

Abstract
Epidemiological studies indicate that parental smoking increases the risk for smoking in children. However, the underlying mechanisms by which parental smoking increases the risk for smoking are not known. The aim of these studies was to investigate if preadolescent tobacco smoke exposure, postnatal days 21–35, affects the rewarding effects of nicotine and nicotine withdrawal in adult rats. The rewarding effects of nicotine were investigated with the conditioned place preference procedure. Nicotine withdrawal was investigated with the conditioned place aversion procedure and intracranial self-stimulation (ICSS). Elevations in brain reward thresholds in the ICSS paradigm reflect a dysphoric state. Plasma nicotine and cotinine levels in the preadolescent rats immediately after smoke exposure were 188 ng/ml and 716 ng/ml, respectively. Preadolescent tobacco smoke exposure lead to the development of nicotine dependence as indicated by an increased number of mecamylamine-precipitated somatic withdrawal signs in the preadolescent tobacco smoke exposed rats compared to the control rats. Nicotine induced similar place preference in adult rats that had been exposed to tobacco smoke or air during preadolescence. Furthermore, mecamylamine induced place aversion in nicotine dependent rats but there was no effect of preadolescent tobacco smoke exposure. Finally, preadolescent tobacco smoke exposure did not affect the elevations in brain reward thresholds associated with precipitated or spontaneous nicotine withdrawal. These studies indicate that passive exposure to tobacco smoke during preadolescence leads to the development of nicotine dependence but preadolescent tobacco smoke exposure does not seem to affect the rewarding effects of nicotine or nicotine withdrawal in adulthood.
Keywords: Tobacco smoke, nicotine, preadolescent, reward, withdrawal, ICSS, rats
1. Introduction
Tobacco addiction is a chronic disorder that is characterized by loss of control over smoking, withdrawal symptoms upon smoking cessation, and relapse after periods of abstinence (American Psychiatric Association, 2000; O’Brien, 2003). Evidence indicates that the positive reinforcing effects of cigarettes play a critical role in the initiation of smoking (Finkenauer et al., 2009; Wise, 1996). The positive reinforcing effects of smoking include mild euphoria, relaxation, and an increased ability to focus and process information (Ague, 1973; Benowitz, 1988; Wesnes and Warburton, 1983). Discontinuation of smoking leads to negative affective symptoms such as depressed mood, anxiety, and a decreased ability to focus (Hughes et al., 1991; Hughes and Hatsukami, 1986). The negative affective symptoms associated with smoking cessation have been suggested to increase the risk for relapse to smoking (Bruijnzeel and Gold, 2005; Koob, 2008).
Extensive evidence indicates that maternal smoking during pregnancy and childhood second hand tobacco smoke exposure increases the risk for a wide array of diseases including psychiatric disorders. Smoking during pregnancy has been associated with attention deficit hyperactivity disorder (Milberger et al., 1998), conduct disorder (e.g., destructive and aggressive behavior)(Braun et al., 2008; Wakschlag et al., 1997), and cognitive deficits (Naeye and Peters, 1984) in the offspring. In addition, the children of mothers who smoked during their pregnancy are more likely to develop a tobacco dependency than the children of mothers who did not smoke (Buka et al., 2003; Kandel et al., 1994; Lieb et al., 2003). Although epidemiological studies indicate that exposure to tobacco smoke constituents during development increases the risk for smoking, many questions remain unanswered. For example, it is not known if there are specific developmental periods during which the brain has a heightened sensitivity to neurochemical perturbations caused by tobacco smoke. The detrimental effects of maternal smoking are mainly attributed to in-utero exposure to tobacco smoke constituents. However, mothers who smoke when they are pregnant continue to smoke after giving birth. Therefore, these children might be exposed to high levels of second hand tobacco smoke. This is supported by the observation that cotinine levels that are higher than those in adult smokers have been detected in infants after they were confined in an enclosed space with smokers (Galanti et al., 1998; Stepans and Fuller, 1999). The children of mothers who breastfeed might be exposed to even higher levels of tobacco smoke constituents postnatally than prenatally as the nicotine level in breast milk is approximately three times as high as that in plasma (Luck and Nau, 1984). Another question that remains unanswered is whether developmental tobacco smoke exposure affects the rewarding effects of smoking, tobacco withdrawal, or both during adolescence and adulthood.
Animal models have been developed to study the neurodevelopmental effects of exposure to neurotoxins and drugs of abuse. When studying the neurodevelopmental effects of chemicals with rodent models it is important to take into account that the development of the rodent brain follows a different time course relative to the time of birth than the development of the human brain. For example, the brain growth spurt/period of synaptogenesis takes places in humans during the third trimester and the first 3–4 postnatal years and in rodents during postnatal week 1–3 (Dobbing and Sands, 1971; Dobbing and Sands, 1973). The developmental period in rodents from postnatal day (PN)10–20 equals early childhood in humans and PN21–35 is equivalent to late childhood early adolescence and is often referred to as preadolescence (Spear, 2000). An advantage of investigating the effects of drugs on development from PN21–35 compared to earlier developmental periods is that rats are weaned around PN21. Previous studies have demonstrated that short periods of stress before PN21 can have complex neurodevelopmental effects that can affect the rewarding effects of drugs of abuse by itself (Brake et al., 2004; Matthews et al., 1999). Evidence indicates that preadolescent exposure to psychostimulants can affect the rewarding effects of stimulants later in life. For example, it has been reported that the administration of methylphenidate to rats (twice a day, PN20–35) decreases the rewarding effects of cocaine in adult rats in the conditioned place preference (CPP) procedure and in a rate-dependent ICSS procedure (Andersen et al., 2002; Mague et al., 2005).
The aim of our experiments was to investigate if exposure to tobacco smoke from PN21–35 affects the rewarding effects of nicotine and the deficit in brain reward function associated with nicotine withdrawal in adult rats. The rewarding effects of nicotine were investigated with the CPP procedure. Nicotine has been shown to induce CPP over a wide range of doses when nicotine is paired with the non-preferred compartment of the test apparatus (biased procedure)(Le Foll and Goldberg, 2005). The negative affective state associated with nicotine withdrawal was investigated with the conditioned place aversion (CPA) procedure and a discrete trial ICSS procedure. Previous research has shown that rats avoid the choice compartment that has been paired with nicotine withdrawal (Suzuki et al., 1996). Nicotine withdrawal also leads to elevations in brain reward thresholds in the ICSS procedure (Bruijnzeel et al., 2007; Epping-Jordan et al., 1998). Elevations in brain reward thresholds are interpreted as a deficit in brain reward function as higher current intensities are required to maintain responding for rewarding electrical stimuli. Developmental exposure to tobacco smoke constituents has been shown to increase the risk for smoking during early adulthood (Buka et al., 2003; Kandel et al., 1994). Therefore, it was hypothesized that preadolescent tobacco smoke exposure potentiates the positive and negative reinforcing effects of nicotine. The present studies may contribute to the understanding of the long-term neurodevelopmental effects of preadolescent tobacco smoke exposure.
2. Methods
2.1. Subjects
Pregnant Wistar rats (Embryonic Day 10) were purchased from Charles River (Raleigh, NC). The pregnant rats were single-housed in a temperature and humidity-controlled vivarium and maintained on a 12 hour light-dark cycle (lights off at 6 PM). The rat pups were weaned at day 21 and the male pups were used for the experiments. All testing occurred at the end of the light cycle. Food and water were available adlibitum in the home cages. All subjects were treated in accordance with the National Institutes of Health guidelines regarding the principles of animal care. Animal facilities and experimental protocols were in accordance with the Association for the Assessment and Accreditation of Laboratory Animal Care (AAALAC) and approved by the University of Florida Institutional Animal Care and Use Committee.
2.2. Drugs
Nicotine hydrogen tartrate salt and mecamylamine hydrochloride were purchased from Sigma (Sigma-Aldrich, St. Louis, MO, USA) and dissolved in sterile saline (0.9% sodium chloride). Research cigarettes (3R4F) were purchased from the University of Kentucky (College of Agriculture, Reference Cigarette Program, Lexington, KY).
2.3. Surgical Procedures
2.3.1 Electrode implantations
At the beginning of all the intracranial surgeries, the rats were anesthetized with an isoflurane/oxygen vapor mixture (1–3% isoflurane) and placed in a model 940 Kopf stereotaxic frame (David Kopf Instruments, Tujunga, CA) with the incisor bar set 5.0 mm above the interaural line. The rats were prepared with stainless steel bipolar electrodes (model MS303/2 Plastics One, Roanoke, VA) 11 mm in length in the medial forebrain bundle at the level of the posterior lateral hypothalamus (AP −0.5 mm, ML ±1.7 mm, DV −8.3 mm from dura). The electrodes and cannulae were permanently secured to the skull using dental cement anchored with four skull screws.
2.3.2. Osmotic minipump implantations
The rats were prepared with osmotic minipumps (model 2ML4, 28 day pumps, Durect Corporation, Cupertino, CA) filled with either saline or nicotine salt dissolved in saline. The pumps were implanted subcutaneously under isoflurane/oxygen (1–3% isoflurane) anesthesia. The nicotine concentration was adjusted to compensate for differences in body weight and to deliver a dose of 9 mg/kg per day of nicotine salt (3.16 mg/kg/day nicotine base).
2.4. Tobacco smoke exposure
The rats were exposed to tobacco smoke in standard polycarbonate rodent cages (38 × 28 × 20 cm; L × W × H) with corncob bedding and a wire top as previously described by our research group (Small et al., 2009). The rats were not restrained (whole body exposure) during the tobacco smoke exposure sessions and water was freely available. The rats were moved to the exposure cages immediately prior the the tobacco smoke exposure session and returned to their home cages after the exposure session. Tobacco smoke was generated using a microprocessor-controlled cigarette smoking-machine (model TE-10, Teague Enterprises, Davis, CA)(Teague et al., 1994). Tobacco smoke was generated by burning filtered 3R4F reference cigarettes using a standardized smoking procedure (35 cm3 puff volume, 1 puff per minute, 2 seconds per puff). Mainstream and sidestream smoke was transported to a mixing and diluting chamber. The smoking machine produced a mixture of approximately 10% mainstream smoke and 90% sidestream smoke; based on total suspended particle matter. The smoke was aged for 2 – 4 minutes and diluted with air to a concentration of about 30 mg of total suspended particles (TSP) per m3 before being introduced into the exposure chambers. Exposure conditions were monitored for carbon monoxide (CO) and total suspended particulate matter. CO levels were assessed using a continuous CO analyzer that accurately measures levels between 0 – 2000 parts per million (Monoxor II, Bacharach, New Kensington, PA USA). Total suspended particle matter in the exposure chambers was determined by measurement of samples collected from the chamber onto pre-weighed filters.
2.5. Intracranial self-stimulation procedure
The experimental setup included twelve operant conditioning chambers that were placed in sound-attenuating chambers (Med Associates, Georgia, VT). The rats were first trained to turn a wheel manipulandum (5 × 7 cm; W × H), which was embedded in a wall of the experimental chamber (Med Associates, Georgia, VT), on a FR1 schedule of reinforcement. Each quarter turn of the wheel resulted in the delivery of a 0.5 second train of 0.1 millisecond cathodal square-wave pulses at a frequency of 100 Hz. After the successful acquisition of responding for stimulation on this FR1 schedule, defined as 100 reinforcements within 10 minutes, the rats were trained on a discrete-trial current-threshold procedure (Kornetsky and Esposito, 1979) as described previously (Markou and Koob, 1992). Each trial began with the delivery of a non-contingent electrical stimulus, followed by a 7.5 second response window during which the animal can respond to receive a second contingent stimulus that is identical to the initial non-contingent stimulus. A response during this 7.5 second response window was labeled a positive response, while the lack of a response was labeled a negative response. During a 2 second period immediately after a positive response, additional responses had no consequences. The inter-trial interval (ITI) that followed either a positive response or the end of the response window (in the case of a negative response) had an average duration of 10 seconds (ranging from 7.5 to 12.5 seconds). Responses that occurred during the ITI resulted in a further 12.5 second delay of the onset of the next trial. During training on the discrete-trial procedure, the duration of the ITI and delay periods induced by time-out responses were gradually increased until animals performed consistently at standard test parameters. Then brain reward thresholds were assessed by using a modification of the psychophysical method of limits. Test sessions consisted of four alternating series of descending and ascending current intensities starting with a descending series. Blocks of three trials were presented to the subject at a given stimulation intensity, and the intensity was altered systematically between blocks of trials by 5 μA steps. The initial stimulus intensity was set 40 μA above the baseline current-threshold for each animal. Each test session typically lasted 30–40 minutes and provided two dependent variables for behavioral assessment: brain reward thresholds and response latencies. The current threshold for a descending series was defined as the midpoint between stimulation intensities that supported responding (i.e., positive responses on at least two of the three trials) and current intensities that failed to support responding. The threshold for an ascending series was defined as the midpoint between stimulation intensities that did not support responding and current intensities that supported responding for two consecutive blocks of trials. Thus, four threshold estimates were recorded and the mean of these values was taken as the final threshold. The time interval between the beginning of the non-contingent stimulus and a positive response was recorded as the response latency. The response latency for each test session was defined as the mean response latency on all trials during which a positive response occurred.
2.6. Place conditioning
Place conditioning tests were conducted in four identical wooden setups. Each setup consisted of two conditioning chambers (45 × 45 × 30 cm; W × L × H) that were connected by a center compartment (15 × 15 × 30 cm; W × L × H). The compartments could be closed off by removable guillotine doors. One of the choice compartments was black with a smooth black floor. The other choice compartment had 5 cm black and 5 cm white stripes and corncob bedding on the floor. The center compartment had a gray floor and gray walls. The behavior of the rats was recorded during the pretest and the posttest with digital camcorders and analyzed with Observer 5.0 software (Noldus Information Technology, Wageningen, The Netherlands). Prior to the onset of the conditioning sessions a 15-minute pretest was conducted to determine the non-preferred side and the preferred side. During this pretest the rats could freely explore the three compartments. The conditioned place preference (CPP) and conditioned place aversion (CPA) sessions were conducted over 8 days. For the CPP experiments, nicotine was administered immediately before the rats were placed in the non-preferred chamber and saline was administered immediately before the rats were placed in the preferred chamber. For the CPA experiments, mecamylamine was administered immediately before the rats were placed in the preferred chamber and saline was administered immediately before the rats were placed in the non-preferred chamber. Drugs and saline were administered on alternate days and the conditioning sessions were 20 minutes. The posttest was conducted 1 day after the last conditioning session and the rats did not receive any drugs immediately prior to the posttest. At the beginning of the posttest the rats were placed into the gray center compartment and the rats were allowed to explore the three compartments for 15 minutes.
2.7. Somatic withdrawal signs
Rats were observed for 10 minutes in a Plexiglas observation chamber (25 × 25 × 45 cm; L × W × H). The rats were habituated to the observation chamber for 5 minutes per day on 2 consecutive days prior to testing. The following somatic signs were recorded based on checklist of nicotine abstinence signs: body shakes, cheek tremors, escape attempts, eye blinks, gasps, genital licks, head shakes, ptosis, teeth chattering, writhes, and yawns (Cryan et al., 2003; Malin et al., 1992; Rylkova et al., 2008). Ptosis was counted once per minute if present continuously. The total number of somatic signs was defined as the sum of the individual occurrences. For the final statistical analyses the signs were divided into the following categories: abdominal constrictions which included gasps and writhes; shakes included head shakes and body shakes; facial fasciculations included cheek tremors and teeth chattering; eye blinks; ptosis; yawns; other signs occurred occasionally and included escape attempts and genital licks.
2.8. Plasma nicotine and cotinine levels
Rats were decapitated and trunk blood was collected in polypropylene tubes. The blood was centrifuged for 10 minutes at 4000g and serum was collected. The samples were stored at −80°C until further processing. A validated high-performance liquid chromatography–tandem mass spectrometry (HPLC/MS/MS) method was used to determine nicotine and cotinine levels. First, proteins which could interfere with the HPLC/MS/MS analysis were precipitated by adding 150 μL methanol to 100 μL plasma. This mixture was vortexed for 30 seconds and then centrifuged at 1500 g for 15 minutes. The clear supernatant (100 μL) was carefully transferred into series 200 Perkin Elmer auto sampler vials for HPLC/MS/MS analysis. Nicotine and cotinine were separated by reversed phase chromatography using a Prodigy 5u, 100 × 4.6 mm, C18 column (Phenomenex, Torrance, CA) that was fitted with a C18 pre-column and an isocratic mobile phase composed of 10 mM ammonium acetate buffer in 75% methanol delivered at 1 mL/min by a series 200 Perkin Elmer HPLC pump (Waltham, MA). The injection volume was 10 μL and the chromatographic run time was 4 minutes. The column eluent was directed to the mass spectrometer by atmospheric pressure ionization (API) source. The mass spectrometer (API 4000 LC-MS-MS system, Applied Biosystems/MDS SCIEX, Foster City, CA) was operated in electro spray positive ion mode (ESI+) and quantitation was performed using multiple-reaction monitoring (MRM). The MRM transitions that were used for the quantification of nicotine and cotinine were m/z 163.1 > 132.0 and m/z 177.1 > 146.1, respectively. High purity nitrogen was used as curtain and collision gas and zero grade air was used as the source gas. The API source was operated at 300°C and the ion spray voltage was set at 5 kV. Data acquisition and quantitation were performed using Analyst software version 1.4.2 (Applied Biosystems/MDS SCIEX, Foster City, CA). During the sample analyses quality control samples were interspaced with test samples to ensure the accuracy and reliability of the assay procedure.
2.9. Experiment 1: Effect of preadolescent tobacco smoke exposure on nicotine-induced conditioned place preference
This experiment was conducted in two parts. In the first sub-experiment, the effect of tobacco smoke exposure (PN21–35) on CPP induced by 0.1 mg/kg of nicotine base was investigated. In the second sub-experiment, the effect of tobacco smoke exposure (PN21–35) on CPP induced by 0.04 mg/kg of nicotine base was investigated. In the first sub-experiment (0.1 mg/kg of nicotine), half the rats were exposed to tobacco smoke (n = 16) and the air-control rats (n = 15) were placed on a cart in the laboratory during the smoke exposure sessions. The control rats were never placed in the laboratory space with the smoking machine in order to prevent exposure to tobacco fumes. The rats in the tobacco group were exposed to tobacco smoke for 4 hours per day and the smoke exposure sessions were conducted between 7:00 AM and 12:00 noon. CPP experiments were conducted in adult animals (> P90). Nicotine was administered immediately before the rats were placed in the non-preferred chamber. The second sub-experiment was the same as the first sub-experiment with the exception that the tobacco smoke exposed rats (n=25) and the air-control rats (n=12) were treated with 0.04 mg/kg of nicotine prior to the conditioning sessions. In order to determine plasma nicotine and cotinine levels, 12 rats were decapitated at two different time points. Blood was collected immediately after tobacco smoke exposure after 7 (n = 6) and 14 days (n = 6) of tobacco smoke exposure.
2.10. Experiment 2: Effect of preadolescent tobacco smoke exposure on nicotine withdrawal-induced conditioned place aversion
Rat pups were exposed to smoke from PN21–35. Half of the rats was exposed to smoke (n = 39) and the other rats (n = 39) were placed on a cart in the laboratory during the smoke exposure sessions. Rats were exposed to smoke for 4 hours per day and the smoke exposure sessions were conducted between 7:00 AM and 12:00 noon. When the rats were adults (> P90), they were prepared with osmotic minipumps that delivered 3.2 mg/kg of nicotine base per day (9 mg/kg of nicotine salt). The conditioning sessions started at least 6 days after the implantations of the minipumps. Mecamylamine (vehicle, 0.33, 1, 3 mg/kg, n = 9–10 per group) was administered immediately before the rats were placed in the preferred chamber.
2.11. Experiment 3: Effect of preadolescent tobacco smoke exposure on nicotine withdrawal-induced elevations in brain reward thresholds
The rats in the tobacco group (n = 20) were exposed to smoke from PN21–35 and the air-control rats (n = 19) were placed on a cart in the laboratory during the smoke exposure sessions. Rats were exposed to smoke for 4 hours per day and the smoke exposure sessions were conducted between 7:00 AM and 12:00 noon. When the rats were adults (> P90), they were prepared with electrodes and trained on the ICSS procedure. After stable brain reward thresholds were achieved (defined as less than 10% variation within a 5 day period) the rats were prepared with nicotine (9 mg/kg/day of nicotine salt/3.16 mg/kg/day nicotine base; tobacco-nicotine pumps n = 13; air-control-nicotine pumps n = 11) or saline pumps (tobacco-saline pumps n = 7, air control-saline pumps n = 8). The nAChR antagonist mecamylamine was used to investigate the effects of precipitated withdrawal on brain reward thresholds and response latencies. Mecamylamine (vehicle, 0.3, 1, 3 mg/kg, sc) was administered according to a Latin square design 5 minutes before the rats were placed in the ICSS test chambers. There was a 48-hour interval between each mecamylamine injection. This time interval allowed the reestablishment/maintenance of nicotine dependence. The plasma elimination half-life of mecamylamine is approximately 1 hour (Debruyne et al., 2003). In order to investigate the effects of preadolescent smoke exposure on spontaneous nicotine withdrawal, the minipumps were removed on day 28 and brain reward thresholds and response latencies were assessed 3, 6, 12, 24, 36, and 48 hours after pump removal.
2.12. Experiment 4: Effects of preadolescent tobacco smoke exposure on mecamylamine-precipitated somatic withdrawal signs
The aim of this experiment was to investigate if preadolescent tobacco smoke exposure would lead to the development of nicotine dependence during the exposure period. Half of the rats (n=10) were exposed to tobacco smoke for 9 consecutive days (PN21–29) and the other half (air-control, n=10) were placed on a cart in the laboratory during the smoke exposure sessions. The time interval was based on previous studies that reported that rats are nicotine dependent after about 7 days of continuous nicotine administration (Bruijnzeel et al., 2007; Epping-Jordan et al., 1998). Rats were exposed to smoke for 4 hours per day and the smoke exposure sessions were conducted between 7:00 AM and 12:00 noon. The nAChR antagonist mecamylamine was used to investigate the effects of precipitated withdrawal on somatic withdrawal signs. Mecamylamine (2 mg/kg, sc) was administered 5 minutes before the rats were placed in the observation chambers.
2.13. Data analyses
In order to analyze the effects of tobacco smoke exposure on body weight gain, the body weights of the rats were expressed as a percentage of the body weights on the day prior to tobacco smoke exposure. The effect of tobacco smoke exposure on body weight gain was analyzed with a two-way repeated-measures analyses of variance (ANOVA) with exposure condition (air or tobacco smoke) as the between subjects factor and time as the within subjects factor. In the CPP experiment, the effects of nicotine conditioning and tobacco smoke exposure on the amount of time spent in the nonpreferred chamber were analyzed with a two-way repeated-measures ANOVA with conditioning as the within subjects factor and exposure condition (air or tobacco smoke) as the between subjects factor. A separate analysis was conducted for each nicotine dose. Then the effects of nicotine dose and tobacco smoke on place preference were analyzed with a two-way ANOVA with nicotine dose and treatment (air or tobacco smoke) as between subjects factors. In the CPA experiment, the effects of mecamylamine and tobacco smoke exposure on place aversion were analyzed with a two-way ANOVA with the dose of mecamylamine and exposure condition (air or tobacco smoke) as the between subjects factors. Comparisons between the pre-conditioning and post-conditioning groups were conducted by using one-way repeated measures ANOVA’s. In the precipitated withdrawal/ICSS experiment, the ICSS parameters (brain reward thresholds and response latencies) were expressed as a percent of the respective animal’s pretest day values. Percent changes in ICSS parameters were analyzed using a three-way repeated-measures ANOVA with the dose of mecamylamine as the within subjects factor and exposure condition (air or tobacco smoke) and pump content (saline or nicotine) as between subjects factors. In the spontaneous withdrawal/ICSS experiment, the ICSS parameters (brain reward thresholds and response latencies) were expressed as a percentage of the respective animal’s pretest values obtained on the day prior to minipump explantation. Percent changes in ICSS parameters were analyzed using a three-way repeated-measures ANOVA with time as the within subjects factor and exposure condition (air or tobacco) and pump content (saline or nicotine) as between subjects factors. Newman-Keuls post hoc tests were conducted when the ANOVA revealed statistically significant effects. The effects of tobacco smoke on mecamylamine-precipitated somatic signs were analyzed with a one-way ANOVA. For all the experiments, the criterion for significance was set at 0.05. The statistical analyses were performed using PASW Statistics version 18.
3. Results
3.1. Experiment 1: Effect of preadolescent tobacco smoke exposure on nicotine-induced conditioned place preference
There were no differences in body weights between the tobacco group and the air-control group prior to the onset of tobacco smoke exposure (Table 1). Exposure to tobacco smoke decreased body weight gain during the 15-day exposure period for the 0.1 mg/kg of nicotine group (Time × Treatment: F14,406=4.71, P<0.0001) and the 0.04 mg/kg of nicotine group (Time × Treatment: F14,322=6.88, P<0.0001). The 0.1 mg/kg dose of nicotine induced CPP in the tobacco group and the air-control group (Figure 1A; Treatment: F1,28=8.19, P<0.008). The 0.04 mg/kg dose of nicotine did not induce CPP in the tobacco smoke exposed rats or in the air-control rats (Figure 1B). Nicotine (0.1 mg/kg) increased the amount of time spent in the initially non-preferred chamber to the same degree in the tobacco group and the air-control group. An additional statistical comparison indicated that the 0.1 mg/kg dose of nicotine was more effective in inducing CPP than the 0.04 mg/kg dose of nicotine (Figure 1C; Dose: F1,52=37.2, P<0.0001). After 7 days of tobacco smoke exposure, the plasma nicotine and cotinine levels were 183.6 ± 42.0 ng/ml and 757.5 ± 78.3 ng/ml, respectively. After 14 days of tobacco smoke exposure, the plasma nicotine and cotinine levels were 192.4 ± 21.6 ng/ml and 674.3 ± 47.2 ng/ml, respectively. Both nicotine and cotinine levels were the same after 7 and 14 days of tobacco smoke exposure. This indicates that the tobacco smoke exposure setup induces a reliable and reproducible increase in plasma nicotine and cotinine levels.
Table 1.
Effect of tobacco smoke exposure (PN21–35) on absolute body weights.
| Experiment | Pre (PN21) | Post (PN35) | Post-Pre | |||
|---|---|---|---|---|---|---|
| Air | Tobacco | Air | Tobacco | Delta | P-value | |
| Expt. 1 (CPP-0.10, n=15–16) | 50.1 ± 1.2 | 50.2 ± 1.1 | 164.0 ± 1.7 | 153.2 ± 1.5 | 10.9 | P<0.0001 |
| Expt. 1 (CPP-0.04, n=12–13) | 54.0 ± 1.0 | 54.1 ± 1.5 | 156.9 ± 2.8 | 153.4 ± 2.8 | 3.6 | P<0.0001 |
| Expt. 2 (CPA, n=39) | 41.4 ± 0.8 | 41.6 ± 0.7 | 134.0 ± 1.8 | 128.3 ± 1.5 | 5.9 | P<0.0001 |
| Expt. 3 (ICSS, n=19–20) | 58.4 ± 2.3 | 59.8 ± 2.7 | 177.0 ± 3.9 | 166.8 ± 4.1 | 11.6 | P<0.0001 |
Data are expressed as means (grams ± S.E.M.). P-values indicate lower body weight gain over the 15-day exposure period in the tobacco smoke exposed rats than in the control rats. Post-Pre Delta (grams) indicates the difference in body weight gain between the tobacco group and the air-control group from PN21–35 ([Air PN35 – Air PN21] - [Tobacco PN35 – Tobacco PN21]).
Abbreviations; N, number of animals per control or tobacco group; CPP, conditioned place preference; CPA, conditioned place aversion; ICSS, intracranial self-stimulation.
Figure 1.



Effect of preadolescent (PN21–35) tobacco smoke exposure on nicotine-induced CPP (A, 0.10 mg/kg, tobacco n = 16, air-control n = 15; B, 0.04 mg/kg, tobacco n = 13, air-control n = 12). Figure 2C depicts the difference in the amount of time spent in the nicotine-paired chamber between the pre-conditioning and post-conditioning session. In figure 1A and 1C, asterisks (** P<0.01) indicate an increased amount of time spent in the nicotine-paired compartment in the posttest than in the pretest.
3.2. Experiment 2: Effect of preadolescent tobacco smoke exposure on nicotine withdrawal-induced conditioned place aversion
Exposure to tobacco smoke decreased body weight gain during the 15-day exposure period (Table 1; Time × Treatment: F14,1064=11.72, P<0.0001). Prior to the implantation of the nicotine pumps there were no differences in body weights between the smoke exposed rats and the air-control rats (Control: 448.1 ± 4.8; Tobacco: 452.9 ± 5.2). Mecamylamine induced CPA in the nicotine-dependent rats but there was no effect of preadolescent tobacco smoke exposure (Figure 2A; Dose: F3,70=3.6, P<0.018). Posthoc comparisons indicated that 3 mg/kg of mecamylamine was more effective in inducing CPA than 0.33 and 1 mg/kg of mecamylamine (Figure 2A). Furthermore, one-way ANOVAs indicated that 3 mg/kg of mecamylamine induced CPA in the tobacco-nicotine group (Figure 2B; F1,9=8.7, P<0.05) and 1 and 3 mg/kg of mecamylamine induced CPA in the air-control-nicotine group (1 mg/kg, F1,9=6.7, P<0.05; 3 mg/kg, F1,9=8.7, P<0.0001; pre-post conditioning comparisons).
Figure 2.


Effect of preadolescent (PN21–35) tobacco smoke exposure on mecamylamine-induced CPA in nicotine dependent rats (n = 9–10 per group). Figure 3A depicts the difference in the amount of time spent in the mecamylamine-paired chamber between the pretest and posttest. In figure 2A, asterisks (** P<0.01) indicate a decreased amount of time spent in the mecamylamine-paired compartment in rats treated with 3 mg/kg of mecamylamine compared to rats treated with vehicle (0 mg/kg of mecamylamine). In figure 2B, asterisks (* P<0.05, ** P<0.01) indicate a decreased amount of time spent in the mecamylamine-paired compartment in the posttest compared to the pretest.
3.3. Experiment 3: Effect of preadolescent tobacco smoke exposure on nicotine withdrawal-induced elevations in brain reward thresholds
Exposure to smoke decreased body weight gain during the 15-day exposure period (Table 1; Time × Treatment: F14,518=4.43, P<0.0001). Prior to implantation of the minipumps there were no differences in brain reward thresholds between any of the groups (Air-Saline: 107.7 ± 11.7; Air-Nicotine: 124.9 ± 9.4; Tobacco-Saline: 129.1 ± 3.7; Tobacco-Nicotine: 131.4 ± 8.6). There were also no differences in response latencies (Air-saline: 3.3 ± 0.1; Air-nicotine: 3.2 ± 0.1; Tobacco-saline: 3.7 ± 0.1; Tobacco-nicotine: 3.3 ± 0.1). The nAChR receptor antagonist mecamylamine elevated the brain reward thresholds of the nicotine-treated rats and did not affect the brain reward thresholds of the saline-treated rats (Figure 3A; Dose × Pump content: F3,105=14.75, P<0.0001). Preadolescent smoke exposure did not alter the effects of mecamylamine on the brain reward thresholds in the nicotine or saline-treated rats. Mecamylamine increased the response latencies of the nicotine-treated rats and did not affect the response latencies of the saline-treated rats (Figure 3B; Dose × Pump content: F3,105=4.50, P<0.005). Preadolescent smoke exposure did not alter the effects of mecamylamine on the response latencies in the nicotine or saline-treated rats. Explantation of the minipumps elevated the brain reward thresholds of the nicotine-treated rats and did not affect the brain reward thresholds of the saline-treated rats (Figure 4A; Time × Pump content: F5,175=6.90, P<0.0001). Preadolescent smoke exposure did not affect the brain reward thresholds after pump explantation in the nicotine or saline-treated rats. Explantation of the minipumps and preadolescent tobacco smoke exposure did not affect the response latencies of the nicotine and saline-treated rats (Figure 4B).
Figure 3.


Effect of preadolescent (PN21–35) tobacco smoke exposure on mecamylamine-precipitated nicotine withdrawal (A, brain reward thresholds; B, response latencies; tobacco-nicotine pumps n = 13, tobacco-saline pumps n = 7; air-control-nicotine pumps n = 11, air-control-saline pumps n = 8). Brain reward thresholds and response latencies are expressed as a percentage of the pretest day values. Asterisks (* P<0.05, ** P<0.01) indicate elevations in brain reward thresholds or increased response latencies compared to those of the corresponding saline-treated control group (0 mg/kg of mecamylamine). Plus signs (+ P<0.05) indicate elevations in brain reward thresholds compared to those of the corresponding control group treated with 1 mg/kg of mecamylamine. Data are expressed as means ± SEM.
Figure 4.


Effect of preadolescent (PN21–35) tobacco smoke exposure on spontaneous nicotine withdrawal (A, brain reward thresholds; B, response latencies; tobacco-nicotine pumps n = 13, tobacco-saline pumps n = 7; air-control-nicotine pumps n = 11, air-control-saline pumps n = 8). Brain reward thresholds and response latencies are expressed as a percentage of the values obtained on the day prior to minipump explantation. Asterisks (* P<0.05, ** P<0.01) indicate elevations in brain reward thresholds compared to those of the corresponding saline-treated control group. Data are expressed as means ± SEM.
3.4 Experiment 4: Effects of preadolescent tobacco smoke exposure on mecamylamine-precipitated somatic withdrawal signs
This experiment was conducted to investigate if preadolescent tobacco smoke exposure leads to the development of nicotine dependence. Mecamylamine precipitated a greater number of somatic signs in the preadolescent rats that were exposed to tobacco smoke for 9 days than in the preadolescent air-control rats (Figure 5; Treatment: F1,19=71.7, P<0.0001). Furthermore, the administration of mecamylamine lead to a greater number of abdominal constrictions (Table 2; Treatment: F1,19=26.4, P<0.0001), eye blinks (Treatment: F1,19=4.8, P<0.05), occurrences of ptosis (Treatment: F1,19=4.9, P<0.05), facial fasciculations (Treatment: F1,19=5.8, P<0.05), and yawns (Treatment: F1,19=12.2, P<0.003) in the tobacco group than in the air-control group. There were no significant differences in the number of shakes or other signs between the tobacco group and the air-control group. This experiment indicates that exposure to tobacco smoke during preadolescence leads to the development of nicotine dependence. Body weights were not systematically recorded as the previous three studies (Experiment 1 – 3) consistently demonstrated that smoke exposure attenuates body weight gain.
Figure 5.
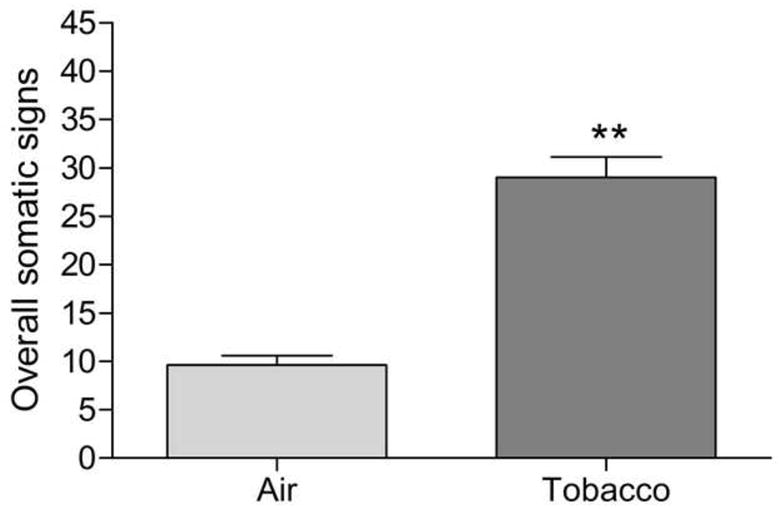
Effect of preadolescent (PN21–29) tobacco smoke exposure on mecamylamine-precipitated somatic withdrawal signs (tobacco n = 10, air-control n = 10). Asterisks (** P<0.01) indicate a greater number of somatic signs in the tobacco group than in the air-control group. Data are expressed as means ± SEM.
Table 2.
Effects of tobacco smoke exposure on mecamylamine-precipitated somatic signs in preadolescent rats.
| Treatment | Air | Tobacco |
|---|---|---|
| Abd. const. | 0.5 ± 0.2 | 9.7 ± 1.8** |
| Eye blinks | 5.2 ± 1.0 | 9.2 ± 1.5* |
| Ptosis | 3.5 ± 0.8 | 6.6 ± 1.1* |
| Facial fasc. | 0.0 ± 0.0 | 1.4 ± 0.6* |
| Yawns | 0.0 ± 0.0 | 1.1 ± 0.3** |
| Shakes | 0.3 ± 0.3 | 1.0 ± 0.5 |
| Other signs | 0.1 ± 0.1 | 0.0 ± 0.0 |
Somatic signs in control rats (n = 10) and rats that were exposed to tobacco smoke for 9 days (P21–29, n = 10). Abdominal constrictions include gasps and writhes; facial fasciculations include cheek tremors and teeth chattering; shakes include head shakes and body shakes; other signs include escape attempts and genital licks. Asterisks (* P<0.05, **P<0.01) indicate more somatic signs in the tobacco group than in the air-control group. Data are expressed as means ± SEM.
4. Discussion
The aim of the present experiments was to investigate the effects of preadolescent tobacco smoke exposure on the rewarding effects of nicotine and nicotine withdrawal. These studies demonstrated that 0.1 mg/kg of nicotine, but not 0.04 mg/kg of nicotine, induced CPP. Preadolescent tobacco smoke exposure did not affect the rewarding effects of nicotine in the CPP procedure. Furthermore, the nAChR antagonist mecamylamine induced CPA in the nicotine-treated rats and there was no effect of preadolescent tobacco smoke exposure on mecamylamine-induced CPA. In the third experiment, it was demonstrated that mecamylamine dose-dependently elevated the brain reward thresholds of the nicotine-treated rats and did not affect the brain reward thresholds of the saline-treated control rats. Explantation of the minipumps also elevated the brain reward thresholds of the nicotine-treated rats and did not affect the brain reward thresholds of the saline-treated control rats. Preadolescent tobacco smoke exposure did not affect the elevations in brain reward thresholds associated with precipitated or spontaneous nicotine withdrawal.
Passive exposure to tobacco smoke lead to a plasma nicotine level of 188 ng/ml and a cotinine level of 716 ng/ml in the preadolescent rats. In the present experiments the rats were exposed to tobacco smoke for 4 hours per day and the TSP level was about 30 mg/m3. It is interesting to note that the plasma nicotine and cotinine levels in the preadolescent rats were in the same range as those of adult animals exposed to higher levels of tobacco smoke. Anderson and colleagues reported that exposure to tobacco smoke with a TSP level of 87 mg/m3 for 6 hours/day leads to a nicotine level of approximately 95 ng/ml and a cotinine level of 790 ng/ml in adult rats (Anderson et al., 2004). In addition, in a previous study we demonstrated that exposure to tobacco smoke with a TSP level of 100 mg/m3 for 4 hours/day leads to a plasma nicotine level of approximately 120 ng/ml and a cotinine level of 570 ng/ml in adult rats (Small et al., 2009). There are several possible explanations for the relatively high nicotine level in the preadolescent rats. First, young animals have a higher oxygen/air intake per unit of body weight compared to older animals (Schefer and Talan, 1996). Therefore, the preadolescent rats might inhale more nicotine per unit of body weight than the older rats. Second, in the present study the rats were rapidly decapitated after the tobacco smoke exposure session and in the other studies blood was collected via an intravenous catheter or from the caudal vena cava of anesthetized rats (Anderson et al., 2004; Small et al., 2009). It can be assumed that it will require less time to decapitate animals and collect blood than to collect blood from a vein. Nicotine is rapidly metabolized, the plasma half-life of nicotine is 20 minutes, and therefore the time point of blood collection relative to the end of the smoke exposure session will affect nicotine levels (Sastry et al., 1995). Plasma nicotine and cotinine levels in heavy smokers are approximately 40 and 300 ng/ml, respectively (Benowitz, 1988; Benowitz et al., 1982; Lawson et al., 1998; Wall et al., 1988). Therefore, the present finding indicates that plasma nicotine and cotinine levels that are the same or higher than those in smokers can be obtained in preadolescent rats by passive exposure to a relatively low level of tobacco smoke.
The present study demonstrated that passive exposure to tobacco smoke leads to the development of nicotine dependence in preadolescent rats as indicated by an increased number of mecamylamine-precipitated somatic signs. This study is in line with previous studies that reported chronic exposure to nicotine via osmotic minipumps, nicotine self-administration, or intermittent exposure to tobacco smoke leads to the development of nicotine dependence (Malin et al., 1994; Paterson and Markou, 2004; Small et al., 2009). The rats displayed 29 somatic signs in the present study and 13.9 and 20.2 somatic signs in a previous tobacco smoke exposure study (Small et al., 2009). It is most likely that the rats in the present study displayed more somatic signs because the plasma nicotine level was higher than in the previous study (188 ng/ml vs. 120 ng/ml) and the rats received a higher dose of mecamylamine (2 mg/kg vs. 1 mg/kg).
Preadolescent tobacco smoke exposure did not affect the rewarding effects of nicotine or nicotine withdrawal later in life. It is unlikely that the lack of an effect of preadolescent tobacco smoke exposure was due to deficiencies in the CPP, CPA, or the ICSS procedures. The present findings showed that 0.1 mg/kg of nicotine, but not 0.04 mg/kg of nicotine, induced CPP. This is in line with a study by Le Foll and Goldberg that demonstrated that 0.1 mg/kg of nicotine but not 0.04 mg/kg induces CPP (Le Foll and Goldberg, 2005). The highest dose of nicotine that did not induce CPP in the study by Le Foll and Goldberg was 0.04 mg/kg of nicotine (Le Foll and Goldberg, 2005). Therefore, it might have been expected that if preadolescent tobacco smoke exposure would have potentiated the rewarding effects of nicotine this would have been reflected in an increase in the rewarding effects of 0.04 mg/kg of nicotine. In the second experiment, it was demonstrated that mecamylamine induced CPA in the nicotine dependent rats. Statistical analysis indicated that 1 and 3 mg/kg of mecamylamine induced CPA in the nicotine-treated air-control group (Figure 3A). This observation is in line with previous studies that reported that mecamylamine in the 1–3 mg/kg dose range induces CPA in nicotine dependent rats (O’Dell et al., 2007; Suzuki et al., 1996). The results of the third experiment demonstrated that mecamylamine and discontinuation of nicotine administration elevated the brain reward thresholds of the nicotine treated rats. Neither mecamylamine nor explantation of the minipumps affected the brain reward thresholds of the saline-treated control rats. This study is in line with previous studies that reported that precipitated or spontaneous nicotine withdrawal leads to a deficit in brain reward function (Bruijnzeel et al., 2007; Epping-Jordan et al., 1998).
The present studies suggest that preadolescent (PN21–35) exposure to tobacco smoke does not affect the rewarding effects of nicotine or nicotine withdrawal in adulthood. We are not aware of any other studies that investigated the long-term effects of tobacco smoke exposure on the rewarding effects of nicotine or nicotine withdrawal. Therefore, we cannot compare the present study with previous studies that investigated the long-term effects of tobacco smoke exposure. In contrast, the long-term effect of the repeated systemic administration of drugs of abuse has been intensively investigated. These studies suggest that the repeated exposure to drugs of abuse leads to sensitized locomotor responses and a potentiation of the rewarding effects of drugs abuse (Robinson and Berridge, 1993; Vanderschuren and Kalivas, 2000). It should be noted, however, that the sensitized locomotor or rewarding effects are not detected under all experimental conditions. For example, the repeated administration of cocaine to adult rats has been shown to potentiate the locomotor activating and rewarding effects of cocaine (Heidbreder et al., 1996; Lett, 1989; Pudiak and Bozarth, 1993). However, in contrast to the aforementioned findings with adult animals, the repeated administration of cocaine to preadolescent animals (P20–35) decreases the rewarding effects of cocaine in the CPP and ICSS procedure in adult animals. Therefore, these studies suggest that the long-term effects of drugs of abuse depend on the developmental period during which the drug is administered (Andersen et al., 2002; Mague et al., 2005). It has also been reported that the administration of nicotine to adolescent rats, PN 34–43, does not affect nicotine-induced CPP (0.3 and 0.6 mg/kg) in adulthood (Adriani et al., 2006). Therefore, the administration of drugs of abuse to adult animals might induce sensitization processes whereas the administration of drugs of abuse to preadolescent animals might have no long-term effects or protect against the rewarding effects of drugs of abuse. Additional studies are warranted to investigate if passive exposure to tobacco smoke during adulthood sensitizes the locomotor and rewarding effects of nicotine in rats. Furthermore, it cannot be ruled out that exposure to tobacco smoke during an earlier developmental period (prenatally or postnatally) would affect the rewarding effects of nicotine during adolescence or later in life. For example, it has been demonstrated that exposure to cocaine from PN11–20 leads to changes in dopamine signaling and dynorphin mRNA levels in adult rats (Busidan and Dow-Edwards, 1999; Dow-Edwards and Hurd, 1998; Zhao et al., 2008). Another variable that might play a role in the development of drug sensitization is the rate of administration. Robinson and colleagues investigated the effects of the rate of intravenous nicotine infusion on the development of locomotor sensitization (Samaha et al., 2005). They demonstrated that the rapid intravenous infusion of nicotine (5 or 25 sec per infusion) but not slow infusion (100 sec) leads to the development of nicotine-induced locomotor sensitization. In the present study, tobacco smoke levels gradually increased and then remained stable for the remainder of the 4-hour exposure sessions. Therefore, it cannot be ruled out that different exposure conditions might have lead to a long term potentiation of the rewarding effects of nicotine. It should also be noted that that the CPP procedure does not provide information about the motivation to self-administer nicotine. Therefore, additional studies are needed to investigate if exposure to tobacco smoke or nicotine during development affects the motivation to self-administer nicotine under fixed and progressive ratio schedules of reinforcement.
In conclusion, these studies extend and corroborate previous studies by demonstrating that passive exposure to a relatively low level of tobacco smoke leads to high nicotine levels and nicotine dependence in preadolescent rats. These studies also suggest that preadolescent tobacco smoke exposure does not affect the rewarding effects of nicotine or nicotine withdrawal later in life. However, it cannot be ruled out that tobacco smoke exposure during a different developmental period or that a different tobacco smoke exposure regimen could have mediated long-term changes in the rewarding effects of nicotine or nicotine withdrawal.
Acknowledgments
This research was funded by a Flight Attendant Medical Research Institute Young Clinical Scientist Award (Grant nr. 52312) and a National Institute on Drug Abuse grant (DA023575) to Adrie Bruijnzeel. Kim Keijzers received salary support from the Dutch Heart Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adriani W, Deroche-Gamonet V, Le MM, Laviola G, Piazza PV. Preexposure during or following adolescence differently affects nicotine-rewarding properties in adult rats. Psychopharmacology (Berl) 2006;184:382–390. doi: 10.1007/s00213-005-0125-1. [DOI] [PubMed] [Google Scholar]
- Ague C. Nicotine and smoking: effects upon subjective changes in mood. Psychopharmacologia. 1973;30:323–328. doi: 10.1007/BF00429191. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. Washington, DC: American Psychiatric Press; 2000. [Google Scholar]
- Andersen SL, Arvanitogiannis A, Pliakas AM, LeBlanc C, Carlezon WA., Jr Altered responsiveness to cocaine in rats exposed to methylphenidate during development. Nat Neurosci. 2002;5:13–14. doi: 10.1038/nn777. [DOI] [PubMed] [Google Scholar]
- Anderson KL, Pinkerton KE, Uyeminami D, Simons CT, Carstens MI, Carstens E. Antinociception induced by chronic exposure of rats to cigarette smoke. Neurosci Lett. 2004;366:86–91. doi: 10.1016/j.neulet.2004.05.020. [DOI] [PubMed] [Google Scholar]
- Benowitz NL. Drug therapy. Pharmacologic aspects of cigarette smoking and nicotine addition. N Engl J Med. 1988;319:1318–1330. doi: 10.1056/NEJM198811173192005. [DOI] [PubMed] [Google Scholar]
- Benowitz NL, Kuyt F, Jacob P., III Circadian blood nicotine concentrations during cigarette smoking. Clin Pharmacol Ther. 1982;32:758–764. doi: 10.1038/clpt.1982.233. [DOI] [PubMed] [Google Scholar]
- Brake WG, Zhang TY, Diorio J, Meaney MJ, Gratton A. Influence of early postnatal rearing conditions on mesocorticolimbic dopamine and behavioural responses to psychostimulants and stressors in adult rats. Eur J Neurosci. 2004;19:1863–1874. doi: 10.1111/j.1460-9568.2004.03286.x. [DOI] [PubMed] [Google Scholar]
- Braun JM, Froehlich TE, Daniels JL, Dietrich KN, Hornung R, Auinger P, Lanphear BP. Association of Environmental Toxicants and Conduct Disorder in U.S. Children: NHANES 2001–2004. Environ Health Perspect. 2008;116:956–962. doi: 10.1289/ehp.11177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruijnzeel AW, Gold MS. The role of corticotropin-releasing factor-like peptides in cannabis, nicotine, and alcohol dependence. Brain Res Brain Res Rev. 2005;49:505–528. doi: 10.1016/j.brainresrev.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Bruijnzeel AW, Zislis G, Wilson C, Gold MS. Antagonism of CRF receptors prevents the deficit in brain reward function associated with precipitated nicotine withdrawal in rats. Neuropsychopharmacology. 2007;32:955–963. doi: 10.1038/sj.npp.1301192. [DOI] [PubMed] [Google Scholar]
- Buka SL, Shenassa ED, Niaura R. Elevated risk of tobacco dependence among offspring of mothers who smoked during pregnancy: a 30-year prospective study. Am J Psychiatry. 2003;160:1978–1984. doi: 10.1176/appi.ajp.160.11.1978. [DOI] [PubMed] [Google Scholar]
- Busidan Y, Dow-Edwards DL. Behavioral sensitization to apomorphine in adult rats exposed to cocaine during the preweaning period: a preliminary study. Pharmacol Biochem Behav. 1999;63:417–421. doi: 10.1016/s0091-3057(99)00003-9. [DOI] [PubMed] [Google Scholar]
- Cryan JF, Bruijnzeel AW, Skjei KL, Markou A. Bupropion enhances brain reward function and reverses the affective and somatic aspects of nicotine withdrawal in the rat. Psychopharmacology (Berl) 2003;168:347–358. doi: 10.1007/s00213-003-1445-7. [DOI] [PubMed] [Google Scholar]
- Debruyne D, Sobrio F, Hinschberger A, Camsonne R, Coquerel A, Barre L. Short-term pharmacokinetics and brain distribution of mecamylamine as a preliminary to carbon-11 labeling for nicotinic receptor investigation. J Pharm Sci. 2003;92:1051–1057. doi: 10.1002/jps.10302. [DOI] [PubMed] [Google Scholar]
- Dobbing J, Sands J. Vulnerability of developing brain. IX. The effect of nutritional growth retardation on the timing of the brain growth-spurt. Biol Neonate. 1971;19:363–378. doi: 10.1159/000240430. [DOI] [PubMed] [Google Scholar]
- Dobbing J, Sands J. Quantitative growth and development of human brain. Arch Dis Child. 1973;48:757–767. doi: 10.1136/adc.48.10.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dow-Edwards DL, Hurd YL. Perinatal cocaine decreases the expression of prodynorphin mRNA in nucleus accumbens shell in the adult rat. Brain Res Mol Brain Res. 1998;62:82–85. doi: 10.1016/s0169-328x(98)00218-6. [DOI] [PubMed] [Google Scholar]
- Epping-Jordan MP, Watkins SS, Koob GF, Markou A. Dramatic decreases in brain reward function during nicotine withdrawal. Nature. 1998;393:76–79. doi: 10.1038/30001. [DOI] [PubMed] [Google Scholar]
- Finkenauer R, Pomerleau CS, Snedecor SM, Pomerleau OF. Race differences in factors relating to smoking initiation. Addict Behav. 2009 doi: 10.1016/j.addbeh.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanti LM, Manigart P, Dubois P. Tobacco smoking and alcohol and drug consumption in a large, young healthy population. Arch Environ Health. 1998;53:156–160. doi: 10.1080/00039896.1998.10545977. [DOI] [PubMed] [Google Scholar]
- Heidbreder CA, Thompson AC, Shippenberg TS. Role of extracellular dopamine in the initiation and long-term expression of behavioral sensitization to cocaine. J Pharmacol Exp Ther. 1996;278:490–502. [PubMed] [Google Scholar]
- Hughes JR, Gust SW, Skoog K, Keenan RM, Fenwick JW. Symptoms of tobacco withdrawal. A replication and extension. Arch Gen Psychiatry. 1991;48:52–59. doi: 10.1001/archpsyc.1991.01810250054007. [DOI] [PubMed] [Google Scholar]
- Hughes JR, Hatsukami D. Signs and symptoms of tobacco withdrawal. Arch Gen Psychiatry. 1986;43:289–294. doi: 10.1001/archpsyc.1986.01800030107013. [DOI] [PubMed] [Google Scholar]
- Kandel DB, Wu P, Davies M. Maternal smoking during pregnancy and smoking by adolescent daughters. Am J Public Health. 1994;84:1407–1413. doi: 10.2105/ajph.84.9.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF. A role for brain stress systems in addiction. Neuron. 2008;59:11–34. doi: 10.1016/j.neuron.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornetsky C, Esposito RU. Euphorigenic drugs: effects on the reward pathways of the brain. Fed Proc. 1979;38:2473–2476. [PubMed] [Google Scholar]
- Lawson GM, Hurt RD, Dale LC, Offord KP, Croghan IT, Schroeder DR, Jiang NS. Application of serum nicotine and plasma cotinine concentrations to assessment of nicotine replacement in light, moderate, and heavy smokers undergoing transdermal therapy. J Clin Pharmacol. 1998;38:502–509. doi: 10.1002/j.1552-4604.1998.tb05787.x. [DOI] [PubMed] [Google Scholar]
- Le Foll B, Goldberg SR. Nicotine induces conditioned place preferences over a large range of doses in rats. Psychopharmacology (Berl) 2005;178:481–492. doi: 10.1007/s00213-004-2021-5. [DOI] [PubMed] [Google Scholar]
- Lett BT. Repeated exposures intensify rather than diminish the rewarding effects of amphetamine, morphine, and cocaine. Psychopharmacology (Berl) 1989;98:357–362. doi: 10.1007/BF00451687. [DOI] [PubMed] [Google Scholar]
- Lieb R, Schreier A, Pfister H, Wittchen HU. Maternal smoking and smoking in adolescents: a prospective community study of adolescents and their mothers. Eur Addict Res. 2003;9:120–130. doi: 10.1159/000070980. [DOI] [PubMed] [Google Scholar]
- Luck W, Nau H. Nicotine and cotinine concentrations in serum and milk of nursing smokers. Br J Clin Pharmacol. 1984;18:9–15. doi: 10.1111/j.1365-2125.1984.tb05014.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mague SD, Andersen SL, Carlezon WA., Jr Early developmental exposure to methylphenidate reduces cocaine-induced potentiation of brain stimulation reward in rats. Biol Psychiatry. 2005;57:120–125. doi: 10.1016/j.biopsych.2004.10.037. [DOI] [PubMed] [Google Scholar]
- Malin DH, Lake JR, Carter VA, Cunningham JS, Hebert KM, Conrad DL, Wilson OB. The nicotinic antagonist mecamylamine precipitates nicotine abstinence syndrome in the rat. Psychopharmacology (Berl) 1994;115:180–184. doi: 10.1007/BF02244770. [DOI] [PubMed] [Google Scholar]
- Malin DH, Lake JR, Newlin-Maultsby P, Roberts LK, Lanier JG, Carter VA, Cunningham JS, Wilson OB. Rodent model of nicotine abstinence syndrome. Pharmacol Biochem Behav. 1992;43:779–784. doi: 10.1016/0091-3057(92)90408-8. [DOI] [PubMed] [Google Scholar]
- Markou A, Koob GF. Construct validity of a self-stimulation threshold paradigm: effects of reward and performance manipulations. Physiol Behav. 1992;51:111–119. doi: 10.1016/0031-9384(92)90211-j. [DOI] [PubMed] [Google Scholar]
- Matthews K, Robbins TW, Everitt BJ, Caine SB. Repeated neonatal maternal separation alters intravenous cocaine self-administration in adult rats. Psychopharmacology (Berl) 1999;141:123–134. doi: 10.1007/s002130050816. [DOI] [PubMed] [Google Scholar]
- Milberger S, Biederman J, Faraone SV, Jones J. Further evidence of an association between maternal smoking during pregnancy and attention deficit hyperactivity disorder: findings from a high-risk sample of siblings. J Clin Child Psychol. 1998;27:352–358. doi: 10.1207/s15374424jccp2703_11. [DOI] [PubMed] [Google Scholar]
- Naeye RL, Peters EC. Mental development of children whose mothers smoked during pregnancy. Obstet Gynecol. 1984;64:601–607. [PubMed] [Google Scholar]
- O’Brien CP. Research advances in the understanding and treatment of addiction. Am J Addict. 2003;12 (Suppl 2):S36–S47. [PubMed] [Google Scholar]
- O’Dell LE, Torres OV, Natividad LA, Tejeda HA. Adolescent nicotine exposure produces less affective measures of withdrawal relative to adult nicotine exposure in male rats. Neurotoxicol Teratol. 2007;29:17–22. doi: 10.1016/j.ntt.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson NE, Markou A. Prolonged nicotine dependence associated with extended access to nicotine self-administration in rats. Psychopharmacology (Berl) 2004;173:64–72. doi: 10.1007/s00213-003-1692-7. [DOI] [PubMed] [Google Scholar]
- Pudiak CM, Bozarth MA. L-NAME and MK-801 attenuate sensitization to the locomotor-stimulating effect of cocaine. Life Sci. 1993;53:1517–1524. doi: 10.1016/0024-3205(93)90559-l. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Brain Res Rev. 1993;18:247–291. doi: 10.1016/0165-0173(93)90013-p. [DOI] [PubMed] [Google Scholar]
- Rylkova D, Boissoneault J, Isaac S, Prado M, Shah HP, Bruijnzeel AW. Effects of NPY and the specific Y1 receptor agonist [D-His(26)]-NPY on the deficit in brain reward function and somatic signs associated with nicotine withdrawal in rats. Neuropeptides. 2008;42:215–227. doi: 10.1016/j.npep.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaha AN, Yau WY, Yang P, Robinson TE. Rapid delivery of nicotine promotes behavioral sensitization and alters its neurobiological impact. Biol Psychiatry. 2005;57:351–360. doi: 10.1016/j.biopsych.2004.11.040. [DOI] [PubMed] [Google Scholar]
- Sastry BV, Chance MB, Singh G, Horn JL, Janson VE. Distribution and retention of nicotine and its metabolite, cotinine, in the rat as a function of time. Pharmacology. 1995;50:128–136. doi: 10.1159/000139274. [DOI] [PubMed] [Google Scholar]
- Schefer V, Talan MI. Oxygen consumption in adult and AGED C57BL/6J mice during acute treadmill exercise of different intensity. Exp Gerontol. 1996;31:387–392. doi: 10.1016/0531-5565(95)02032-2. [DOI] [PubMed] [Google Scholar]
- Small E, Shah HP, Davenport JJ, Geier JE, Yavarovich KR, Yamada H, Sabarinath SN, Derendorf H, Pauly JR, Gold MS, Bruijnzeel AW. Tobacco smoke exposure induces nicotine dependence in rats. Psychopharmacology (Berl) 2009 doi: 10.1007/s00213-009-1716-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear LP. The adolescent brain and age-related behavioral manifestations. Neurosci Biobehav Rev. 2000;24:417–463. doi: 10.1016/s0149-7634(00)00014-2. [DOI] [PubMed] [Google Scholar]
- Stepans MB, Fuller SG. Measuring infant exposure to environmental tobacco smoke. Clin Nurs Res. 1999;8:198–218. doi: 10.1177/10547739922158269. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Ise Y, Tsuda M, Maeda J, Misawa M. Mecamylamine-precipitated nicotine-withdrawal aversion in rats. Eur J Pharmacol. 1996;314:281–284. doi: 10.1016/s0014-2999(96)00723-6. [DOI] [PubMed] [Google Scholar]
- Teague SV, Pinkerton KE, Goldsmith M, Gebremichael A, Chang S, Jenkins RA, Moneyhun JH. A sidestream cigarette smoke generation and exposure system for environmental tobacco smoke studies. Inhalation Toxicology. 1994;6:79–93. [Google Scholar]
- Vanderschuren LJ, Kalivas PW. Alterations in dopaminergic and glutamatergic transmission in the induction and expression of behavioral sensitization: a critical review of preclinical studies. Psychopharmacology (Berl) 2000;151:99–120. doi: 10.1007/s002130000493. [DOI] [PubMed] [Google Scholar]
- Wakschlag LS, Lahey BB, Loeber R, Green SM, Gordon RA, Leventhal BL. Maternal smoking during pregnancy and the risk of conduct disorder in boys. Arch Gen Psychiatry. 1997;54:670–676. doi: 10.1001/archpsyc.1997.01830190098010. [DOI] [PubMed] [Google Scholar]
- Wall MA, Johnson J, Jacob P, Benowitz NL. Cotinine in the serum, saliva, and urine of nonsmokers, passive smokers, and active smokers. Am J Public Health. 1988;78:699–701. doi: 10.2105/ajph.78.6.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesnes K, Warburton DM. Smoking, nicotine and human performance. Pharmacol Ther. 1983;21:189–208. doi: 10.1016/0163-7258(83)90072-4. [DOI] [PubMed] [Google Scholar]
- Wise RA. Neurobiology of addiction. Curr Opin Neurobiol. 1996;6:243–251. doi: 10.1016/s0959-4388(96)80079-1. [DOI] [PubMed] [Google Scholar]
- Zhao N, Wang HY, Dow-Edwards D. Cocaine exposure during the early postnatal period diminishes medial frontal cortex Gs coupling to dopamine D1-like receptors in adult rat. Neurosci Lett. 2008;438:159–162. doi: 10.1016/j.neulet.2008.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]