

Abstract
Hepatic stellate cells (HSC) are the liver mesenchymal cell type which responds to hepatocellular damage and participates in wound healing. Although HSC myofibroblastic trans-differentiation (activation) is implicated in excessive extracellular matrix deposition, molecular understanding of this phenotypic switch from the viewpoint of cell fate regulation is limited. Recent studies demonstrate the roles of anti-adipogenic morphogens (Wnt, Necdin, Shh) in epigenetic repression of the HSC differentiation gene Pparγ as a causal event in HSC activation. These morphogens have positive cross-interactions which converge to epigenetic repression of Pparγ involving the methyl-CpG binding protein MeCP2. However, these morphogens expressed by activated HSC may also participate in cross-talk between HSC and hepatoblasts/hepatocytes to support liver regeneration, and their aberrant regulation may contribute to liver tumorigenesis. Implications of HSC-derived morphogens in these possibilities are discussed.
Keywords: necdin, Pparγ, Shh, Wnt
Historical perspective of fat-storing phenotype of hepatic stellate cells (HSC)
HSC are the major mesenchymal cell type in the liver with several known functions including vitamin A storage, control of sinusoidal vascular tone, mesenchymal-epithelial interaction, and wound healing. Although the storage of lipid-soluble retinyl esters is primarily considered to represent the most notable characteristic of differentiated HSC, they store other lipids, particularly neural lipids.1 This lipid-storing phenotype was recognized 60 years ago by the work of Professor Toshio Ito and co-workers who termed the cells “fat-storing cells.”2-4 They are the first to recognize that fat content in HSC increases in response to injection of insulin and glucose, a key functional feature of adipocytes.2 Since then, this intriguing aspect of HSC was in large left unexplored.
As myofibroblastic trans-differentiation (activation) of HSC became recognized as one of the critical events in liver fibrogenesis, the mechanisms of this cellular alternation have become a popular area of investigation. This has led to the generation of an explosive amount of new information including the identification of mediators, gene regulation and intracellular signaling that control the expression of activation-associated molecules such as collagens, cytokines (transforming growth factor-β, platelet-derived growth factor [PDGF]), monocyte chemotactic protein-1 (MCP-1), extracellular matrix degradation enzymes and inhibitors (matrix metalloproteinase [MMP], tissue inhibitors of metalloproteinases [TIMP]), nicotinamide adenine dinucleotide phosphate (reduced) oxidase, renin-angiotensin system, and toll-like receptor.5 However, there is insufficient fundamental understanding of the HSC activation from the viewpoint of cell fate or lineage regulation. This question obviously cannot be addressed without understanding the embryonic origin of HSC. HSC express many neuronal or glial cell markers, and their neuroectoderm origin was proposed, with a subsequent failure to validate this notion using the Wnt1-Cre and ROSA26 reporter mice.6 This finding logically favored a hypothesis of mesoderm-derived multipotent mesenchymal progenitor cells (MMPC) as the origin of HSC, particularly because MMPC also give rise to neural cells in addition to other mesenchymal lineages for smooth muscle cells, chondrocytes, osteoblasts, and adipocytes whose markers are also expressed by HSC.7 Consistent with this notion, recent studies by Asahina et al. demonstrate that HSC are derived from mesoderm and at least in part via septum transversum and mesothelium.8,9
A unique finding in cell fate regulation of different mesenchymal cell types derived from MMPC is that they undergo trans-differentiation within their lineages in culture upon addition of mediators. As HSC are derived from the mesoderm most likely via MMPC, we entertained the notion that HSC trans-differentiation may also reside in these mesenchymal lineages.
Adipogenic regulation of HSC
We further hypothesized that HSC trans-differentiation may be similar to adipocyte and preadipocytic fibroblast de-differentiation. This hypothesis is based on several similarities found between two processes. First, both differentiated HSC and adipocytes share a fat-storing phenotype, as discussed above in reference to Professor Ito’s work. Second, intracellular lipids are lost when HSC are trans-differentiated and when adipocytes are de-differentiated. Third, both differentiated HSC and adipocytes express type IV collagen while trans-differentiated HSC and preadipocytic fibroblasts primarily express interstitial collagens. Fourth, HSC are known to express adipocyte-specific genes including leptin,10 adiponectin,11 and adipsin.12 Last, the mediators known to suppress adipocyte differentiation such as tumor necrosis factor (TNF)α, leptin, PDGF, and MCP-1, also activate HSC. Based on these similarities, our laboratory proposed more than a decade ago that there is a regulatory commonality between adipocytes and HSC differentiation.13 Central to this proposal is the expression and regulation of the master adipogenic transcription factor peroxisome proliferator-activated receptor (PPAR)γ, which is essential for both adipocyte14 and HSC12,15 differentiation. PPARγ promotes the storage of intracellular fat including retinyl esters in HSC14 while suppressing α1(I) collagen gene via the inhibition of p300-facilitated NF-I binding to its promoter.16 Based on these findings and the efficacy of PPARγ ligands shown in animal models of liver fibrosis,13,17,18 PPARγ is considered a potential therapeutic target for liver fibrosis.
Anti-adipogenic morphogens linking to epigenetic mechanisms of HSC activation
Wnt
Wnt are a highly conserved family of secreted glycoproteins that regulate cellular differentiation and proliferation by binding to the Frizzled receptor-low density lipoprotein receptor-related protein (LRP) 5/6 co-receptor complex. Activation of the canonical Wnt pathway results in phosphorylation and inhibition of glycogen synthase kinase 3 and allows stabilization of cytosolic β-catenin and its translocation to the nucleus, where it serves as a co-activator for T cell factor/lymphocyte enhancer factor (TCF/LEF) family of transcription factors. Typical β-catenin target genes include those involved in cell proliferation (c-myc, c-jun, cyclin D1). It is also important to recognize that β-catenin interacts with many other transcription factors including PPARγ, Smads, NF-κB, cAMP response element-binding protein, retinoic acid receptor and hypoxia inducible factor 1α, to render inductive or repressive effects on target gene transcription and to exert diverse biological effects in different cell types.19,20
Canonical Wnt signaling induced by Wnt1 and Wnt10b inhibits adipogenesis via suppression of the adipogenic transcription factors CCAAT-enhancer-binding protein α and PPARγ.21,22 Similarly, canonical Wnt signaling is activated in HSC trans-differentiation. All components of Wnt signaling are induced including Wnt ligands, Frizzled receptor 1 and 2 and the LRP5 co-receptor, and the nuclear level of β-catenin is increased. TCF promoter activity (as a measurement of canonical Wnt pathway) in activated HSC is inhibited with Dickkopf-1 (Dkk-1), the Wnt co-receptor antagonist, and with Chibby, a nuclear protein that blocks β-catenin interaction with TCF.23 Activated canonical Wnt signaling suppresses the expression and promoter activation of Pparγ and promotes the trans-differentiation process for HSC. The forced expression of Dkk-1 blocks HSC activation, their collagen expression and contractility while reversing activated HSC to their quiescent phenotype in culture.23 This reversal is associated with and caused by the restored expression and activity of PPARγ. Further adenovirally expressed Dkk-1 attenuates cholestatic liver fibrosis induced by bile duct ligation in mice.23 We have observed typical Wnt target genes such as cyclin D to be repressed by Dkk-1 in reversal of activated HSC, but it is currently unknown what genes are targeted by the Wnt-β-catenin pathway and directly contribute to HSC activation and liver fibrogenesis. It also remains to be determined whether β-catenin’s effect involves transcriptional regulation via a TCF/LEF-dependent manner or a manner involving other transcription factors.
Necdin
Necdin, a member of the melanoma antigen family (MAGE) of proteins, inhibits the differentiation of adipocytes24 but promotes that of neurons,25 skeletal and smooth muscle cells.26,27 Our recent study demonstrates that necdin is selectively expressed by HSC among the different liver cell types and its expression is induced by activation in vitro and in vivo.28 Necdin silencing with small hairpin RNA, reverses culture-activated HSC to their quiescent morphology much as Wnt antagonism does, and this reversal is again dependent on restored PPARγ expression. Necdin silencing also suppresses the expression of Wnt3a and Wnt10b, two canonical Wnt expressed by activated HSC and TCF/β-catenin-dependent promoter activation. We have found that Wnt10b, one of canonical Wnt expressed by activated HSC, is a direct target of necdin and the necdin-Wnt pathway causes HSC trans-differentiation via the epigenetic repression of Pparγ involving the methyl-CpG binding protein MeCP2.28 This epigenetic regulation involves the induction and recruitment of MeCP2 to the Pparγ promoter and concomitant H3K27 dimethylation and tri-methylation in the 3′ exons of Pparγ, resulting in the formation of a repressive chromatin structure, as recently demonstrated by Mann et al.29 Intriguingly, this study also demonstrates the MeCP2-mediated induction of EZH2, a H3K27 methyltransferase of the polycomb repressive complex 2 (PRC2), responsible for H3K27 dimethylation and tri-methylation.29 Most recently, this paradigm of the MeCP2-EZH2 regulatory relay has been characterized in neuronal differentiation where the MeCP2-mediated epigenetic repression of miR137 is shown to result in EZH2 induction.30
Sonic hedgehog (Shh)
Shh is one of the major three hedgehog ligand family members that are known for their role in cell fate regulation, morphogenesis, mesenchymal–epithelial interactions in embryos and adults. Shh expression and signaling is upregulated in activated HSC and serve as their survival mechanism.31 The Shh pathway may also facilitate interactions with bone marrow-derived mesenchymal stem cells, which may migrate into the liver during wound healing.32 Degenerating hepatocytes may produce hedgehog (Hh) ligands to enhance the proliferation of myofibroblasts, which may be critical in the progression of nonalcoholic steatohepatitis.33 Our unpublished results suggest that there are positive cross-interactions among necdin, Wnt, and Shh pathways in the activation of HSC, all converging to the epigenetic repression of Pparγ and anti-adipogenic HSC trans-differentiation (Fig. 1).
Figure 1.
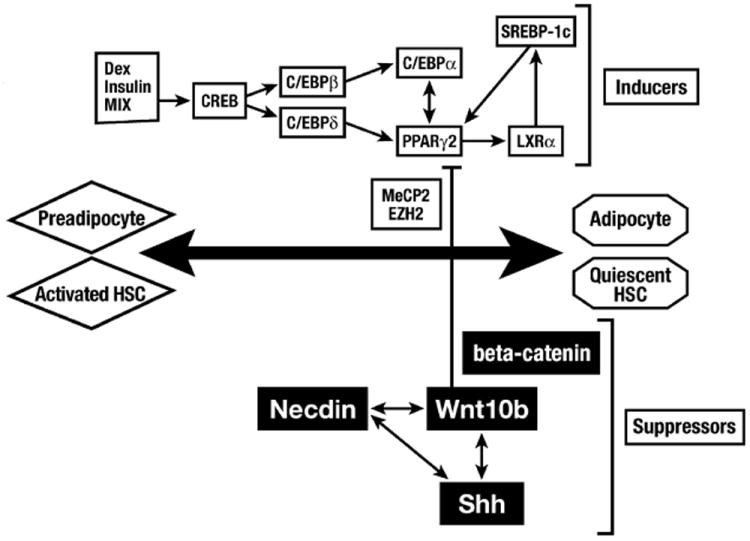
The transcriptional regulation required for adipocyte differentiation is also essential for hepatic stellate cell (HSC) differentiation or quiescence. Morphogens, necdin, Wnt and Shh positively cross-interact and epigenetically repress the master adipogenic gene Pparγ involving the methyl-CpG binding protein MeCP2 and polycomb repressive complex 2 (PRC2) H3K27 methyltransferase, EZH2.
Notch
Notch signaling is also implicated in HSC activation. The Notch intracellular domain (NICD), cleaved upon the activation of Notch by γ-secretase, activates NF-κB via the recruitment of transcriptional co-activators and histone acetylase such as p300.34 Hepatocytes that express the Notch ligand Jagged1 may interact with HSC via Notch to achieve cellular cross-talk much as Wnt and Hh do in a wound healing response.35 How Notch interacts with other morphogens in HSC activation is currently unknown.
Morphogens and liver regeneration
Morphogens are required for morphogenesis, as the term itself signifies. Therefore, it is expected that adult tissue regeneration may also be controlled by morphogens. Indeed, Wnt36 and Hh37 signaling are involved in liver regeneration after a partial hepatectomy. β-catenin also cooperates with HGF to produce a mitogenic response in the liver. This is achieved by the tyrosine phosphorylation of β-catenin by c-Met activation, the dissociation of β-catenin from its complex with c-Met, the nuclear translocation of β-catenin, and the subsequent activation of canonical Wnt signaling at the transcriptional level.38 Serine/threonine protein kinase CK2 phosphorylates transcription factors and regulators that induce proliferative genes. For instance, CK2 phosphorylates c-Jun to enhance its DNA binding,39 c-Myc to stabilize it,40 and IκB for its degradation and NF-κB activation.41 Relevant to our discussion, CK2 also phosphorylates β-catenin at Thr393 to potentially prevent its degradation.42 CK2 also activates AKT by phosphorylation at Ser129, and p-AKT in turn phosphorylates β-catenin at Ser552 to enhance its nuclear translocation and transcriptional activity.43 Although it is yet to be determined whether these regulatory mechanisms involving β-catenin participate in liver regeneration, it can be assumed that β-catenin may orchestrate liver regeneration via the integration of other mitogenic pathways and transcriptional regulation. Mesenchymal–epithelial interactions are integral to morphogenesis, and morphogens released by the mesenchyme serve as key signals for these interactions. The roles of HSC in this specific area of liver regeneration have to be scrutinized further. Moreover, cross-regulation among the morphogens will need to be examined in the context of hepatocyte/hepatoblast-HSC cross-talk.
Morphogens in chronic liver disease
Can morphogens be therapeutic targets for chronic liver disease? This is a rather complex question. HSC-derived morphogens may have different roles in different phases of chronic liver disease. For instance, the upregulation of morphogens and their signaling are involved in HSC activation, which may contribute to liver fibrogenesis in response to hepatocellular damage. At the same time, the morphogens may facilitate cross-talk between HSC and hepatic progenitor cells or hepatocytes to possibly stimulate a regenerative response. Thus, blocking morphogens’ actions to inhibit excessive fibrogenesis may impair liver regeneration. To complicate the matter, these morphogens are also implicated in liver tumorigenesis,44-46 which is a common end-stage consequence of chronic liver disease (Fig. 2). Obviously, aberrant regulations of morphogens must be involved in excessive pathological responses in the evolution and progression of chronic liver disease, and their elucidation seems to be a critical prerequisite for the identification of more precise therapeutic targets.
Figure 2.
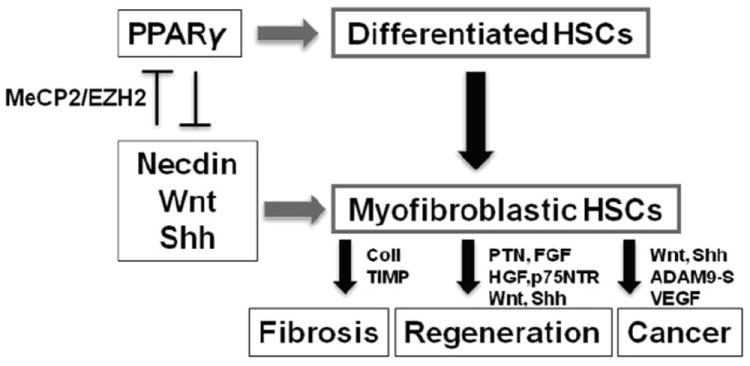
The activation of hepatic stellate cells, which is in part caused by the epigenetic repression of Pparγ by morphogens, may play different roles in the evolution of chronic liver disease from liver fibrosis, regeneration and cancer. Mediators shown are representatives: coll, collagens; TIMP, tissue inhibitor of metalloptroteinase; PTN, pleiotrophin, FGF, fibroblast growth factor, HGF, hepatocyte growth factor; p75NTR, p75 neurotrophin receptor; VEGF, vascular endothelial growth factor.
Acknowledgments
This review article is based in part on work supported by the National Institute on Alcohol Abuse and Alcoholism grants, P50AA11999 (HT), R24AA12885 (HT), U01AA018663 (HT), R01AA018857 (KM), R01AA020753 (KA) and Medical Research Service of Department of Veterans Affairs (HT). The authors also acknowledge the contributions made by their laboratory members to published work described in this article.
Biography

Hidekazu Tsukamoto
Footnotes
Conflict of interest: no conflict of interest has been declared by the authors.
References
- 1.Yamada M, Blaner WS, Soprano DR, Dixon JL, Kjeldbye HM, Goodman DS. Biochemical characteristics of isolated rat liver stellate cells. Hepatology. 1987;7:1224–9. doi: 10.1002/hep.1840070609. [DOI] [PubMed] [Google Scholar]
- 2.Kano K, et al. Uber den EingluB der Insulin-Traubenzucker-Injektion auf die “Fettspeicherungsellen” der Leber. Arch Histol Jpn. 1952;4:13–24. [Google Scholar]
- 3.Sunaga Y. Uber den EinfluB der Insulin-Traubenzucker-Injektion auf die “Fettspeicherungsellen” der Leber. Arch Histol Jpn. 1954;7:241–9. [Google Scholar]
- 4.Ito T, et al. Studies on the “fat-storing cells” (Fettspeicherungszellen) in the liver. Arch Anat Nippon. 1956;31:10–15. [Google Scholar]
- 5.Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–72. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cassiman D, Barlow A, Vander BS, Libbrecht L, Pachnis V. Hepatic stellate cells do not derive from the neural crest. J Hepatol. 2006;44:1098–104. doi: 10.1016/j.jhep.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 7.Geerts A. History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin Liver Dis. 2001;21:311–35. doi: 10.1055/s-2001-17550. [DOI] [PubMed] [Google Scholar]
- 8.Asahina K, Tsai SY, Li P, et al. Mesenchymal origin of hepatic stellate cells, submesothelial cells, and perivascular mesenchymal cells during mouse liver development. Hepatology. 2009;49:998–1011. doi: 10.1002/hep.22721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Asahina K, Zhou B, Pu WT, Tsukamoto H. Septum transversum-derived mesothelium gives rise to hepatic stellate cells and perivascular mesenchymal cells in developing mouse liver. Hepatology. 2011;53:983–95. doi: 10.1002/hep.24119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Potter JJ, Womack L, Mezey E, Anania FA. Transdifferentiation of rat hepatic stellate cells results in leptin expression. Biochem Biophys Res Commun. 1998;244:178–82. doi: 10.1006/bbrc.1997.8193. [DOI] [PubMed] [Google Scholar]
- 11.Potter JJ, Mezey E. Acetaldehyde increases endogenous adiponectin and fibrogenesis in hepatic stellate cells but exogenous adiponectin inhibits fibrogenesis. Alcohol Clin Exp Res. 2007;31:2092–100. doi: 10.1111/j.1530-0277.2007.00529.x. [DOI] [PubMed] [Google Scholar]
- 12.She H, Xiong S, Hazra S, Tsukamoto H. Adipogenic transcriptional regulation of hepatic stellate cells. J Biol Chem. 2005;280:4959–67. doi: 10.1074/jbc.M410078200. [DOI] [PubMed] [Google Scholar]
- 13.Miyahara T, Schrum L, Rippe R, et al. Peroxisome proliferator-activated receptors and hepatic stellate cell activation. J Biol Chem. 2000;275:35715–22. doi: 10.1074/jbc.M006577200. [DOI] [PubMed] [Google Scholar]
- 14.Spiegelman BM, Flier JS. Adipogenesis and obesity: rounding out the big picture. Cell. 1996;87:377–89. doi: 10.1016/s0092-8674(00)81359-8. [DOI] [PubMed] [Google Scholar]
- 15.Hazra S, Xiong S, Wang J, et al. Peroxisome proliferator-activated receptor gamma induces a phenotypic switch from activated to quiescent hepatic stellate cells. J Biol Chem. 2004;279:11392–401. doi: 10.1074/jbc.M310284200. [DOI] [PubMed] [Google Scholar]
- 16.Yavrom S, Chen L, Xiong S, Wang J, Rippe RA, Tsukamoto H. Peroxisome proliferator-activated receptor gamma suppresses proximal alpha1(I) collagen promoter via inhibition of p300-facilitated NF-I binding to DNA in hepatic stellate cells. J Biol Chem. 2005;280:40650–9. doi: 10.1074/jbc.M510094200. [DOI] [PubMed] [Google Scholar]
- 17.Marra F, Efsen E, Romanelli RG, et al. Ligands of peroxisome proliferator-activated receptor gamma modulate profibrogenic and proinflammatory actions in hepatic stellate cells. Gastroenterology. 2000;119:466–78. doi: 10.1053/gast.2000.9365. [DOI] [PubMed] [Google Scholar]
- 18.Galli A, Crabb DW, Ceni E, et al. Antidiabetic thiazolidinediones inhibit collagen synthesis and hepatic stellate cell activation in vivo and in vitro. Gastroenterology. 2002;122:1924–40. doi: 10.1053/gast.2002.33666. [DOI] [PubMed] [Google Scholar]
- 19.Le NH, Franken P, Fodde R. Tumour-stroma interactions in colorectal cancer: converging on β-catenin activation and cancer stemness. Br J Cancer. 2008;98:1886–93. doi: 10.1038/sj.bjc.6604401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lehwald N, Tao G-Z, Jang KY, Sorkin M, Knoefel WT, Sylvester KG. Wnt-β-catenin signaling protects against hepatic ischemia and reperfusion injury in mice. Gastroenterology. 2011;141:707–18. doi: 10.1053/j.gastro.2011.04.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bennett CN, Ross SE, Longo KA, et al. Regulation of Wnt signaling during adipogenesis. J Biol Chem. 2002;277:30998–1004. doi: 10.1074/jbc.M204527200. [DOI] [PubMed] [Google Scholar]
- 22.Ross SE, Hemati N, Longo KA, et al. Inhibition of adipogenesis by Wnt signaling. Science. 2000;289:950–3. doi: 10.1126/science.289.5481.950. [DOI] [PubMed] [Google Scholar]
- 23.Cheng JH, She H, Han YP, et al. Wnt antagonism inhibits hepatic stellate cell activation and liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2008;294:G39–49. doi: 10.1152/ajpgi.00263.2007. [DOI] [PubMed] [Google Scholar]
- 24.Tseng YH, Butte AJ, Kokkotou E, et al. Prediction of preadipocyte differentiation by gene expression reveals role of insulin receptor substrates and necdin. Nat Cell Biol. 2005;7:601–11. doi: 10.1038/ncb1259. [DOI] [PubMed] [Google Scholar]
- 25.Kuwajima T, Nishimura I, Yoshikawa K. Necdin promotes GABAergic neuron differentiation in cooperation with Dlx homeodomain proteins. J Neurosci. 2006;26:5383–92. doi: 10.1523/JNEUROSCI.1262-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuwajima T, Taniura H, Nishimura I, Yoshikawa K. Necdin interacts with the Msx2 homeodomain protein via MAGE-D1 to promote myogenic differentiation of C2C12 cells. J Biol Chem. 2004;279:40484–93. doi: 10.1074/jbc.M404143200. [DOI] [PubMed] [Google Scholar]
- 27.Brunelli S, Tagliafico E, De Angelis FG, et al. Msx2 and necdin combined activities are required for smooth muscle differentiation in mesoangioblast stem cells. Circ Res. 2004;94:1571–8. doi: 10.1161/01.RES.0000132747.12860.10. [DOI] [PubMed] [Google Scholar]
- 28.Zhu NL, Wang J, Tsukamoto H. The necdin-Wnt pathway causes epigenetic peroxisome proliferator-activated receptor gamma repression in hepatic stellate cells. J Biol Chem. 2010;285:30463–71. doi: 10.1074/jbc.M110.156703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mann J, Chu DC, Maxwell A, et al. MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology. 2010;138:705–14. doi: 10.1053/j.gastro.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szulwach KE, Li X, Smrt RD, et al. Cross talk between microRNA and epigenetic regulation in adult neurogenesis. J Cell Biol. 2010;189:127–41. doi: 10.1083/jcb.200908151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang L, Wang Y, Mao H, et al. Sonic hedgehog is an autocrine viability factor for myofibroblastic hepatic stellate cells. J Hepatol. 2008;48:98–106. doi: 10.1016/j.jhep.2007.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin N, Tang Z, Deng M, et al. Hedgehog-mediated paracrine interaction between hepatic stellate cells and marrow-derived mesenchymal stem cells. Biochem Biophys Res Commun. 2008;372:260–5. doi: 10.1016/j.bbrc.2008.05.029. [DOI] [PubMed] [Google Scholar]
- 33.Jung Y, Diehl AM. Non-alcoholic steatohepatitis pathogenesis: role of repair in regulating the disease progression. Dig Dis. 2010;28:225–8. doi: 10.1159/000282092. [DOI] [PubMed] [Google Scholar]
- 34.Oakley F, Mann J, Ruddell RG, Pickford J, Weinmaster G, Mann DA. Basal expression of IkappaBalpha is controlled by the mammalian transcriptional repressor RBP-J (CBF1) and its activator Notch1. J Biol Chem. 2003;278:24359–70. doi: 10.1074/jbc.M211051200. [DOI] [PubMed] [Google Scholar]
- 35.Sawitza I, Kordes C, Reister S, Haussinger D. The niche of stellate cells within rat liver. Hepatology. 2009;50:1617–24. doi: 10.1002/hep.23184. [DOI] [PubMed] [Google Scholar]
- 36.Monga SP, Pediaditakis P, Mule K, Stolz DB, Michalopoulos GK. Changes in WNT/beta-catenin pathway during regulated growth in rat liver regeneration. Hepatology. 2001;33:1098–109. doi: 10.1053/jhep.2001.23786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ochoa B, Syn WK, Delgado I, et al. Hedgehog signaling is critical for normal liver regeneration after partial hepatectomy in mice. Hepatology. 2010;51:1712–23. doi: 10.1002/hep.23525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Apte U, Zeng G, Muller P, et al. Activation of Wnt/beta-catenin pathway during hepatocyte growth factor-induced hepatomegaly in mice. Hepatology. 2006;44:992–1002. doi: 10.1002/hep.21317. [DOI] [PubMed] [Google Scholar]
- 39.Lin A, Frost J, Deng T, et al. Casein kinase II is a negative regulator of c-Jun DNA binding and AP-1 activity. Cell. 1992;70:777–89. doi: 10.1016/0092-8674(92)90311-y. [DOI] [PubMed] [Google Scholar]
- 40.Channavajhala P, Seldin DC. Functional interaction of protein kinase CK2 and c-Myc in lymphomagenesis. Oncogene. 2002;21:5280–8. doi: 10.1038/sj.onc.1205640. [DOI] [PubMed] [Google Scholar]
- 41.Pando MP, Verma IM. Signal-dependent and -independent degradation of free and NF-kappa B-bound IkappaBalpha. J Biol Chem. 2000;275:21278–86. doi: 10.1074/jbc.M002532200. [DOI] [PubMed] [Google Scholar]
- 42.Song DH, Dominguez I, Mizuno J, Kaut M, Mohr SC, Seldin DC. CK2 phosphorylation of the armadillo repeat region of beta-catenin potentiates Wnt signaling. J Biol Chem. 2003;278:24018–25. doi: 10.1074/jbc.M212260200. [DOI] [PubMed] [Google Scholar]
- 43.Ponce DP, Maturana JL, Cabello P, et al. Phosphorylation of AKT/PKB by CK2 is necessary for the AKT-dependent up-regulation of beta-catenin transcriptional activity. J Cell Physiol. 2011;226:1953–9. doi: 10.1002/jcp.22527. [DOI] [PubMed] [Google Scholar]
- 44.de La CA, Romagnolo B, Billuart P, et al. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci U S A. 1998;95:8847–51. doi: 10.1073/pnas.95.15.8847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Devereux TR, Anna CH, Foley JF, White CM, Sills RC, Barrett JC. Mutation of beta-catenin is an early event in chemically induced mouse hepatocellular carcinogenesis. Oncogene. 1999;18:4726–33. doi: 10.1038/sj.onc.1202858. [DOI] [PubMed] [Google Scholar]
- 46.Yanai H, Nakamura K, Hijioka S, et al. Dlk-1, a cell surface antigen on foetal hepatic stem/progenitor cells, is expressed in hepatocellular, colon, pancreas and breast carcinomas at a high frequency. J Biochem. 2010;148:85–92. doi: 10.1093/jb/mvq034. [DOI] [PubMed] [Google Scholar]