

Abstract
Repetitive extragenic palindromic (REP) sequences are a ubiquitous feature of bacterial genomes. Recent work shows that REPs are remnants of a larger mobile genetic element termed a REPIN. REPINs consists of two REP sequences in inverted orientation separated by a spacer region and are thought to be non-autonomous mobile genetic elements that exploit the transposase encoded by REP-Associated tYrosine Transposases (RAYTs). Complimentarity between the two ends of the REPIN suggests that the element forms hairpin structures in single stranded DNA or RNA. In addition to REPINs, other more complex arrangements of REPs have been identified in bacterial genomes, including the genome of the model organism Pseudomonas fluorescens SBW25. Here, we summarize existing knowledge and present new data concerning REPIN diversity. We also consider factors affecting the evolution of REPIN diversity, the ease with which REPINs might be co-opted by host genomes and the consequences of REPIN activity for the structure of bacterial genomes.
Keywords: miniature inverted-repeat transposable element, MITE, RAYTs, REP-associated tyrosine transposase, repetitive sequences, transposase, transposon
REPINs: A New Class of Mobile Bacterial DNA
Repetitive extragenic palindromic (REP) sequences are a common feature of bacterial genomes.1-5 The possibility that REPs might be selfish genetic elements was suggested on first discovery in Escherichia coli.2,3,6 However, their short length (~20 nucleotides), plus absence of plausible mechanism for within genome dissemination, meant this idea received limited support. Over the next 30 years the “selfish element hypothesis” further paled, in part, thanks to numerous studies that provided evidence that REPs located at particular locations and in specific genomes perform a diverse range of cellular processes.1,6-9 However, the fact that the distribution and abundance of REPs can vary substantially among even closely related strains4,10 suggests that the range of functional roles is likely to be incidental, arising from, for example, co-option or genetic accommodation.11
Recently we provided evidence that REP sequences are part of a selfish genetic element.10 The element consists of two REP sequences (a REP doublet) in inverted orientation. Evidence supporting this hypothesis was derived from analysis of the genome of the model organism Pseudomonas fluorescens SBW25 and includes the following: (1) demonstration that the distribution of REP doublets is comparable to expectations under a randomly generated null model (whereas the distribution of REP singlets shows a significant departure from random); (2) demonstration that REP sequences found as REP doublets show higher sequence conservation compared with REP sequences existing as singlets (this suggests that REP doublets are under selection as opposed to singlets, which are most likely non-functional decaying remnants of REP doublets); and (3) identification of excisions of REP doublets (but not of single REP sequences) from population sequencing data from the SBW25 genome.
The discovery of excision events—events that are likely to define transposition intermediates—not only supports the hypothesis that REP doublets are a unit of selection, but also indicates that these elements are actively moving in the genome. Sequence characteristics of the excised element also suggest a likely transposition mechanism reminiscent of the mechanism of IS605 transposition.12 Such mechanistic similarity makes sense given the similarities between IS605 and RAYTs [the entities thought responsible for providing transposase function to REPINs (see below)].5 Given the likely evolutionary significance of the REP doublet, the entity was designated a REPIN (REP doublet forming hairpin). A schematic of the 89 bp element is shown in Figure 1.
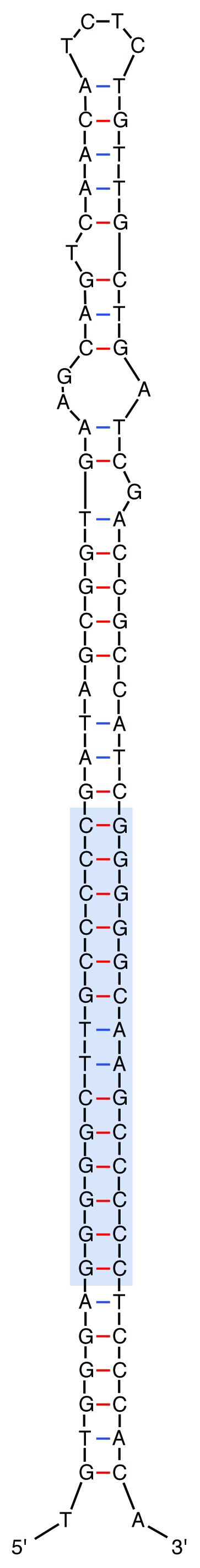
Figure 1. Secondary structure predicted for a GI REPIN. The secondary structure shows the almost perfect hairpin formed by a GI REP doublet. Blue box indicates the position of the short imperfect palindromes (REPs). Secondary structure predicted by the mfold web server (http://mfold.rna.albany.edu/?q=mfold/DNA-Folding-Form).22
While our initial analyses were based on a single (P. fluorescens SBW25) chromosome, the study of REPINs was extended to 18 selected bacterial genomes including Escherichia coli K-12 DH10B, Salmonella enterica serovar Paratyphi A AKU 12601, Thioalkalivibrio HL-EbGR7, Nostoc. punctiforme PCC73102 and all fully sequenced Pseudomonas genomes. REP sequences were identified based on their proximal association with REP-associated tyrosine transposases (RAYTs),10 which are implicated in REPIN dispersal. In these 18 genomes, REP sequences adjacent to RAYTs were found to exist as doublets with specific features characteristic of REPINs. REPINs therefore appear to be widely distributed elements.
REPIN Diversity within the SBW25 Genome
Comparisons of the frequency of the most abundant 16-mers from SBW25, with randomly assembled genomes, and with the closely related genome of P. fluorescens Pf0–1, revealed at least 96 different over-represented 16-mers. Using a grouping algorithm, these 96 different 16-mers were found to belong to just three distinct groups, termed GI, GII and GIII (Table 1). The three groups were named in order of their abundance in the SBW25 genome, with GI being the most abundant (618 occurrences). All three sequence groups occur predominantly in extragenic space and contain an imperfect palindromic core, thus possessing all features of repetitive extragenic palindromic sequences (REPs).
Table 1. Short repetitive sequence groups in the SBW25 genome.
| Groupa | Sequenceb | Occurrences | Palindromic corec |
|---|---|---|---|
| I |
GTGGGAGGGGGCTTGC |
618 |
GGGGGCTTGCCCCC |
| II |
GTGAGCGGGCTTGCCC |
241 |
GCGGGCTTGCCCCGC |
| III | GAGGGAGCTTGCTCCC | 208 | GGGAGCTTGCTCCC |
a 16-mers were assigned to one of three groups (GI, GII and GIII) using a grouping algorithm.
b Sequence of the most common 16-mer from each group.
c Each GI, GII and GIII sequence either contains or overlaps an imperfect palindrome (the palindromic core). Table reproduced from Bertels and Rainey.10
Since REPINs are exclusively formed by REP sequences of the same group, they can also be categorized into GI, GII and GIII REPINs. Nevertheless, REPINs show considerable within group diversity, both with regard to the length of the spacer sequence between the individual REP units, but also with respect to the specific sequence of the spacer region.
In terms of the diversity in spacer length, GI REPINs in SBW25 show 11 different inter-REP spacings; five different spacings are found within GII REPINs and eight within GIII REPINs (Table 2). Perhaps more surprising—given that there are hundreds of REPINs in the SBW25 genome—is the fact that no two REPINs are identical at the level of the DNA sequence that comprises the spacer region. Nonetheless, the spacer region of all REPINs is organized so as to form a hairpin structure.
Table 2. Characteristics of REPINs found in the SBW25 genome.
| REPIN | Distance between REPs | Number of occurrences within SBW25 | REPIN orientationa | REP conservation within REPIN (%) ± std devb |
|---|---|---|---|---|
| GI |
34 35 36 |
1 11 3 |
AA-TT |
94.56 ± 0.4 |
| |
41 42 43 |
6 13 9 |
AA-TT |
98.22 ± 0.06 |
| |
51 52 53 |
1 8 12 |
AA-TT |
94.34 ± 0.7 |
| |
69 70 71 72 |
9 11 138 59 |
TT-AA |
97.19 ± 0.8 |
| GII |
18 19 20 21 22 |
2 1 4 0 5 |
TT-AA |
89.52 ± 1.2 |
| |
72 |
4 |
TT-AA |
94.32 ± 1.3 |
| |
107 108 109 110 111 112 |
1 1 19 54 17 4 |
TT-AA |
98.21 ± 0.07 |
| GIII |
64 65 66 67 68 |
3 7 7 4 1 |
TT-AA |
97.63 ± 0.06 |
| |
77 78 79 |
2 2 18 |
TT-AA |
99.43 ± 1e-12 |
| 104 105 |
10 8 |
TT-AA | 95.21 ± 1.0 |
a Shows the two bases that are observed in the center of each palindrome (either AA or TT see Table 1) contained within a REPIN.
In addition to the diversity of inter-REP spacings and diversity of the spacer sequence, GI REPINs also exist in two different orientations as evident by the arrangement of the central AA or TT motif (the presence of either the AA or TT motif (in all SBW25 REP sequences) ensures that each REP palindrome is imperfect). Since REPINs consist of two inverted REP sequences, there are two possible doublet configurations: either TT-AA (common) (as in most GI, all GII and GIII doublets) or AA-TT (rare) (as found in a minority of GI doublets) (Fig. 2A and B). Interestingly, GI doublets in the TT-AA configuration are flanked by multiple conserved ‘A’s and ‘T’s at the 5′ and 3′ end, respectively, which may reflect co-option of the REP doublet for transcription attenuation.8 The region flanking GI doublets in the AA-TT orientation are devoid of runs of ‘A’s or ‘T’s, however, runs of ‘A’ and ‘T’ nucleotides directly flanking REP sequences are observed inside the doublet (Fig. 2A and B). This suggests that the AA-TT configuration evolved from the 3′ and 5′ REP sequences of two co-localized TT-AA GI doublets (Fig. 2C).

Figure 2. REP sequence orientation within GI doublets. (A) Alignment of 101 GI REP doublets from SBW25 (seven are shown) that are found at a distance of 71 bp to each other. REP sequences within the doublet are found in opposite orientations and are divided by a less conserved spacer sequence. Each REP sequence consists of a palindrome, a 5′ and a 3′ flanking sequence. The bases in the center of each palindrome indicate the orientation within the doublet. TT is found in the center of the first palindrome and AA in the center of the second, hence, the shown doublet is of type AA-TT. Three conserved As and Ts are found at the 5′ and 3′ end respectively, indicating the co-option of this REP doublet class as transcription terminator. (B) Alignment of the less commonly found AA-TT GI doublet conformation separated by 43 bp. Note that the conserved As and Ts at the 5′ and 3′ end of the alignment do not exist. However, As are found at the 5′ end of the bc sequence and at the 3′ end of the b sequence similar to GI doublets in TT-AA orientation. (C) A potential scenario for the evolution of AA-TT GI doublets from TT-AA GI doublets. An accidental transposition of the 3′ and 5′ end of two co-localized TT-AA GI doublet could have been sufficient to create the new AA-TT REP doublet type.
REPIN diversity becomes more complex when the focus shifts to higher order arrangements (multiple REPINs in close proximity). In SBW25 there are various structurally different arrangements. The least common arrangement consists of co-localized REPINs. Such co-localized REPINs are no more frequently found than expected by random chance. The most common arrangement consists of highly structured tandemly repeated REPINs.10 In SBW25 these comprise as many as five tandem REPIN repeats. Such organisations have also been observed before in E. coli,13 but no mechanistic or evolutionary explanation for their formation has been put forward.
REPINs and RAYTs
REPINs, if mobile elements, as suggested, are too short to encode their own transposition machinery. Their movement thus requires exploitation of transposase activity encoded by some other fully autonomous element. Strongly implicated are the so-named REP-associated tyrosine transposases (RAYTs): transposases that are distantly related to the IS200 family of insertion sequences and which are typically flanked by multiple REPINs.
There are several notable features of RAYTs and their associated REPINs. First, RAYT-encoding genes are typically short (~500 bp), and while highly conserved residues are apparent5 [among which are the HUH and Y motifs, which are essential for transposition by TnpA of IS60512 (a member of the IS200 family)], overall, the genes show substantive diversity (e.g., 22 RAYTs from the sequenced Pseudomonas genomes show just 57% and 53% pairwise identity at the nucleotide level and amino acid level, respectively). Second, each RAYT has a specific association with a particular sequence type of REPIN. This is evident in the genome of SBW25, which harbours three distinct RAYTs, each RAYT in the SBW25 genome is associated with a specific family (GI, GII or GIII) of REPINs. Third, RAYTs are only ever present as single copy entities (in those genomes harbouring more than a single RAYT each RAYT is distinctly different, e.g., the three RAYTs in SBW25 are as different from each other as they are from any RAYT chosen at random from the total population of Pseudomonas RAYTs). This last fact is particularly curious, because it begs an explanation for the maintenance of RAYTs in bacterial genomes.
The raison d’etre for transposons and related elements is to disproportionately increase their representation within a given host genome (and to disseminate horizontally wherever possible).14,15 However, extinction is the long-term fate of most transposons given that selection is relatively impotent when it comes to purging deleterious mutations in transposases. This is because transposons encoding defective transposases (non-autonomous transposons) can exploit transposase function encoded by functional (autonomous) transposons. The weakness of purifying selection means that non-autonomous elements are expected to increase in frequency—even to the point where they may drive the functional family extinct.16
The fact that RAYTs are present as just single copy entities is indicative of their incapacity for transposition. If RAYTs cannot transpose, then selection cannot act to maintain RAYT function—even if RAYT function is required for the movement of REPINs. This means that either RAYTs are the non-functional remains of once active transposons, or they are maintained by virtue of some complex relationship with the host cell, or with the REPINs or a combination of both.
Examination of the RAYT-encoding genes provides little evidence in support of the hypothesis that they are fossilized remnants. Indeed, evolutionary analyses of dN/dS show that approximately 60% of codons have a significant excess of synonymous substitutions and are thus subject to negative (purifying) selection. This suggests that the amino acid sequence of RAYTs is constrained by virtue of function, and that whatever that function might be, that it is important for cell viability.
While dN/dS analyses fail to find evidence of positive selection, the substantive diversity at non conserved codons makes such analyzes problematic. Indeed, high levels of polymorphism at non-conserved codons are to be expected where genes are subject to strong diversifying selection. Tests using evolutionary fingerprinting17 reveal that a number of sites have in fact experienced strong diversifying selection. Together, the signature of both purifying and diversifying selection suggest that RAYTs are, on one hand, functionally constrained, yet also subject to positive selection. Such a signature is reminiscent of genes involved in interactions with hosts, or more generally, genes involved in co-evolutionary processes.18,19
One possible explanation for the maintenance of RAYTs is the existence of some kind of addiction system, which ensures that cells containing defective RAYTs (the defect being caused by a spontaneous mutation) are killed and thus eliminated. Such a scenario would be akin to well described plasmid addiction systems.20 To test this hypothesis the three RAYT genes from SBW25 were deleted from the genome, however, the resulting mutant was fully viable (Zhang XX, Bertels F and Rainey PB, unpublished). This suggests that addiction is not responsible for the maintenance of RAYT function.
An alternative possibility is that the RAYT, in addition to mobilizing REPINs, performs some function that not only benefits the REPIN, but is also important for cellular physiology. Just what this function might be is currently unknown, however, it is possible that the RAYT-encoded transposase physically interacts with REPINs at their extragenic chromosomal locations and by virtue of this interaction (perhaps DNA binding) plays some kind of regulatory function, possibly aiding fine tuning of patterns of gene expression. It is even possible that different REPs and REPIN structures may have different affinities for RAYT binding and thus differentially affect the levels of expression of neighboring genes. Implicit in this idea is the notion that REPINs and their associated RAYTs can be readily co-opted for cellular function. This is an intriguing and not unrealistic possibility, especially given the convenient placement of protein (RAYT) binding sites in the extragenic space of hundreds of genes and the known effects of REPs and REPINs on the expression of neighboring genes.1,6-9
Origins of REPIN Diversity
As mentioned above, and described in our earlier publication,10 REPIN diversity is apparent at different organisational levels. The existence of three different REPINs (GI, GII and GIII) and their associated RAYTs (in both the SBW25 genome and other genomes investigated) points to a specificity of association reminiscent of co-evolution between the two entities.
It is possible to envisage both antagonistic and mutualistic models of coevolution. For example, if REPINs evolved from single REP sequences that flanked an ancestral IS200-like element, as previously suggested10 [imperfect palindromes (similar to REPs) flanking IS200 are essential for transposition]—and did so via a simple duplication event—then in little more than a single step, this could have resulted in enslavement of the transposase and generation of a non-autonomous mobile entity. An ensuing arms race between host (transposase) and parasite (REPIN) is thus plausible. Such processes could be responsible for both the various components of RAYT and REPIN diversity, but also for the high specificity between REPIN and RAYT.
A less antagonistic model comes when considering the possibility that REPINs, inserted into appropriate extragenic locations, might act as binding sites for the RAYT-encoded protein; that together the protein-DNA interaction might be readily co-opted by the host as a means of fine tuning or modulating gene expression.
Coevolution under this more mutualistic model is readily envisaged by virtue of the fact that a RAYT / REPIN containing population of cells is likely to experience different environmental conditions. If there is an association between REPIN and RAYT that is functionally maintained because of benefit to the host cell, then it is likely that this association evolves with changes in environment. Such interactions could, as in the antagonistic model, account for both REPIN / RAYT diversity, plus specificity; the lack of conservation in the spacer region (other than to maintain hairpin structure) could also be explained by the need for the nature of interaction between RAYT and REPIN to be tuned separately for each locus in order to ensure that the association is not costly (and perhaps even beneficial).
Under both co-evolutionary models the genome is expected to harbour REPINs of different ages with older REPINs showing evidence of mutational decay. To this end we examined the average pairwise identity of REPs found in REPINs of each class with different inter-REP spacers. In each instance we found evidence of families with specific spacings that have greater or lesser sequence conservation (Table 2). This is as expected under a co-evolutionary model.
A distinctly different explanation for REPIN diversity draws on the possibility of interactions among different RAYTs. For example, GI RAYTs most likely transpose GI REPINs with a 71 bp spacer (101 occurrences). GII RAYTs most likely transpose GII REPINs with a 110 bp spacer (50 occurrences). Perhaps REPINs can sometimes be substrates for more than a single RAYT, which may lead to different spacer distances and thus different specificities. Against this thesis however is the fact that no hybrid REPINs exist in the SBW25 genome, i.e., no REPINs where the two REP sequences that comprise a given REPIN are derived from different REP groups.
Consequences of REPIN Activity
Transposons are well known for the multiplicity of effects, both direct and indirect, that they reap on both genome architecture and evolution.21 Here we have suggested that REPINs and their associated RAYTs have properties that might be readily co-opted to perform a diverse range of cellular functions, but it is likely that there are numerous additional consequences. One is the formation of long palindromic sequences.
The SBW25 genome contains a variety of long palindromic REP singlets that can be explained as a consequence of REPIN activity. REP sequences typically consist of three regions: a 5′ flanking sequence (a), a central palindrome and a 3′ flanking sequence (b). The genome of SBW25 contains numerous long palindromic sequences (20bp - 28bp) with the general structure a-palindrome-ac (where ac is the complement of a) and b-palindrome-bc (Fig. 3A). The existence of these long palindromic REP sequences can be accounted for by REPIN excision events (Fig. 3B). For example palindromes of the structure a-palindrome-ac can be formed by excision of the central REPIN sequence—from all three REPIN groups—whereas palindromes of the structure b-palindrome-bc can be formed by excision of the central REPIN sequence from a subset of GI REPINs only, namely, those GI REPINs that occur in the rare AA-TT orientation (Table 2 and Fig. 3). In support of this model is the fact that the most common long palindromic REP singlet is derived from the most common REPIN, whereas the long palindromic REP singlet derived from the rare AA-TT GI REPIN is rare. Further support comes from the discovery of a single REPIN excision event from population sequencing in which the REPIN was excised at the 3′ end of the 5′ palindrome and at the 3′ end of the 3′ palindrome, leaving a long palindromic REP sequence behind (Fig. 4).

Figure 3. Unusual long palindromic GI, GII and GIII sequences and their potential evolution from REP doublets found in the SBW25 genome. (A) Shown are all four long palindromic GI, GII and GIII sequences together with their frequency found in the SBW25 genome. Note that two configurations are found for GI sequences and only one for both GII and GIII sequences. (B) Shows how long palindromic REP singlets could arise from REP doublets through the excision of the central sequence. Hence, REP doublets found in AA-TT orientation would produce a-palindrome-ac REPs (left) and REP doublets found in TT-AA orientation bc-palindrome-b REPs (right).

Figure 4. Incomplete symmetric excision event of a REP doublet detected in Illumina sequencing data. The first line of the alignment shows the genomic sequence of SBW25 from position 598,553 to position 598,634. The second line of the alignment shows part of the sequence read that maps perfectly to the corresponding genome sequence apart from the excision in the center of the read. The cartoon below the alignment shows the general composition of the GI REP doublet. The last line in the picture shows the remaining REP sequence found in the sequence read. It only contains flanking sequence (a), the central palindrome and flanking sequence (ac).
Conclusion
REPINs and their associated RAYTs are a common feature of bacterial genomes, yet as indicated here, there is much about their origin, maintenance and means of dissemination (both with and between genomes) that is currently unknown. Critical for progress are studies that shed light on the mechanism of RAYT-mediated REPIN mobilization and the basis of the REPIN-RAYT association, along with knowledge of the functional significance of the interaction for the host cell. From an evolutionary perspective it is of interest to know whether REPINs are derived from REP sequences flanking ancestral IS200-like elements as has been suggested. Also worthy of study are the dynamics of REPIN movement along with the spectrum of mutational effects wrought by transposition. Future work may even shed light on regulation of REPIN movement and with this there might emerge possibilities for controlling microbial infection through manipulation of selfish element behavior.
Acknowledgments
FB acknowledges a post-graduate scholarship from the Allan Wilson Centre; PBR is supported by a James Cook Research Fellowship from the Royal Society of New Zealand.
Glossary
Abbreviations:
- RAYT
REP-associated tyrosine transposase
- REP
repetitive extragenic palindrome
- REPIN
REP doublet forming hairpin
- MITE
miniature inverted repeat transposable element
Footnotes
Previously published online: www.landesbioscience.com/journals/mge/article/18610
References
- 1.Aranda-Olmedo I, Tobes R, Manzanera M, Ramos J, Marques S. Species-specific repetitive extragenic palindromic (REP) sequences in Pseudomonas putida. Nucleic Acids Res. 2002;30:1826–33. doi: 10.1093/nar/30.8.1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stern MJ, Ames G, Smith N, Robinson E, Higgins C. Repetitive extragenic palindromic sequences: a major component of the bacterial genome. Cell. 1984;37:1015–26. doi: 10.1016/0092-8674(84)90436-7. [DOI] [PubMed] [Google Scholar]
- 3.Gilson E, Clément JM, Brutlag D, Hofnung M. A family of dispersed repetitive extragenic palindromic DNA sequences in E. coli. EMBO J. 1984;3:1417–21. doi: 10.1002/j.1460-2075.1984.tb01986.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Silby MW, Cerdeno-Tarraga A, Vernikos G, Giddens S, Jackson R, Preston G, et al. Genomic and genetic analyses of diversity and plant interactions of Pseudomonas fluorescens. Genome Biol. 2009;10:R51. doi: 10.1186/gb-2009-10-5-r51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nunvar J, Huckova T, Licha I. Identification and characterization of repetitive extragenic palindromes (REP)-associated tyrosine transposases: implications for REP evolution and dynamics in bacterial genomes. BMC Genomics. 2010;11:44. doi: 10.1186/1471-2164-11-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Higgins CF, McLaren RS, Newbury SF. Repetitive extragenic palindromic sequences, mRNA stability and gene expression: evolution by gene conversion? A review. Gene. 1988;72:3–14. doi: 10.1016/0378-1119(88)90122-9. [DOI] [PubMed] [Google Scholar]
- 7.Stern MJ, Prossnitz E, Ames GF. Role of the intercistronic region in post-transcriptional control of gene expression in the histidine transport operon of Salmonella typhimurium: involvement of REP sequences. Mol Microbiol. 1988;2:141–52. doi: 10.1111/j.1365-2958.1988.tb00015.x. [DOI] [PubMed] [Google Scholar]
- 8.Espéli O, Moulin L, Boccard F. Transcription attenuation associated with bacterial repetitive extragenic BIME elements. J Mol Biol. 2001;314:375–86. doi: 10.1006/jmbi.2001.5150. [DOI] [PubMed] [Google Scholar]
- 9.Gilson L, Mahanty HK, Kolter R. Genetic analysis of an MDR-like export system: the secretion of colicin V. EMBO J. 1990;9:3875–84. doi: 10.1002/j.1460-2075.1990.tb07606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bertels F, Rainey PB. Within-Genome Evolution of REPINs: a New Family of Miniature Mobile DNA in Bacteria. PLoS Genet. 2011;7:e1002132. doi: 10.1371/journal.pgen.1002132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kidwell MG. Transposable elements and host genome evolution. Trends Ecol Evol. 2000;15:95–9. doi: 10.1016/S0169-5347(99)01817-0. [DOI] [PubMed] [Google Scholar]
- 12.Ton-Hoang B, Guynet C, Ronning DR, Cointin-Marty B, Dyda F, Chandler M. Transposition of ISHp608, member of an unusual family of bacterial insertion sequences. EMBO J. 2005;24:3325–38. doi: 10.1038/sj.emboj.7600787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gilson E, Saurin W, Perrin D, Bachellier S, Hofnung M. Palindromic units are part of a new bacterial interspersed mosaic element (BIME) Nucleic Acids Res. 1991;19:1375–83. doi: 10.1093/nar/19.7.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Charlesworth B, Sniegowski P, Stephan W. The evolutionary dynamics of repetitive DNA in eukaryotes. Nature. 1994;371:215–20. doi: 10.1038/371215a0. [DOI] [PubMed] [Google Scholar]
- 15.Bichsel M, Barbour AD, Wagner A. The early phase of a bacterial insertion sequence infection. Theor Popul Biol. 2010;78:278–88. doi: 10.1016/j.tpb.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 16.Hartl DL, Lozovskaya ER, Lawrence JG. Nonautonomous transposable elements in prokaryotes and eukaryotes. Genetica. 1992;86:47–53. doi: 10.1007/BF00133710. [DOI] [PubMed] [Google Scholar]
- 17.Pond SLK, Murrell B, Fourment M, Frost SDW, Delport W, Scheffler K. A random effects branch-site model for detecting episodic diversifying selection. Mol Biol Evol. 2011;28 doi: 10.1093/molbev/msr125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laine AL. Role of coevolution in generating biological diversity: spatially divergent selection trajectories. J Exp Bot. 2009;60:2957–70. doi: 10.1093/jxb/erp168. [DOI] [PubMed] [Google Scholar]
- 19.Aguileta G, Refrégier G, Yockteng R, Fournier E, Giraud T. Rapidly evolving genes in pathogens: methods for detecting positive selection and examples among fungi, bacteria, viruses and protists. Infect Genet Evol. 2009;9:656–70. doi: 10.1016/j.meegid.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 20.Gerdes K, Rasmussen PB, Molin S. Unique type of plasmid maintenance function: postsegregational killing of plasmid-free cells. Proc Natl Acad Sci USA. 1986;83:3116–20. doi: 10.1073/pnas.83.10.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burt A, Trivers R. Genes in Conflict: The Biology of Selfish Genetic Elements. Cambridge, Massachusetts: Belknap Press of Harvard University Press 2006 [Google Scholar]
- 22.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–15. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]