

During a long discussion of our favorite enzyme mechanisms, Gene Sander advised me that the easiest way to obtain a true picture of the transitory steps of a reaction would be to “hire a 2-Å postdoc.” In the mid-'70s, when this whimsical conversation took place, molecular and structural biology was just beginning to exert an effect on mechanistic enzymology. Whereas the advances of the past 25 years have been impressive in terms of our understanding of hundreds of additional enzymes and pathways, there remains a need to provide details of chemical conversions within enzyme active sites. In most cases, we have excellent structural information that provides a static picture of how a single substrate or a single product may bind to the catalytic surface, but still lack definition of the sequence of events that takes place between those extremes. Further understanding of the processes taking place during catalysis is required to design ever more specific inhibitors to block the action of key enzymes from pathogenic organisms. In this issue of PNAS, Mobashery and colleagues (1) have taken us closer to the center of the final reaction in the synthesis of bacterial cell walls, by providing a new high-resolution (1.2 Å) structure of an analog of the two-substrate reaction.
The use of the β-lactams in antibacterial therapy (2) has been one of the success stories of the “Pharmaceutical Century.” These antibacterial agents bind to two types of proteins in bacterial cells, collectively known as penicillin-binding proteins, that differ in molecular weight. The low molecular weight group includes both β-lactamases and the d-Ala-d-Ala-peptidases. The latter form family S12 of clan SE of the serine peptidase family in the MEROPS nomenclature (3). Family S12 is characterized by a folding pattern (Fig. 1) that can be described by two domains, an α/β domain plus a largely α-helical domain. The conserved catalytic residues include Ser-62, Lys-65, and Tyr-159 (using the numbering of Streptomyces strain R61 d-Ala-d-Ala carboxypeptidase/transferase, EC 3.4.16.4). This enzyme catalyzes the two-step process of completing the cross-linking of bacterial cell walls by attacking the d-Ala-d-Ala peptide bond of the peptidoglycan precursor, forming an acyl enzyme intermediate, then transferring the acyl group to the diaminopimelate moiety of a second peptidoglycan. The β-lactam antibiotics act by mimicking the first substrate of the normal reaction to form a stable acyl enzyme that cannot be attacked by the second substrate. Whereas such acyl enzyme intermediates have been observed previously, it has not been possible to study the final step of the process, because binding of the second substrate is prevented by the hindered structure of the acyl enzyme. Mobashery and colleagues (1) have devised a way to overcome this limitation, by using some clever chemistry to make a modified cephalosporin that combines mimics of both participants in the reaction. This derivative forms an acyl enzyme intermediate that provides a glimpse of the Michaelis-Menten complex for the second step of the cross-linking reaction.
Figure 1.
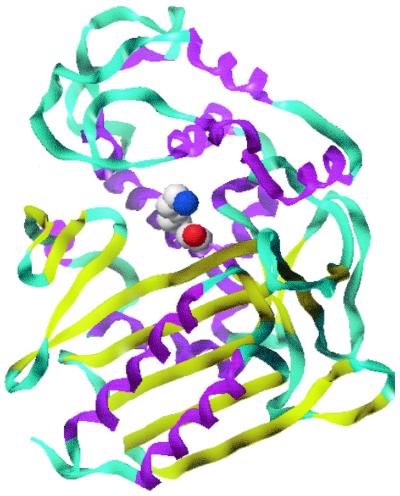
Ribbon representation of the structure of Streptomyces R61 d-Ala-d-Ala carboxypeptidase/transpeptidase [prepared by using file 3PTE from the Brookhaven Databank and the program sybyl (Tripos Associates, St. Louis)]. β structure is shown as yellow ribbons, and α structure is shown as purple spirals. The α/β domain can be seen on the right hand side and the all-α domain is on the left. The side chains of Ser-62 and Lys-65 are seen in space-fill representation at the interface between the two domains.
Others (4) have observed the structure of acyl enzymes involving penicillin derivatives; however, in those cases the structure represents a dead-end complex. Further attack by nucleophiles is prevented by steric hindrance of approach. Attempts to bind other molecules representing the second component of the normal reaction have failed. In the derivative prepared by Lee and colleagues (1), additional structural features have been built into the cephalosporin. In essence, they have built a single molecule that contains components of both substrates of the normal reaction, tethered by the metathiazan moiety at the center (Fig. 2). Thus, after attack of Ser-62 on the four-membered lactam ring, the entire molecule remains bound to the enzyme. The additional pentapeptide structure, or “DAP surrogate” in the author's terminology, provides additional hydrogen bonding interactions with remote parts of the active site. These additional interactions give positioning keys that force the structure to retain conformational information, allowing the comparisons to what would be the final attack step. The approach taken here, using a larger inhibitor analog to learn more about how things bind to an active site, requires both synthetic skill and considerable courage, because the resulting structure could easily be too large or have bond angles inconsistent with a tight fit in the binding cleft. Thus, the design of the structure to be prepared must be undertaken with a prior knowledge of the active site geometry.
Figure 2.

(Upper) Hypothetical Michaelis-Menten complex between the acyl enzyme, derived from attack of the enzyme on the penultimate d-Ala residue, and the diaminopimelate. (Lower) Schematic of the complex reported by Lee et al. (1), with red lines representing the extra bonds formed that differentiate this structure from that in the Upper complex. The carbon atom labeled β replaces the β-carbon of d-Ala.
The process of aminolysis of an ester can proceed through a tetrahedral intermediate or simply involve direct displacement of the leaving group. The difference between the two is the length of time the entering nitrogen and the leaving oxygen are both bonded to the carbonyl carbon. Enzymes seem to be designed to stabilize intermediates along a reaction pathway by arranging a constellation of atoms capable of hydrogen bonding to polar atoms. A second experimental method to gather additional information on the microscopic steps during an enzymatic reaction is the use of “time resolved x-ray crystallography” (5). It is possible to envision a future refinement of the derivative used by Lee et al. If the two halves of the structure were joined by a linkage that was, for example, sensitive to radiation of a specific wavelength, it could be possible to form the complex, as in this study, determine the starting structure, and then flash with the appropriate wavelength to release the tether in a fast reaction. Combining this step with rapid x-ray data collection using Laue diffraction methods might provide additional views of the progress of the attack of the nucleophile on the acyl enzyme.
The new information derived from the structure reported here should trigger new strategies for drug design. By revealing new contact points in the active site that are important for interactions of the natural substrates, it may now be possible to plan the synthesis of a new class of compounds that could effectively block newly emerging bacteria that are resistant to penicillin-based drugs.
Footnotes
See companion article on page 1427.
References
- 1.Lee W, McDonough M A, Kotra L P, Li Z-H, Silvaggi N R, Takeda Y, Kelly J A, Mobashery S. Proc Natl Acad Sci USA. 2001;98:1427–1431. doi: 10.1073/pnas.98.4.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miller J B. The Pharmaceutical Century: Ten Decades of Drug Discovery. Washington, DC: Am. Chem. Soc.; 2000. pp. 53–67. [Google Scholar]
- 3.Frere J M. In: Handbook of Proteolytic Enzymes. Barrett A J, Rawlings N D, Woessner J F, editors. San Diego: Academic; 1998. pp. 427–430. [Google Scholar]
- 4.Kuzin A P, Liu H, Kelly J A, Knox J R. Biochemistry. 1995;34:9532–9540. doi: 10.1021/bi00029a030. [DOI] [PubMed] [Google Scholar]
- 5.Hajdu J, Neutze R, Sjorgen T, Edman K, Szoke A, Wilmouth R, Wilmot C M. Nat Struct Biol. 2000;7:1006–1012. doi: 10.1038/80911. [DOI] [PubMed] [Google Scholar]