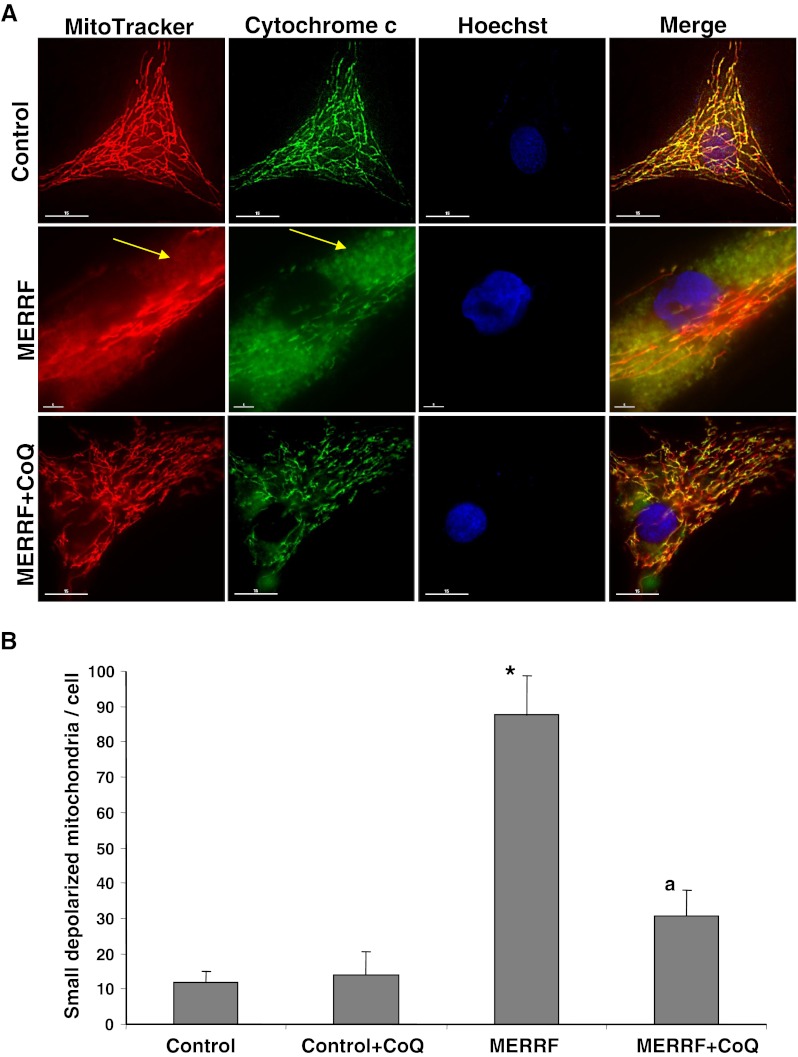
Mitochondrial membrane potential. (A) Representative images of MitoTracker and cytochrome c staining in control and myoclonic epilepsy with ragged-red fibers (MERRF) fibroblasts cultured in the presence or absence of coenzyme Q10 (CoQ) (100 μM). Yellow arrows indicate small depolarized mitochondria. (B) Quantification of depolarized mitochondria by fluorescence imaging analysis in 50 randomly selected cells from control and MERRF cultures. Mitochondrial specificity of MitoTracker staining was assessed by examining colocalization of MitoTracker fluorescence with cytochrome c. Scale bar = 15 μm. *p < 0.01 between control and MERRF fibroblasts. ap < 0.05 between the presence and the absence of CoQ.
Effect of CoQ on Mitochondrial ROS Production in MERRF Fibroblasts
As it is well established that CoQ deficiency and mitochondrial dysfunction is associated with increased mitochondrial ROS production; we examined ROS and H2O2 levels in control and MERRF fibroblasts using the mitochondrial superoxide indicator, MitoSOX Red. At the same time, we estimated mitochondrial mass with NAO, and determined the ratio of MitoSOX signal to NAO fluorescence. ROS production was increased approximately 2.5-fold in MERRF fibroblasts (Fig. 3A). The inclusion of 100 μM CoQ in the culture medium caused a considerable reduction in ROS levels in MERRF cultures, but had no effect in control cultures (Fig. 3A).
Fig. 3.
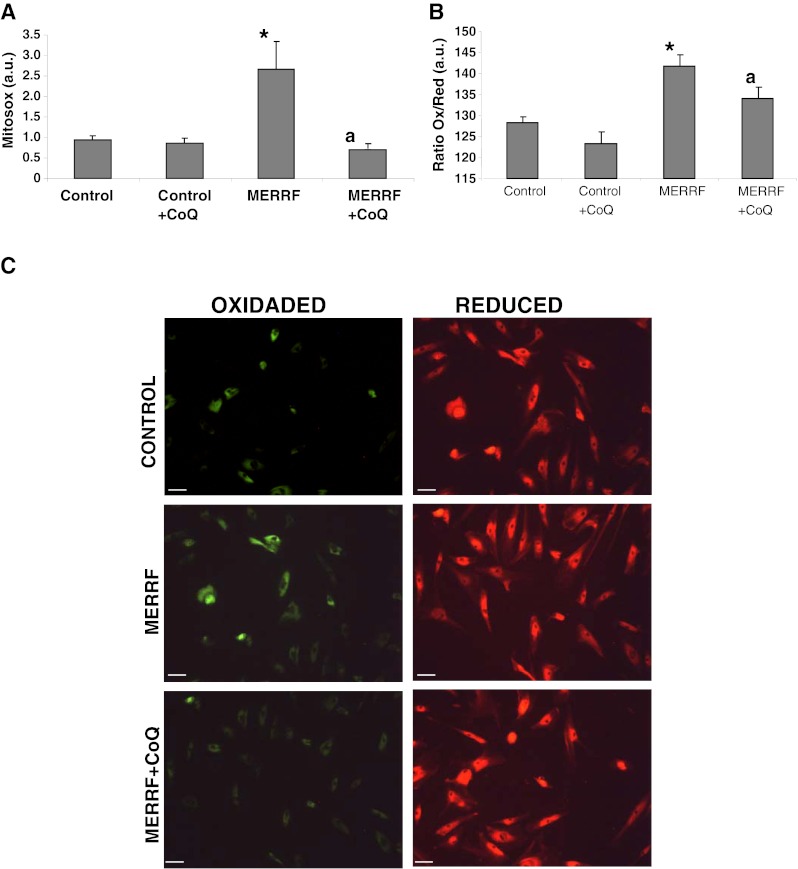
Mitochondrial reactive oxygen species (ROS) generation and lipid peroxidation in myoclonic epilepsy with ragged-red fibers (MERRF) fibroblasts. (A) Mitochondrial ROS generation in fibroblasts cultured for 72 h in normal growth medium or medium supplemented with coenzyme Q10 (CoQ) (100 μM) prior to analysis. Results are expressed as the ratio of MitoSOX signal to 10-N-nonyl acridine orange signal. The mean ± SD of 3 independent experiments are shown. (B) Quantification of lipid peroxidation in control and MERRF fibroblasts in the presence or absence of CoQ (100 μM) for 72 h. Data represent the ratio oxidized lipid/reduced lipid. (C) C11-Bodipy staining. Red fluorescence represents nonoxidized lipids, and green fluorescence represents oxidized lipids. Scale bar = 30 μm. For the control cells, the data are the mean ± SD for experiments on 2 different control cells. Data represent the mean ± SD of 3 separate experiments. *p < 0.01 between control and MERRF fibroblasts. ap < 0.01, between the presence and the absence of CoQ. a.u. = arbitrary units
Then we examined lipid peroxidation as a marker of oxidative stress-induced membrane damage in control and MERRF fibroblasts. The ratio of oxidized to reduced lipids was significantly increased in MERRF fibroblasts compared to controls (Fig. 3B), suggesting oxidative stress damage, but this was partially prevented by treatment with 100 μM CoQ (Fig. 3B).
Mitochondrial Degradation in MERRF Fibroblasts
Recent evidence suggests the involvement of ROS in autophagy [24]. To determine if autophagy is increased in MERRF fibroblasts, first we analyzed the expression of the genes ATG12, BECLIN1, and LC3, which encode proteins involved in autophagy. Compared to control fibroblast cultures, the expression of all 3 of these genes was significantly increased in MERRF fibroblasts, with a 13-fold increase in ATG12, a 14-fold increase in BECLIN1, and a 22-fold increase in LC3 expression in MERRF fibroblasts (Fig. 4A). Addition of 100 μM CoQ to the culture medium resulted in a decrease in the expression of these genes in MERRF cells, but had no effect in control cells (Fig. 4A). The amount of BECLIN1 protein was also increased in MERRF cells when compared to control cells (Fig. 4B, C). CoQ supplementation (100 μM) resulted in a decrease in the amount of this protein in both control and MERRF cells (Fig. 4A, B). We also investigated the conversion of LC3-I (microtubule-associated light chain 3) to LC3-II, as the amount of the latter is closely correlated with the number of autophagosomes. The ratio of LC3-II to LC3-1 was significantly increased in MERRF fibroblasts (Fig. 4B, C), indicating enhanced autophagosome formation in MERRF cells. Supplementation of the culture medium with 100 μM CoQ resulted in a significant decrease in the relative amount of LC3-II in MERRF cultures, but it was without an effect in the control cells. As autophagosome formation involves an ubiquitin-like conjugation system in which Atg12 is covalently bound to Atg5, we also measured the amount of ATG12-ATG5 conjugate. The amount of ATG12-ATG5 was significantly increased in MERRF fibroblasts, but decreased to control levels with 100 μM CoQ supplementation (Fig. 4B, C). The amount of actin, used as a reference protein, was similar in all cultures (Fig. 4B, C).
Fig. 4.

Expression of autophagic proteins. (A) The expression levels of ATG12, BECLIN1, and LC3 mRNA in control and myoclonic epilepsy with ragged-red fibers (MERRF) fibroblasts measured by real time polymerase chain reaction. (B) The amount of LC3-I (upper band) and LC3-II (lower band), ATG12 and BECLIN1 protein were determined in the control and MERRF fibroblast cultures by Western blotting. The ATG12 band represents the Atg12-Atg5 conjugated form. Fibroblast cultures were grown in normal culture medium or in medium supplemented with coenzyme Q10 (CoQ) (100 μM) for 72 h. Actin was used as a loading control. (C) The amount of various proteins estimated by densitometry. Actin was used as a loading control. For the control cells, the data are the mean ± SD for experiments on 2 different control cell lines. Data, expressed as arbitrary units (a.u.) represent the mean ± SD of 3 separate experiments. *p < 0.01 between control and MERRF fibroblasts. ap < 0.01, between the presence and the absence of coenzyme Q10 (CoQ).
Lysosomal Markers in MERRF Fibroblasts
To further verify that autophagy was activated in MERRF fibroblasts, we measured the expression of the lysosomal indicators β-galactosidase and cathepsin D, and quantified the amount of acidic vacuoles. The amount of β-galactosidase was approximately twofold higher in MERRF fibroblast cultures than in controls (Fig. 5A, B). Addition of 100 μM CoQ to the culture medium resulted in a significant reduction in the level of β-galactosidase in MERRF fibroblasts, but had no effect on the amount of β-galactosidase in control cells (Fig. 5A, B).
Fig. 5.
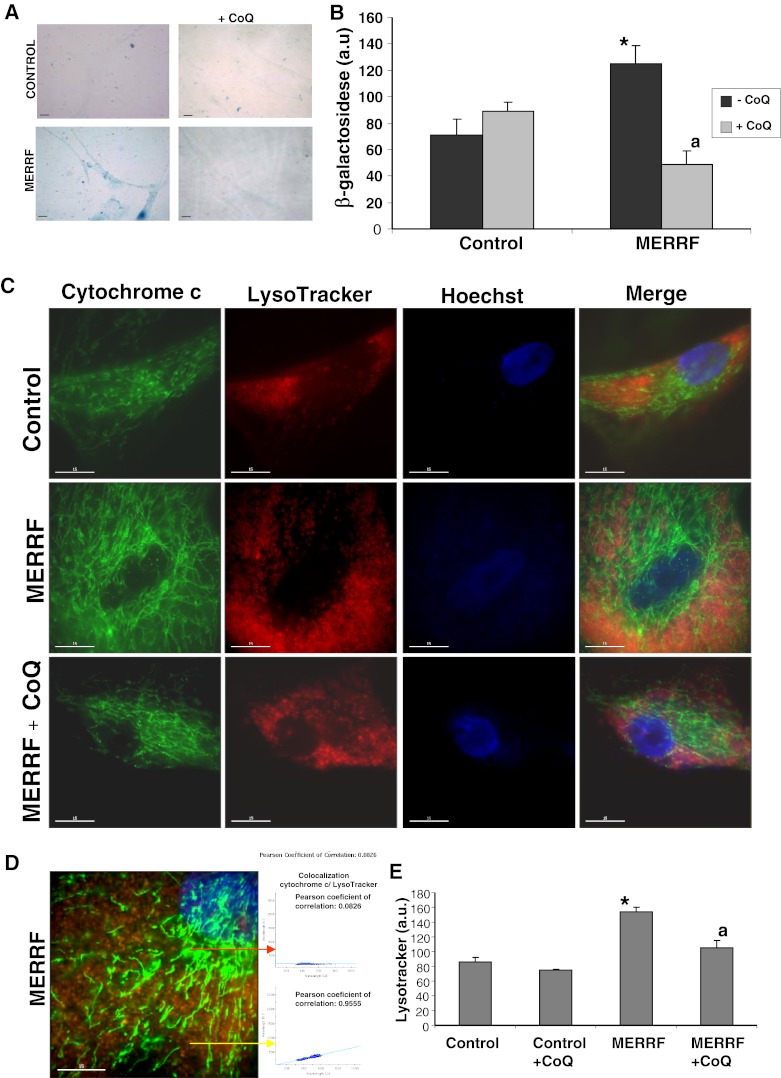
Autophagic markers in myoclonic epilepsy with ragged-red fibers (MERRF) and control fibroblasts. (A) Representative images of β-galactosidase staining in control and patient fibroblasts, visualized by light microscopy. Cells were cultured in normal medium or in the presence of coenzyme Q10 (CoQ) (100 μM) for 72 h. Scale bar = 15 μm. (B) β-galactosidase staining of control and MERRF fibroblast cultures quantified using ImageJ software. (C) LysoTracker staining in control and MERRF fibroblasts visualized by fluorescence microscopy. The effect of CoQ supplementation (100 μM) for 72 h was also evaluated. Scale bar = 15 μm. (D) Magnification of a small area in a MERRF fibroblast. Arrows show autophagolysosomes with LysoTracker and cytochrome c colocalization. The colocalization of both markers was assessed by DeltaVision software (Applied Precision, Issaquah, WA) calculating the Pearson coefficient of correlation. Scale bar = 15 μm. (E) Quantification of acidic vacuoles in control and patient fibroblast by LysoTracker staining and flow cytometry analysis. Cells were cultured in the presence or absence of CoQ, as in (B). Results are expressed in arbitrary units (a.u.). For the control cells, the data are the mean ± SD for experiments conducted on 2 different control cell lines. Data represent the mean ± SD of 3 separate experiments. *p < 0.01 between control and MERRF fibroblasts. ap < 0.01 between the presence and the absence of CoQ
We used LysoTracker staining and confocal microscopy to estimate the number of lysosomes. The intensity of LysoTracker staining was significantly higher in MERRF fibroblasts than in controls (Fig. 5C, D). To elucidate whether autophagy in MERRF fibroblasts could be attenuated by improving mitochondrial function via CoQ supplementation, we quantified the number of lysosomes in the various cultures following 100 μM CoQ supplementation. CoQ treatment was associated with a significant reduction in the intensity of LysoTracker staining in MERRF cells (Fig. 5C), indicating that lysosomal activity was reduced following CoQ treatment. The number of lysosomes in MERRF and control cells was also compared using LysoTracker staining followed by flow cytometry analysis. Using this technique, a twofold increase in LysoTracker staining was observed in MERRF cells. CoQ supplementation resulted in a decrease in staining in MERRF fibroblasts, but was without effect in the control cells.
Cathepsin D was visualized by immunostaining. The number of cathepsin D positive cells was eightfold higher in fibroblasts from MERRF patient 1 than in control fibroblasts (Fig. 6A). Supplementation with 100 μM CoQ led to a small decrease in cathepsin D positive cells in control cultures, and a significant twofold decrease in cathepsin D positive cells in MERRF cultures (Fig. 6A). The increased amount of cathepsin D in MERRF fibroblasts and its reduction by CoQ treatment was confirmed by Western blotting (Fig. 6B).
Fig. 6.
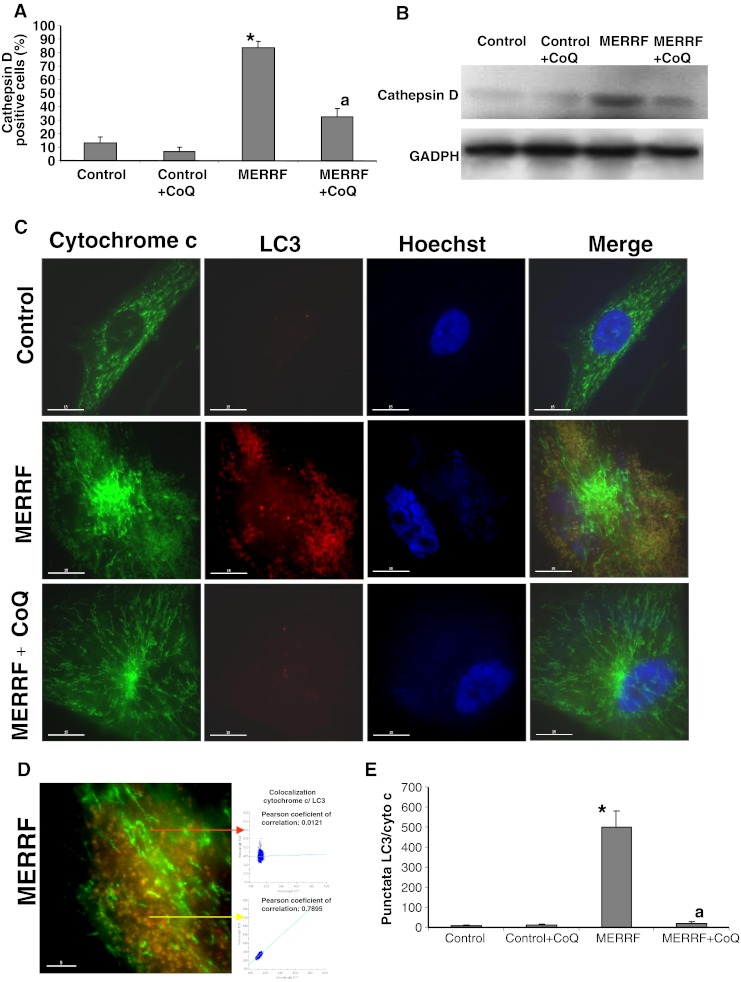
Expression of lysosomal and autophagosome markers in myoclonic epilepsy with ragged-red fibers (MERRF) and control fibroblasts. (A) Quantification of cathepsin positive cells in control and MERRF cells cultured with or without coenzyme Q10 (CoQ) supplementation (100 μM for 72 h). Cathepsin D staining was visualized by fluorescence microscopy. (B) The amount of cathepsin D protein determined in control and MERRF fibroblast cultures by Western blotting control and MERRF fibroblast cultures were grown in normal culture medium or in medium supplemented with CoQ (100 μM) for 72 h. Actin was used as a loading control. *p < 0.01 between control and MERRF fibroblasts. ap < 0.01 between the presence and the absence of CoQ. (C) Image analysis of LC3 and cytochrome c immunostained control and MERRF fibroblasts. Cells were fixed and immunostained with anti-LC3 (autophagosome marker) and cytochrome c (mitochondrial marker) and examined by fluorescence microscopy. Colocalization of both markers was assessed using DeltaVision software (Applied Precision, Issaquah, WA). (D) Magnification of a small area in a MERRF fibroblast. Arrows show autophagosomes with LC3 and cytochrome c colocalization. The colocalization of both markers was assessed by the DeltaVision software (Applied Precision) calculating the Pearson coefficient of correlation. Scale bar = 5 μm. (E) Quantification of LC3/cytochrome c puntacta in control and MERRF fibroblasts incubated with or without 100 μM CoQ (n = 100 cells). *p < 0.01 between control and MERRF fibroblasts. ap < 0.01, between the presence and the absence of CoQ. GADPH = glyceraldehyde 3-phosphate dehydrogenase
To determine whether selective mitochondrial degradation or mitophagy was increased in MERRF fibroblasts, we performed immunofluorescence double staining with antibodies to LC3 and cytochrome c (Fig. 6C, D). LC3 staining was hardly detectable in control cells (Fig. 6C), whereas in the MERRF fibroblasts, a few normal tubular mitochondria negative for LC3 (colocalization, r = −0.1034) were apparent, along with many small, fragmented mitochondria, which were positive for LC3 (colocalization, r = 0.8687) (Fig. 6C, D). The population of small, rounded mitochondria colocalized with LC3 staining, (r = 0.7584), whereas the tubular mitochondria did not (r = 0.0253) (Fig. 6C-E).
Mitophagy activation in MERRF fibroblasts was also confirmed by checking that the expression levels of mitochondrial proteins complex I (30 kDa subunit), complex II (30 kDa subunit), complex III (core 1 subunit), complex IV (COX II subunit), and porin were reduced and, on the contrary, Golgi (Golgi marker), endoplasmic reticulum (PDI), and peroxisome (catalase) proteins expression levels were not affected compared to controls (Fig. 7A, B).
Fig. 7.
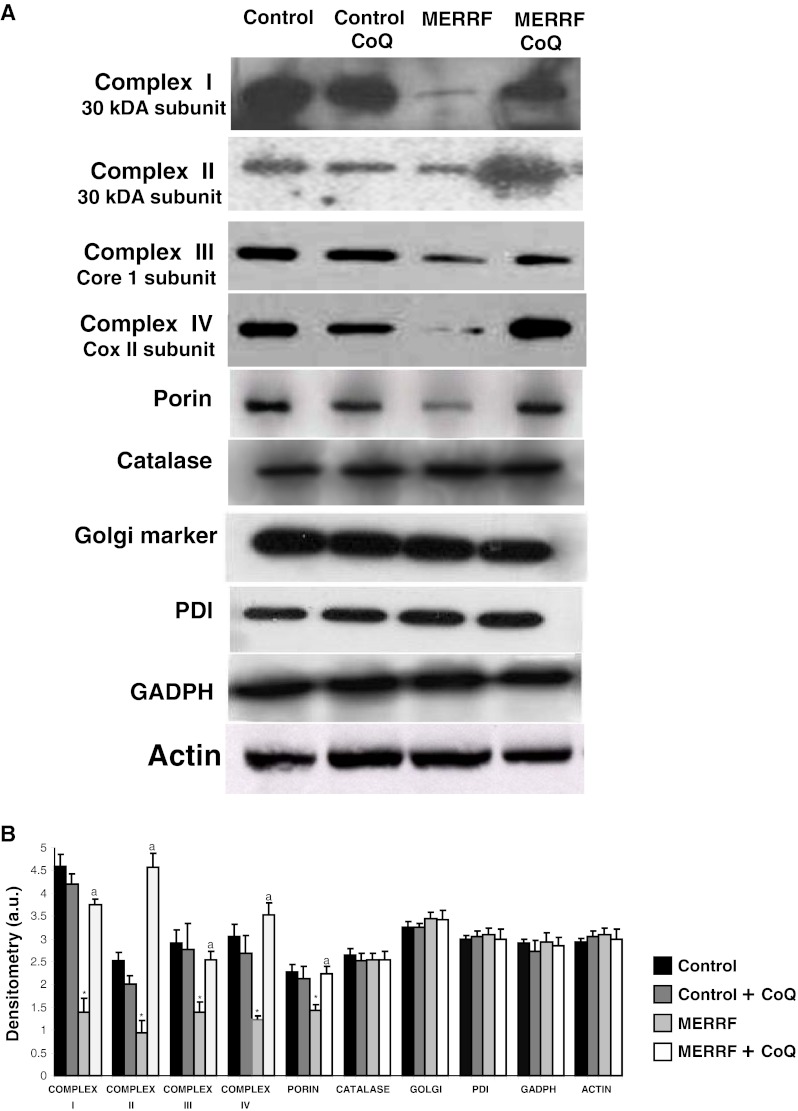
Mitophagy in myoclonic epilepsy with ragged-red fibers (MERRF) fibroblasts. (A) Western blot analysis of mitochondrial proteins of complex I (30 kDa subunit), complex II (30 kDa subunit), complex III (core 1 subunit), complex IV (COX II subunit), and porin-, Golgi (Golgi marker), endoplasmic reticulum (PDI), and peroxisome (catalase) proteins in MERRF fibroblasts. Fibroblast protein extracts (50 μg) were separated on a 12.5% sodium dodecyl sulfate polyacrylamide gel and immunostained with antibodies against porin, cytochrome c, catalase, PDI, and Golgi marker. Glyceraldehyde 3-phosphate dehydrogenase (GADPH) and actin were used as loading control. (B) Densitometry of Western blotting was performed using the ImageJ software. Data, expressed as arbitrary units (a.u.), represent the mean ± SD of 3 separate experiments. *p < 0.01 between control and MERRF fibroblasts. ap < 0.01, between the presence and the absence of coenzyme Q10 (CoQ)
Effect of Coenzyme Q Supplementation on Transmitochondrial Cybrids Harboring the A8344G Mutation
To confirm that CoQ deficiency and autophagy activation was the result of the mutation in mtDNA and not the product of a concomitant nuclear gene defect, CoQ levels, ATP content, and cell growth were measured in a control cybrid clone and in a cybrid clone harboring the m.8344A > G mutation. The amount of CoQ and ATP were 54% and 62% lower, respectively in the MERRF cybrid clone than in the control cybrid clone (Fig. 8A, B), and cell proliferation was also significantly reduced in the MERRF cybrid (Fig. 8C). CoQ supplementation (100 μM) caused a significant threefold increase in ATP levels, and a twofold increase in the rate of cell proliferation in the MERRF clone, but had either no effect or minimal effect on these parameters in the control clone (Fig. 8C, D). Conversely, mitochondrial ROS production and the number of lysosomes, were increased eightfold and twofold, respectively in the MERRF cybrid (Fig. 8D, E), indicating oxidative stress and autophagy activation. Supplementation with 100 μM CoQ significantly decreased both ROS production and LysoTracker staining in MERRF cybrid cells, but had no effect in the control cybrid cells (Fig. 8D, E).
Fig. 8.
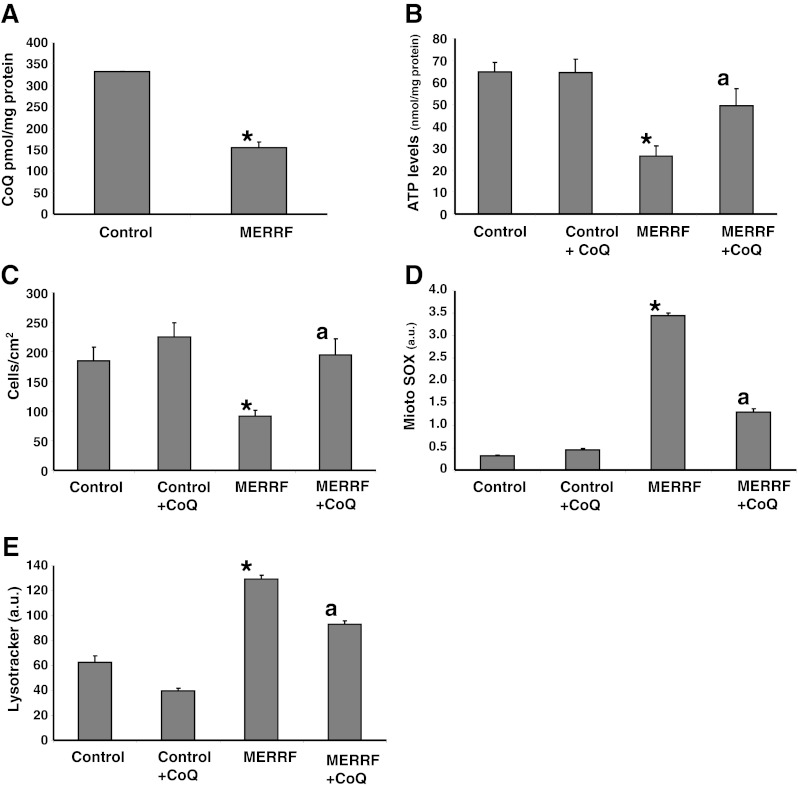
Coenzyme Q10 (CoQ) levels, adenosine-5'-triphosphate (ATP) levels, rate of cellular proliferation, mitochondrial reactive oxygen species (ROS) production, and lysosomal mass in transmitochondrial cybrids. (A) CoQ levels, (B) ATP content, (C) cellular proliferation rate, (D) mitochondrial ROS production, and (E) quantification of acidic vacuoles in control and mutant transmitocondrial cybrids expressing high levels of the m.8344A > G mutation. Cells were grown in the presence or absence of CoQ (100 μM for 72 h). Data represent the mean ± SD of 3 separate experiments arbitrary units (a.u.). *p < 0.01 between control and mutant cybrids. ap < 0.01 between the presence and the absence of CoQ. MERRF = myoclonic epilepsy with ragged-red fibers
To verify autophagy activation in MERRF cybrids, we examined the expression of autophagic proteins by Western blotting. The ratio LC3-II/LC3I and amount of ATG5-ATG12 were increased 2- and 1.8- fold, respectively, in the MERRF cybrid (Fig. 9A, B). CoQ supplementation (100 μM) caused the amount of these proteins to decrease to control levels in the MERRF cybrid, but had little effect on the expression of these proteins in the control (Fig. 9A, B). On the contrary, expression levels of complexes II, III, and IV were reduced by 3.5-, 2.2-, and 1.3-fold, respectively, in MERRF cybrids, but increased to control levels following supplementation with 100 μM CoQ (Fig. 9A, B).
Fig. 9.
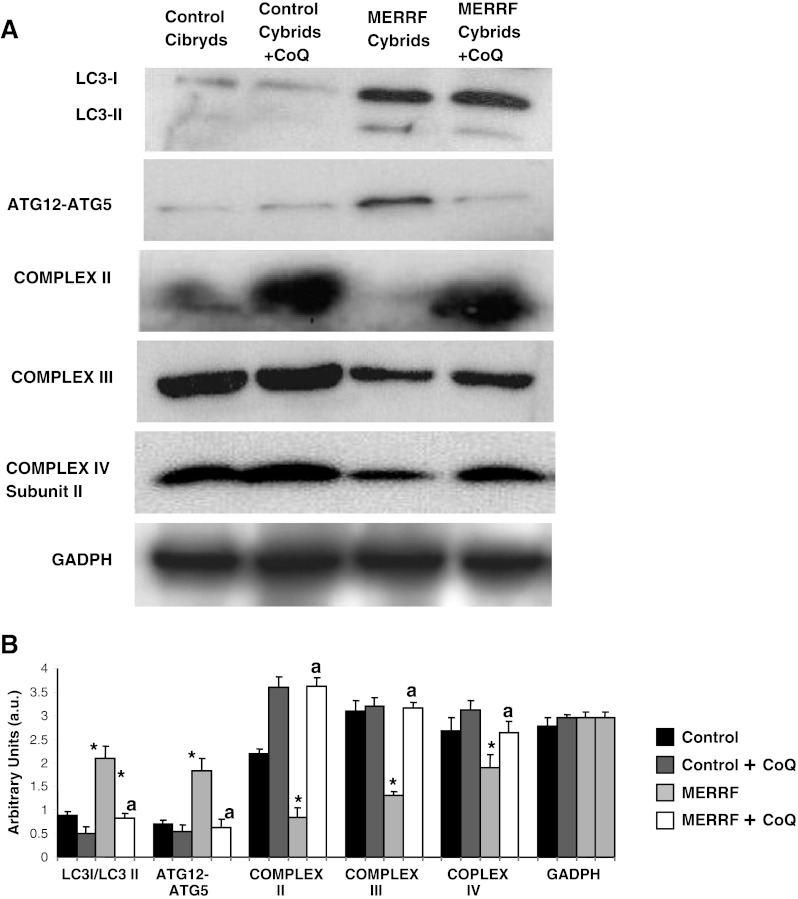
Western blot analysis of autophagic and mitochondrial proteins in myoclonic epilepsy with ragged-red fibers (MERRF) cybrids. (A) Western blot analysis of cybrids grown in normal culture medium or in medium supplemented with coenzyme Q10 (CoQ) (100 μM) for 72 h prior to analysis. Cybrid protein extracts (50 μg) were separated on a 12.5% SDS polyacrylamide gel and immunostained with antibodies against LC3, ATG12, complex III (core 1 subunit) and complex II (30 kDa subunit). Glyceraldehyde 3-phosphate dehydrogenase (GADPH) was used as a loading control. (B) Densitometry of Western blot performed using ImageJ software arbitrary units (a.u.). For control cells, the data are the mean ± SD for experiments on 2 different control cell lines. Data, expressed as a.u., represent the mean ± SD of 3 separate experiments. *p < 0.01, between control and mutant cybrids. ap < 0.01 between the presence and the absence of CoQ
To investigate the presence of mitophagy in MERRF and control cybrids we immunolocalized LC3 and cytochrome c, markers of autophagosomes and mitochondria, respectively (Fig. 10A, B). In control cells, cytochrome c staining revealed a tubular network of mitochondria, whereas LC3 was barely detectable. In MERRF cybrids, a few normal tubular mitochondria negative for LC3 (colocalization, r = −0.0145) were observed, along with many small, fragmented mitochondria that colocalized with LC3 (colocalization, r = 0.8841) (Fig. 10B, C). After 100 μM CoQ supplementation, immunostaining revealed a tubular network of mitochondria, which did not colocalize with LC3 in the MERRF cybrid cells (Fig. 9A).
Fig. 10.
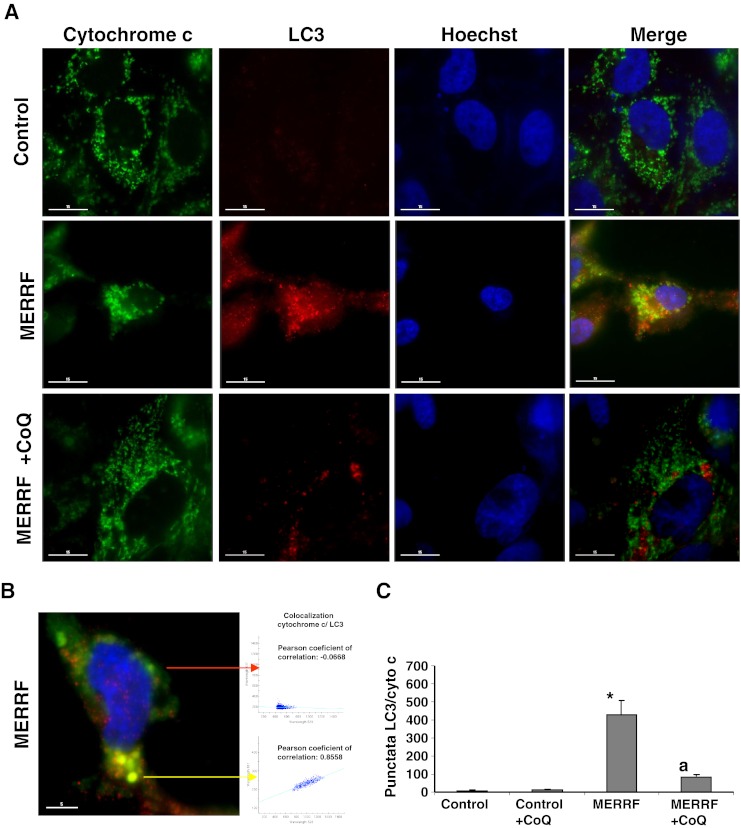
Image analysis of LC3 and cytochrome c immunostained control and myoclonic epilepsy with ragged-red fibers (MERRF) cybrids. (A) Fluorescent analysis of control and MERRF cybrids immunostained with anti-LC3 (autophagosome marker) and cytochrome c (mitochondrial marker) Colocalization of both markers was assessed by the DeltaVision software (Applied Precision, Issaquah, WA). (B) Magnification of a small area in a MERRF cybrid cell. Arrows show autophagolysosomes with LC3 and cytochrome c colocalization. The colocalization of both markers was assessed using DeltaVision software (Applied Precision). (C) Quantification of LC3/cytochrome c puntacta in control and MERRF cybrids incubated with or without 100 μM coenzyme Q10 (CoQ) (n = 100 cells). *p < 0.01 between control and mutant cybrids. ap < 0.01 between the presence and the absence of CoQ
Effect of Coenzyme Q Supplementation in 2 Further Fibroblast Cultures Harboring the m.8344A > G Mutation
To confirm the beneficial effects of CoQ supplementation on MERRF cellular physiology, we estimated ROS levels, H2O2 content, ATP levels, and the number of lysosomes before and after 100 μM CoQ treatment in MERRF fibroblasts (MERRF-1), and two additional fibroblasts cell lines (MERRF-2 and MERRF-3) harboring the A8344G mutation. ROS levels and H2O2 content were significantly increased in fibroblasts cultures established from MERRF-1, MERRF-2, and MERRF-3 patients (Fig. 11A). The inclusion of 100 μM CoQ in the culture medium caused a considerable reduction in ROS levels and H2O2 content in MERRF cultures, but it had no effect in control cultures (Fig. 11A, B). The amount of cellular ATP was significantly reduced in MERRF-1, MERRF-2, and MERRF-3 fibroblast cultures (Fig. 11C). Treatment with CoQ had no effect on ATP levels in control cells, but resulted in an increase in ATP to control values in MERRF cultures (Fig. 11C), suggesting that CoQ is effective in improving cell bioenergetics in MERRF cellular models.
Fig. 11.
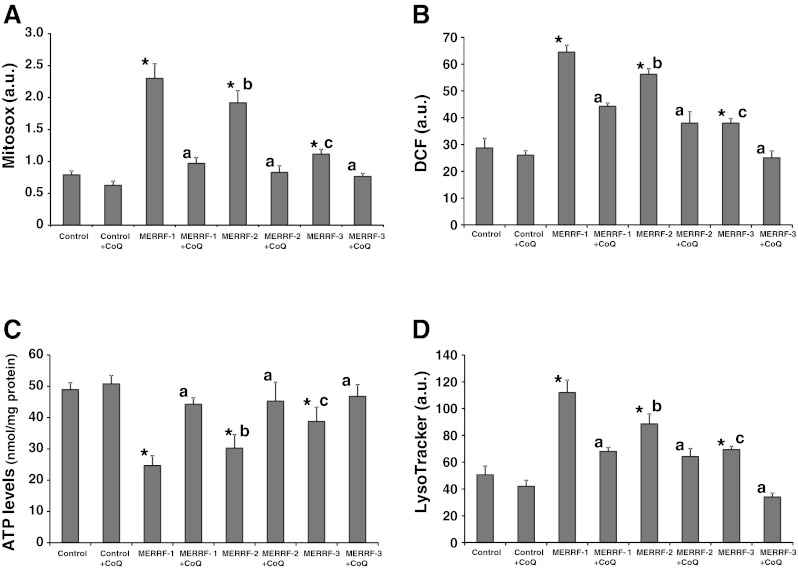
Effect of coenzyme Q10 (CoQ) supplementation in reactive oxygen species (ROS) levels, adenosine-5'-triphosphate (ATP) levels and LysoTracker staining, and in fibroblasts cell lines derived from 2 additional myoclonic epilepsy with ragged-red fibers (MERRF) patients. (A) Mitochondrial ROS generation in control and MERRF fibroblasts (MERRF-1, MERRF-2, and MERRF-3) cultured for 72 h in normal growth medium or medium supplemented with CoQ (100 μM) prior to analysis. Results are expressed as the ratio of MitoSOX signal to NAO signal. (B) H202 levels in control and MERRF fibroblasts cultured for 72 h in normal growth medium or medium supplemented with CoQ (100 μM) prior to analysis. H202 levels were measured using CMH2-DCFDA and flow cytometry analysis, as described (see “Material and Methods” for details). (C) ATP levels in control and MERRF fibroblasts grown in the absence or presence of CoQ (100 μM) for 72 h. (D) Quantification of acidic vacuoles in control and MERRF fibroblasts by LysoTracker staining and flow cytometry analysis. Cells were cultured in the presence or absence of 100 μM CoQ. For the control cells, the data are the mean ± SD for experiments conducted on 2 different control cell lines. Data represent the mean ± SD of 3 separate experiments arbitrary units (a.u.). *p < 0.01 between control and MERRF fibroblasts. ap < 0.01 between the presence and the absence of CoQ. bp < 0.01 between MERRF-2 and MERRF-1 fibroblasts. cp < 0.01 between MERRF-3 and MERRF-2 fibroblasts. DCF=Dichlorofluorescein
The number of lysosomes was significantly increased in MERRF cell lines, with a 1.8-fold increase in LysoTracker red signal in MERRF-1, a 1.5 fold increase in MERRF-2, and a 1.2-fold increase in MERRF-3 (Fig. 11D). CoQ treatment significantly decreased LysoTraker staining to control levels in MERRF cell lines, but only had a moderate effect in control cells (Fig. 11D), suggesting that CoQ is effective in preventing autophagy activation in MERRF cellular models. Interestingly, oxidative stress, ATP levels, and lysosomal activity were correlated with the proportion of A8344G mutated mtDNA in MERRF fibroblasts. MERRF-1 fibroblasts with high heteroplasmy (57%) had significant higher ROS levels and lysosomal activity, and lower ATP levels than MERRF-2 fibroblasts (35% of heteroplasmy) and MERRF-3 fibroblasts (8% of heteroplasmy).
Discussion
In this work, we studied the pathophysiology of the m.8344A > G mutation in primary cultured fibroblasts derived from patients with MERRF syndrome. The MERRF fibroblasts showed reduced expression and activity of the mitochondrial respiratory chain complexes, as has been previously reported [12]. Importantly, the amount of CoQ, an essential electron carrier of the mitochondrial respiratory chain, was also reduced in MERRF fibroblasts, and a large proportion of the mitochondrial population were depolarized, both of which may further compromise normal mitochondrial function and cellular energy metabolism. Reduced ΔΨm will also affect protein import into mitochondria, thus having a secondary effect on critical components of the mitochondrial respiratory chain, such as CoQ, whose biosynthesis depends on mitochondrial proteins encoded by the nucleus, further amplifying mitochondrial disorganization. Ten nuclear genes involved in CoQ biosynthesis have been identified in humans and disease-causing mutations, which lead to primary CoQ deficiency, have been identified in some of these genes [25]. Secondary CoQ deficiency has been identified in a wide range of diseases, including mitochondrial diseases, neurodegenerative diseases, such as Parkinson’s disease, cancer, fibromyalgia, and in patients undergoing statin treatment [22, 26, 27]. Currently, primary and secondary CoQ deficiencies are the focus of some attention, due to the high prevalence of the latter, and the potential for treatment. Detection of secondary CoQ deficiency in mitochondrial diseases is of great importance because CoQ supplementation could be potentially beneficial for the patients, and there is at present, no other effective treatment option.
As well as decreased respiratory chain activity and loss of mitochondrial membrane potential, we identified mitochondrial structural abnormalities, increased ROS production, and H2O2 content, increased lipid peroxidation, and increased expression of autophagic proteins in cells harboring the m.A8344A > G mutation. We observed that CoQ supplementation reversed these pathophysiological changes in MERRF fibroblasts and cybrids. CoQ is the most widely used therapeutic agent in patients with mitochondrial disease because of its well-documented role in energy metabolism. The putative beneficial effects of CoQ supplementation include: enhanced electron transport and ATP production, antioxidant, regulation of redox signaling, stabilization of the mitochondrial permeability transition pore, protecting against autophagy, and apoptotic cell loss [20, 28, 29]. Overall, reports of the efficacy of CoQ treatment in mitochondrial disorders have been mixed. Inconsistencies in clinical studies, including differences in methodology, outcome measures, dosages, duration of treatment, and drug combinations, make it difficult to formulate a clear picture of the effects of CoQ in patients with MERRF syndrome.
Increased ROS production and oxidative stress is a common consequence of mitochondrial dysfunction and CoQ deficiency [20, 29]. We identified a significant increase in ROS generation and H2O2 content in MERRF fibroblasts, which could be ameliorated by CoQ treatment. It has been proposed that excessive ROS can cause the opening of nonspecific high conductance permeability transition pores (PT) in the mitochondrial inner membrane, inducing mitochondrial permeability transition [30], and leading to the simultaneous collapse of ΔΨm. The opening of PT pores causes mitochondria to become permeable to all solutes up to a molecular mass of approximately 1500 Da [31, 32]. Moreover, a possible involvement of the PT in the induction of autophagy of altered mitochondria has been postulated. Thus, oxidative stress may play a prominent role in the induction of the mitochondrial damage, mitochondrial permeability transition, and autophagy activation, which we observed in MERRF fibroblasts. The elimination of dysfunctional mitochondria could play a crucial role in protecting cells from the damage caused by perturbed mitochondrial function and the release of potentially proapoptotic molecules; our finding that the autophagic protein LC3 colocalized with structurally abnormal mitochondria, but not with normal tubular mitochondria in MERRF fibroblasts, suggests that mitophagy specifically targets dysfunctional mitochondria in these cells. In this regard, it has been shown that the voltage-dependent anion channel (VDAC), which is a major component of the PT pore complex on the outer mitochondrial membrane, is more susceptible to oxidative damage in MERRF cybrids than in wild-type cybrids [15]. A recent study showed that Parkin-dependent ubiquitylation of VDAC is a cardinal feature of mitophagy activation in a model of Parkinson’s disease [33].
CoQ deficiency and mitophagy activation were also demonstrated in cybrids harboring the m.8344A > G mutation, indicating that both processes arise as a consequence of this mutation, not of a concomitant nuclear mutation. In addition, CoQ treatment restored the pathophysiological alterations found in MERRF cybrids. In conclusion, our studies in cell cultures, which harbors the m.8344A > G mutation, provide robust experimental evidence that CoQ supplementation could be beneficial in the treatment of patients with MERRF.
Electronic supplementary material
ICMJE form for disclosure of potential conflicts of interest (PDF 586 kb)
Acknowledgments
This work was supported by FIS PI10/00543 grant, FIS EC08/00076 grant, Ministerio de Sanidad, Spain and Fondo Europeo de Desarrollo Regional (FEDER-Unión Europea), SAS 111242 grant, Servicio Andaluz de Salud-Junta de Andalucía, Proyecto de Investigación de Excelencia de la Junta de Andalucía CTS-5725, International Coenzyme Q Association Grant and by AEPMI (Asociación de Enfermos de Patología Mitocondrial), FEEL (Fundación Española de Enfermedades Lisosomales) and Federación Andaluza de Fibromialgia y Fatiga Crónica (ALBA Andalucía). Mario de la Mata is granted with a fellowship from Colegio Oficial de Farmacéuticos de Sevilla. We also thank Elena Garcia from Hospital Vall d´Hebron (Barcelona, Spain) for her help with the cybrid clones. This group is founded by the Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), ISCIII.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
References
- 1.Wallace DC. Mitochondrial DNA sequence variation in human evolution and disease. Proc Natl Acad Sci U S A. 1994;91:8739–8746. doi: 10.1073/pnas.91.19.8739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shoffner JM, Lott MT, Lezza AM, et al. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA(Lys) mutation. Cell. 1990;61:931–937. doi: 10.1016/0092-8674(90)90059-N. [DOI] [PubMed] [Google Scholar]
- 3.Erol I, Alehan F, Horvath R, et al. Demyelinating disease of central and peripheral nervous systems associated with a A8344G mutation in tRNALys. Neuromuscul Disord. 2009;19:275–278. doi: 10.1016/j.nmd.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 4.Wallace DC, Zheng XX, Lott MT, et al. Familial mitochondrial encephalomyopathy (MERRF): genetic, pathophysiological, and biochemical characterization of a mitochondrial DNA disease. Cell. 1988;55:601–610. doi: 10.1016/0092-8674(88)90218-8. [DOI] [PubMed] [Google Scholar]
- 5.Ozawa M, Goto Y, Sakuta R, et al. The 8,344 mutation in mitochondrial DNA: a comparison between the proportion of mutant DNA and clinico-pathologic findings. Neuromuscul Disord. 1995;5:483–488. doi: 10.1016/0960-8966(95)00009-C. [DOI] [PubMed] [Google Scholar]
- 6.Blakely EL, Trip SA, Swalwell H, et al. A new mitochondrial transfer RNAPro gene mutation associated with myoclonic epilepsy with ragged-red fibers and other neurological features. Arch Neurol. 2009;66:399–402. doi: 10.1001/archneurol.2008.576. [DOI] [PubMed] [Google Scholar]
- 7.Virgilio R, Ronchi D, Bordoni A, et al. Mitochondrial DNA G8363A mutation in the tRNA Lys gene: clinical, biochemical and pathological study. J Neurol Sci. 2009;281:85–92. doi: 10.1016/j.jns.2009.01.025. [DOI] [PubMed] [Google Scholar]
- 8.Hammans SR, Sweeney MG, Brockington M, et al. The mitochondrial DNA transfer RNA(Lys)A– > G(8344) mutation and the syndrome of myoclonic epilepsy with ragged red fibers (MERRF) Relationship of clinical phenotype to proportion of mutant mitochondrial DNA. Brain. 1993;116(pt 3):617–632. doi: 10.1093/brain/116.3.617. [DOI] [PubMed] [Google Scholar]
- 9.Enriquez JA, Chomyn A, Attardi G. MtDNA mutation in MERRF syndrome causes defective aminoacylation of tRNA(Lys) and premature translation termination. Nat Genet. 1995;10:47–55. doi: 10.1038/ng0595-47. [DOI] [PubMed] [Google Scholar]
- 10.Yasukawa T, Suzuki T, Ishii N, et al. Defect in modification at the anticodon wobble nucleotide of mitochondrial tRNA(Lys) with the MERRF encephalomyopathy pathogenic mutation. FEBS Lett. 2000;467:175–178. doi: 10.1016/S0014-5793(00)01145-5. [DOI] [PubMed] [Google Scholar]
- 11.James AM, Sheard PW, Wei YH, et al. Decreased ATP synthesis is phenotypically expressed during increased energy demand in fibroblasts containing mitochondrial tRNA mutations. Eur J Biochem. 1999;259:462–469. doi: 10.1046/j.1432-1327.1999.00066.x. [DOI] [PubMed] [Google Scholar]
- 12.James AM, Wei YH, Pang CY, et al. Altered mitochondrial function in fibroblasts containing MELAS or MERRF mitochondrial DNA mutations. Biochem J. 1996;318(pt 2):401–407. doi: 10.1042/bj3180401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chomyn A, Meola G, Bresolin N, et al. In vitro genetic transfer of protein synthesis and respiration defects to mitochondrial DNA-less cells with myopathy-patient mitochondria. Mol Cell Biol. 1991;11:2236–2244. doi: 10.1128/mcb.11.4.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boulet L, Karpati G, Shoubridge EA. Distribution and threshold expression of the tRNA(Lys) mutation in skeletal muscle of patients with myoclonic epilepsy and ragged-red fibers (MERRF) Am J Hum Genet. 1992;51:1187–1200. [PMC free article] [PubMed] [Google Scholar]
- 15.Wu SB, Ma YS, Wu YT, et al. Mitochondrial DNA mutation-elicited oxidative stress, oxidative damage, and altered gene expression in cultured cells of patients with MERRF syndrome. Mol Neurobiol. 2010;41:256–266. doi: 10.1007/s12035-010-8123-7. [DOI] [PubMed] [Google Scholar]
- 16.Quinzii C, Naini A, Salviati L, et al. A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am J Hum Genet. 2006;78:345–349. doi: 10.1086/500092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Sci Dig (NY) 1989;246:500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- 18.Rustin P, Chretien D, Bourgeron T, et al. Biochemical and molecular investigations in respiratory chain deficiencies. Clin Chim Acta. 1994;228:35–51. doi: 10.1016/0009-8981(94)90055-8. [DOI] [PubMed] [Google Scholar]
- 19.Lowry OH, Rosebrough NJ, Farr AL, et al. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 20.Rodriguez-Hernandez A, Cordero MD, Salviati L, et al. Coenzyme Q deficiency triggers mitochondria degradation by mitophagy. Autophagy. 2009;5:19–32. doi: 10.4161/auto.5.1.7174. [DOI] [PubMed] [Google Scholar]
- 21.Lee BY, Han JA, Im JS, et al. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell. 2006;5:187–195. doi: 10.1111/j.1474-9726.2006.00199.x. [DOI] [PubMed] [Google Scholar]
- 22.Sacconi S, Trevisson E, Salviati L, et al. Coenzyme Q10 is frequently reduced in muscle of patients with mitochondrial myopathy. Neuromuscul Disord. 2010;20:44–48. doi: 10.1016/j.nmd.2009.10.014. [DOI] [PubMed] [Google Scholar]
- 23.Vander Heiden MG, Chandel NS, Williamson EK, et al. Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997;91:627–637. doi: 10.1016/S0092-8674(00)80450-X. [DOI] [PubMed] [Google Scholar]
- 24.Scherz-Shouval R, Shvets E, Fass E, et al. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quinzii CM, Lopez LC, Naini A, et al. Human CoQ10 deficiencies. BioFactors (Oxford, England) 2008;32:113–118. doi: 10.1002/biof.5520320113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cordero MD, De Miguel M, Moreno Fernandez AM, et al. Mitochondrial dysfunction and mitophagy activation in blood mononuclear cells of fibromyalgia patients: implications in the pathogenesis of the disease. Arthritis Res Ther. 2010;12:R17. doi: 10.1186/ar2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Molyneux SL, Young JM, Florkowski CM, et al. Coenzyme q10: is there a clinical role and a case for measurement? Clin Biochem Rev. 2008;29:71–82. [PMC free article] [PubMed] [Google Scholar]
- 28.Armstrong JS, Whiteman M, Rose P, et al. The coenzyme Q10 analog decylubiquinone inhibits the redox-activated mitochondrial permeability transition: role of mitcohondrial [correction mitochondrial] complex III. J Biol Chem. 2003;278:49079–49084. doi: 10.1074/jbc.M307841200. [DOI] [PubMed] [Google Scholar]
- 29.Cotan D, Cordero MD, Garrido-Maraver J, et al. Secondary coenzyme Q10 deficiency triggers mitochondria degradation by mitophagy in MELAS fibroblasts. FASEB J. 2011;25:2669–2687. doi: 10.1096/fj.10-165340. [DOI] [PubMed] [Google Scholar]
- 30.Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Archives of biochemistry and biophysics. 2007;462:245–253. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Forte M, Bernardi P. Genetic dissection of the permeability transition pore. J Bioenerg Biomembr. 2005;37:121–128. doi: 10.1007/s10863-005-6565-9. [DOI] [PubMed] [Google Scholar]
- 32.Zoratti M, Szabo I. The mitochondrial permeability transition. Biochim Biophys Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]
- 33.Geisler S, Holmstrom KM, Skujat D, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
ICMJE form for disclosure of potential conflicts of interest (PDF 586 kb)