

Abstract
The IMPDH/GMPR family of (β/α)8 enzymes presents an excellent opportunity to investigate how subtle changes in enzyme structure change reaction specificity. IMP dehydrogenase (IMPDH) and GMP reductase (GMPR) bind the same ligands with similar affinities and share a common set of catalytic residues. Both enzymes catalyze a hydride transfer reaction involving a nicotinamide cofactor hydride, and both reactions proceed via the same covalent intermediate. In the case of IMPDH, this intermediate reacts with water while in GMPR it reacts with ammonia. In both cases, the two chemical transformations are separated by a conformational change. In IMPDH, the conformational change involves a mobile protein flap while in GMPR the cofactor moves. Thus reaction specificity is controlled by differences in dynamics, which in turn are controlled by residues outside the active site. These findings have some intriguing implications for the evolution of the IMPDH/GMPR family.
key terms: guanine nucleotide biosynthesis, IMP dehydrogenase, GMP reductase, monovalent cation activation, drug resistance, water activation, ammonia selectivity, enzyme evolution
Introduction
How structure determines function remains one of the central challenges in modern biochemistry. This problem is brought sharply into focus with need to annotate genomic sequences. However, the growing appreciation of how drastically reaction outcomes can change with subtle structural modifications gives rise to the gnawing suspicion that most such assignments rest on rickety foundations.
The (β/α)8 barrel, also known as the TIM barrel, is the most common and versatile enzyme fold (Glasner et al., 2006, Soskine and Tawfik, 2010, Zalatan and Herschlag, 2009, Nagano et al., 2002, Gerlt and Raushel, 2003, Wise and Rayment, 2004). Approximately thirty (β/α)8 barrel protein superfamilies are listed in the current SCOP and CATH databases (Lo Conte et al., 2002, Orengo et al., 1997), catalyzing over twenty-five different reactions (Anantharaman et al., 2003). Therefore these proteins present a particularly thorny annotation problem. One of these superfamilies is comprised of two enzymes, inosine monophosphate dehydrogenase (IMPDH) and guanosine monophosphate reductase (GMPR). These two enzymes catalyze very similar reactions (Figure 1; (Hedstrom, 2009)). IMPDH catalyzes the NAD+-dependent oxidation of IMP into XMP while GMPR catalyzes the NADPH-dependent reduction of GMP to IMP and ammonia. IMPDH and GMPR utilize the same catalytic residues and bind the same ligands with similar affinities (Table 1). Both reactions involve the same covalent intermediate, but why this intermediate partitions in different directions is only beginning to be understood. This review will summarize recent insights into the mechanistic and structural determinants of reaction specificity in IMPDH/GMPR.
Figure 1.

Purine Nucleotide Metabolism.
Table 1.
Ligand affinities for IMPDH and GMPR.
| Ligand | IMPDH | GMPR |
|---|---|---|
| GMP (μM) | 2–90 a | 3–21 (Km) c,d,e,f |
| IMP (μM) | 1.7–100 (Km) a | 8–140 c,d |
| XMP (μM) | 30–100 a | 0.01–28 c,f,h |
| MZP (μM) | 0.0005–0.008 a | 0.015 c |
| RVP (μM) | 0.1–0.6 a | 98 c |
| 2-Cl-IMP (μM) | 48 (Km) a | 7.9 (Km) c |
(Antonino and Wu, 1994).
Overview of the guanine nucleotide pathways
IMP is the product of the de novo purine biosynthetic pathway (Figure 1). Most organisms can also produce IMP via salvage pathways, either by a phosphoribosylation reaction with hypoxanthine or a kinase reaction with inosine. IMP is converted into both adenine and guanine nucleotides. The IMPDH-catalyzed production of XMP is the first committed and rate-limiting step in guanine nucleotide biosynthesis. GMP synthetase (GMPS) catalyzes the subsequent conversion of XMP to GMP. This reaction requires the hydrolysis of ATP to AMP and pyrophosphate as well as the hydrolysis of glutamine to glutamate. The IMPDH/GMPS pathway is widely accepted as the only biosynthetic route to guanine nucleotides, and this pathway appears to be present in all but the handful of protozoan parasites that salvage precursors from their hosts (Morrison et al., 2007, Carlton et al., 2007). More intriguingly, approximately 20 organisms appear to contain an IMPDH gene but to lack a gene encoding GMPS (The SEED, http://theseed.uchicago.edu/FIG/index.cgi accessed June 11, 2011). These organisms may have another route to GMP.
GMPR appears to have a much more modest role in cellular metabolism. This enzyme catalyzes the only recognized route to recycle guanine nucleotides into adenine nucleotides (Figure 1). The apparently opposing metabolic roles of IMPDH and GMPR dictate that their expression is coordinated- conditions that increase IMPDH activity generally decrease GMPR expression. In mammalian cells, IMPDH expression decreases and GMPR expression increases during differentiation, and the guanine nucleotide pool decreases accordingly (Snyder et al., 1973, Weber et al., 1992). Proliferation requires an expansion of the guanine nucleotide pool, and is therefore accompanied by an increase in IMPDH expression (Jackson et al., 1975, Liu et al., 2008). In many organisms, IMPDH expression is induced when guanine nucleotides are low (Kuehner and Brow, 2008, Thomas and Drabble, 1984). GMPR expression is not as well characterized. In bacteria, GMPR is induced in response to excess guanine and when glutamine concentrations are low (Kessler and Gots, 1985). Curiously, GMPR is also induced during thermogenesis (Salvatore et al., 1998).
IMPDH-targeted drugs and GMPR
IMPDH is a validated drug target for immunosuppressive (Ratcliffe, 2006), anticancer (Chen and Pankiewicz, 2007, Olah et al., 2006) and antiviral chemotherapy (Nair and Shu, 2007), and a promising target for antibiotics (Hedstrom et al., 2011). IMPDH and GMPR bind IMP and GMP with similar affinities (Table 1), which suggests that GMPR may also be inhibited by IMPDH-targeted drugs. Inhibitors that act as IMP analogs are generally potent inhibitors of both enzymes, e.g., mizoribine monophosphate (MZP), the active metabolite of the immunosuppressive drug mizoribine, and ribavirin monophosphate, the active metabolite of the antiviral drug ribavirin (Figure 2 and Table 1; (Patton et al., 2011)). What role GMPR inhibition plays in the pharmacology of these drugs remains to be established. Tiazofurin and benzamide riboside are converted into NAD analogs, TAD and BAD, respectively, which are potent inhibitors of IMPDH (Figure 2; (Pankiewicz et al., 2004)). It is likely the NADP analogs of these compounds would inhibit GMPR, but this does not appear to have been tested. Non-nucleoside drugs that target IMPDH are perhaps less likely to inhibit GMPR. The potent IMPDH inhibitor mycophenolic acid (MPA) does not inhibit GMPR (our unpublished experiments). At present, no GMPR-targeted therapy exists, although GMPR inhibitors may have applications in the treatment of leishmaniasis (Spector et al., 1984, Looker et al., 1986).
Figure 2.

Structures of IMPDH targeted drugs.
Overview of the structures of IMPDH and GMPR
Both IMPDH and GMPR are homotetramers, with the catalytic (β/α)8 barrels arranged in square planar geometry (Figure 4). The active site is found at the C-terminal ends of the β sheets. The catalytic Cys residue is found in the characteristic sequence motif GIGPGSICTT on the loop between β6 and α6 the Cys loop (Figure 5A) This residue is Cys319 in IMPDH (Tritrichomonas foetus numbering will be used unless otherwise noted) and Cys188 in GMPR (human GMPR2 numbering). The second Thr in the Cys loop motif is also a key catalytic residue (Thr321 in IMPDH, Thr188 in GMPR), as is the Glu residue that is its hydrogen bonding partner (Glu431 and Glu289 in GMPR).
Figure 4.

The structures of IMPDH and GMPR. The structures of S. pyogenes IMPDH (1zfj) and human GMPR2 (1c6q) are shown. The left panels show the tetramers, with alternating subunits colored in light and dark gray. The right panels show the lower left subunit, with a 180 degree rotation from the left panel. IMP is shown in spacefill. A color version of this figure is available online.
Figure 5.

Active site conservation in IMPDH/GMPR. The barrel domain of T. foetus IMPDH (1lrt), is shown containing IMP and the NADH analog tiazofurin adenine dinucleotide (TAD). The catalytic Cys319 and the residues that interact with the adenine ring of TAD, Arg241 and Trp269, are depicted in sticks. Residues 416–431, which form part of the mobile flap, are disordered in this structure. A color version of this figure is available online.
Not surprisingly, the IMP/GMP site is highly conserved in both enzymes, and this conservation extends to the nicotinamide portion of the cofactor binding site (Figure 5A). However, the adenosine portions of the cofactors bind in different regions of the barrel domain in IMPDH and GMPR (described in more detail below). A mobile flap extends between β8 and α8; both the sequence and length of this flap are quite variable (Figure 5B). The flap has very different roles in each enzyme. In IMPDH, the flap binds in the same site as the cofactor during the hydrolysis reaction. The IMPDH flap contains a conserved Arg418-Tyr419 dyad that is involved in the activation of water. This second functional constraint makes the divergence of the adenosine portion of the cofactor binding site even more surprising in the case of IMPDH (Figure 5A and B). This divergence may be a response to the presence of naturally occurring IMPDH inhibitors (Kohler et al., 2005). In GMPR, the flap interacts with the 2′-phosphate of NADPH via a conserved Tyr285-Arg286 dyad.
IMPDH and GMPR share an additional structural feature, an allosteric binding site for monovalent cations such as K+. The Cys loop forms half of the monovalent cation binding site, while the other half is contributed by the end of a C-terminal alpha helix from an adjacent monomer (Figure 5C). These residues are not conserved, and may account for the differences in monovalent cation specificity and other catalytic properties among enzymes from different sources.
Most IMPDHs contain an additional subdomain inserted between α2 and β3 that is not found in GMPRs (Figure 4). This subdomain contains two cystathionine beta synthetase (CBS) domains (also known as Bateman domains (Bateman, 1997, Ignoul and Eggermont, 2005)). Removal of the CBS domains does not perturb enzymatic activity (Nimmesgern et al., 1999, Gan et al., 2002). CBS domains are found in several unrelated proteins, including protein kinases and CLC channels, where they bind adenine nucleotides (Janosik et al., 2001, Scott et al., 2004, Jamsen et al., 2007, Jentsch, 2008). While one laboratory has reported a similar function for the CBS domains of IMPDH (Scott et al., 2004), others have been unable to confirm this finding (Mortimer and Hedstrom, 2005, Pimkin and Markham, 2008, Carr et al., 1993, Holmes et al., 1974). The presence of the adenine nucleotide binding site in the (β/α)8 domain of IMPDH obviously complicates these experiments. In addition, while the overall fold of the CBS domains is clearly maintained, there is little sequence identity, so it is quite possible that the CBS domains have different functions in different proteins. Deletion of the CBS domains disrupts the coordinated regulation the adenine and guanine nucleotide pools in E. coli (Pimkin and Markham, 2008, Pimkin et al., 2009). Others have suggested that the CBS domains are involved in the regulation of translation and transcription (Cornuel et al., 2002, McLean et al., 2004, Mortimer and Hedstrom, 2005, Bowne et al., 2006, Mortimer et al., 2008, Park and Ahn, 2010), though the exact role of IMPDH in these contexts remains to be elucidated.
IMPDH transcripts from various organisms suggest that additional domains can be appended to the N- and C-termini. The best characterized of these are variants of mammalian IMPDH1 that are produced in the retina as the result of alternative mRNA splicing (Spellicy et al., 2007, Gunter et al., 2008). The functions of these additional domains are unknown.
The mechanism of the IMPDH reaction
The mechanistic challenge of both the IMPDH and GMPR reactions is “how can a single active site catalyze two different chemical transformations?” In the case of IMPDH, these reactions are: (1) a hydride transfer reaction that produces NADH and the covalent intermediate E-XMP* and (2) a hydrolysis reaction that releases XMP (Figure 3). In contrast, GMPR catalyzes: (1) deamination of GMP to form E-XMP* followed by (2) hydride transfer to produce IMP. These enzymes use complementary strategies involving conformational changes to solve this problem.
Figure 3.

The mechanisms of IMPDH and GMPR.
IMPDH is a well-characterized enzyme. More than twenty IMPDH genes have been expressed and activity confirmed, and more than 25 crystal structures exist for IMPDHs from eight different organisms, including many different complexes of substrates and inhibitors. Steady-state, pre-steady-state, isotope effect, inhibitor characterization and mutagenesis experiments have been reported for IMPDHs from many different sources (Hedstrom, 2009). Though several early reports concluded that substrate binding was ordered, later isotope effect and ligand binding experiments revealed that IMP and NAD+ bind randomly (Heyde et al., 1976, Wang and Hedstrom, 1997, Digits and Hedstrom, 1999, Xiang et al., 1996). This discrepancy can be explained by the use of product inhibition experiments, which are unreliable when an intermediate accumulates, as occurs during the IMPDH reaction. The hydride transfer reaction likely occurs in two steps, first the attack of the catalytic Cys319 on C2 of IMP, followed by expulsion of the hydride to NAD+ (Tritrichomonas foetus numbering will be use throughout), though no experiments address this point. Hydride transfer is fast in most IMPDHs. Mutation of Thr321 decreases the rate of hydride transfer, suggesting that this residue may serve to activate Cys319 (Guillen Schlippe and Hedstrom, 2005). NADH releases, leaving the covalent intermediate E-XMP*. This step is partially rate-limiting in some IMPDHs. The mobile flap (residues 412–432) folds into the vacant cofactor binding site, bringing the conserved Arg418Tyr419 dyad into position to activate water for the hydrolysis of E-XMP*. A solvent isotope effect is generally observed on kcat, suggesting that the conformational change is fast and hydrolysis is rate-limiting (Riera et al., 2008). Thus IMPDH has two conformations as defined by the position of the flap: an open conformation for the hydride transfer step and a closed conformation for the hydrolysis step.
All enzymes that perform hydrolysis reactions have some strategy to activate water, but the general base catalyst has been difficult to identify in IMPDH. The x-ray crystal structure of the E•MZP complex captured a transition state-like arrangement of the Cys319 and a putative catalytic water (Figure 6A). The water molecule interacts with Arg418, Tyr419 and Thr321. None of these residues are usually candidates for general base catalysts, though precedence is available for each (see examples in (Guillén Schlippe and Hedstrom, 2005B)). Given that hydrolysis is rate-limiting in IMPDH, the substitution of the general base catalyst would be expected to decrease the value of kcat by a factor of 100–1000. Only the substitution of Arg418 has an effect of this magnitude, suggesting that this residue is the primary player in the activation of water (Table 2). Mutations of Arg418 and Tyr419 only affect the hydrolysis reaction, while the mutation of Thr321 perturbs both hydride transfer and hydrolysis (Guillen Schlippe and Hedstrom, 2005, Guillen Schlippe et al., 2004). Importantly, while the mutation of Arg418 also perturbs flap closure, this effect is not sufficient to account for the decrease in kcat. Chemical rescue experiments are also consistent with Arg418 acting as a general base (Guillén Schlippe and Hedstrom, 2005a).
Figure 6.

The activation of water in IMPDH. A. Interactions of the catalytic with water in the E•MZP complex (1pvn). Figure from (Min et al., 2008). B. The Arg418 pathway for water activation. C. The Thr321 pathway for water activation.
Table 2.
Kinetic parameters for mutants of T. foetus IMPDH and E. coli GMPR.
| Catalytic residue | kcat (s−1) | Comments | ||
|---|---|---|---|---|
| IMPDH (IMP) | GMPR (GMP) f | GMPR f (2-Cl-IMP) f | ||
| wild-type | 1.9 b (3.8) b | 0.35 (≤0.0008) | 0.40 (0.08) | parentheses denote reaction with acetyl pyridine cofactor a |
| Cys | ≤ 0.001 b | ≤ 0.0001 | ≤ 0.0001 | IMPDH: Cys319Ser GMPR: Cys186Ala |
| Thr | 0.18 c | 0.00088 | 0.021 | IMPDH: Thr321Ala GMPR: Thr188Ala |
| Glu | 1.4 d | 0.00038 | 0.027 | IMPDH: Glu GMPR: Glu289Gln |
| Arg418Ala | 0.004 e | n.a. | n.a. | general base that activates water in IMPDH; counterpart is cofactor amide in GMPR |
| Tyr419Phe | 0.22 e | n.a. | n.a. | part of conserved motif with Arg418, but only minor role in the activation of water |
n.a., not applicable.
APAD reaction: for IMPDH, reaction with APAD+, for GMPR, reaction with APADH.
Combined molecular mechanics/quantum mechanics simulations provided further insight into how water is activated during the IMPDH reaction (Min et al., 2008). The lowest energy pathway to products involves a neutral Arg418 abstracting a proton from water (Figure 6B). If the pKa of Arg418 is “normal”, i.e., ~12.5, the calculated energy barrier for this reaction is similar to that observed experimentally. However, if Arg418 is protonated, water is activated via a proton relay involving Thr321 and Glu431 (Figure 6C). The calculated energy barrier for this reaction is similar to that observed when Arg419 is mutated. These findings suggest that IMPDH primarily uses the Thr321 pathway to activate water at low pH, and the Arg418 pathway dominates at high pH. The substitution of Glu341 with Gln disables the Thr321 pathway, and shifts the pH rate profile to the right, as predicted by the simulations. Thus IMPDH appears to have two sets of catalytic machinery to activate water.
While the closure of the flap is clearly required for the hydrolysis reaction, less dramatic changes in the conformation of the Cys319 loop appear to occur throughout the catalytic cycle (Hedstrom and Gan, 2006). X-ray crystal structures find the Cys319 loop in at least three different conformations in various ligand complexes (two of these conformations can be seen in Figure 5A and B). If these complexes mimic distinct states in the catalytic cycle, then the Cys319 has different conformations during each stage of the reaction. The results of alanine scanning mutagenesis of three residues in the Cys319 loop, Arg322, Glu323 and Gln324, are consistent with this view (Josephine et al., 2010). All three mutations increased the equilibrium of the hydride transfer reaction, but decreased the equilibrium between the open and closed conformations of the flap. These results indicate that the mutations stabilize E-XMP*open, but not E-XMP*closed (Figure 7), as might be expected if different conformations of the Cys319 loop are required for the hydride transfer and hydrolysis reactions. Further, these results suggest that the conformation of the Cys319 loop gates closure of the flap.
Figure 7.
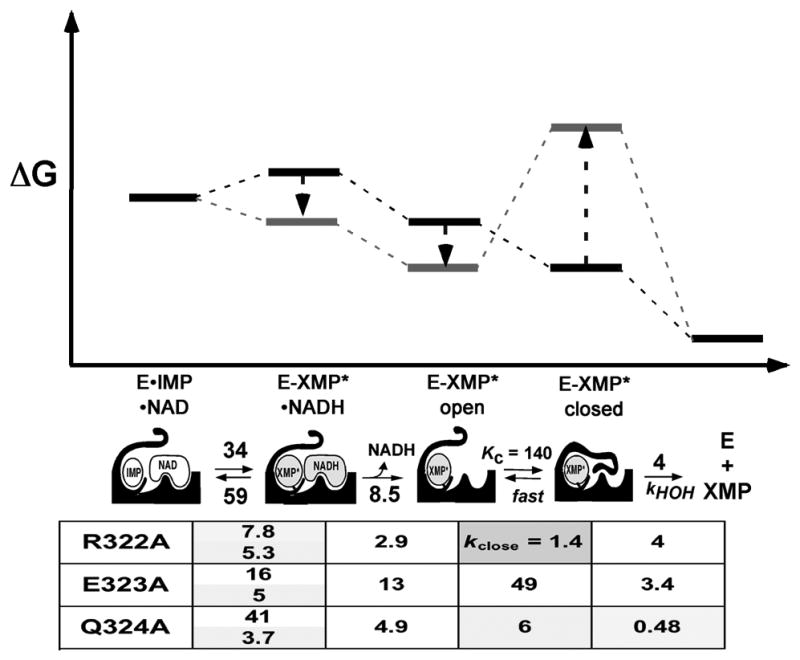
Effects of mutations in the Cys319 loop on the IMPDH reaction. Figure modified from (Josephine et al., 2010). In the energy diagrams, the dark bars show the relative positions the various complexes in the wild-type enzyme, and the lighter bars show the direction of change with the mutations. The table is shaded according to the magnitude of the difference in the parameter from the wild-type enzyme: no shading, factor of 1–2; light, factor of 2–20; dark, factor >20.
The conformation of the Cys319 loop, and its influence of the flap, are also factors in the mechanism of monovalent cation activation. The Cys319 loop forms half of the K+ binding site-the other half is formed by the end of a C-terminal helix from the adjacent subunit (residues 485–487; Figure 5C). Monovalent cations are found in only a handful of IMPDH structures: from Chinese hamster, E-XMP*•MPA•K+ (1jr1; (Sintchak et al., 1996)), and from T. foetus, E•MZP•K+ (1pvn; (Gan et al., 2003)), E•RVP•Na+ (1me8; (Prosise et al., 2002)) and E•RVP•MPA•Na+ (1me7; (Prosise et al., 2002)). No monovalent cations are observed in other complexes of T. foetus IMPDH, including E•IMP, E•IMP•TAD and E•XMP•NAD+ (1me9, 1meh and 1lrt, respectively; (Prosise and Luecke, 2003, Gan et al., 2002, Prosise et al., 2002), suggesting that K+ transiently associates during the catalytic cycle, perhaps only binding to E-XMP* complexes. This hypothesis was inconsistent with steady state kinetic characterization of IMPDHs from other sources, which indicated that K+ is required for substrate/cofactor binding (Heyde et al., 1976, Xiang et al., 1996, Kerr et al., 2000)). Unfortunately, T. foetus IMPDH contains a second K+ site in addition to the conserved monovalent site, so it is not possible to address this question directly in this enzyme. However, a detailed investigation of the kinetics of Cryptosporidium parvum IMPDH reaction revealed that the presence K+ increases the rate of association of NAD+ to the E•IMP complex (Riera et al., 2011). Thus K+ must be present throughout the catalytic cycle. Further, this finding casts doubt on the catalytic relevance of the x-ray crystal structures of cofactor complexes, since none of the currently available structures contain a monovalent cation.
The presence of K+ increases the value of kcat for IMPDH reactions by >40-fold, yet the evidence suggests that K+ has little effect on either chemical transformation in C. parvum IMPDH. In the absence of K+, burst of NADH production is observed in the steady state, and no isotope effect is found when 2-[2H]-IMP is the substrate, indicating that hydride transfer remains fast. The release of NADH is slower in the absence of K+, but not slow enough to account for kcat. No solvent isotope effect is observed in the absence of K+, which indicates that the hydrolysis step is also not rate-limiting. These observations suggest that the rate-limiting step is a conformational change following NADH release, i.e., the closure of the flap. Importantly, the rate of the conformational change is the key variable, not the shift in the equilibrium between open and closed conformations- since this step is rate-limiting, it cannot be at equilibrium. Thus the presence of K+ accelerates flap closure.
The question of how K+ can promote the rate of conformational exchange was investigated in a series of alchemical free energy simulation and potential of mean force calculations using the orthogonal space random walk strategy (Riera et al., 2011). Models of the E-XMP*•K+ complex of C. parvum IMPDH in the open and closed states were constructed based on the E•MZP and E•RVP•MPA structures of T. foetus IMPDH (1pvn and 1meh, respectively). These models can explain the monovalent cation specificity of C. parvum IMPDH. When a simulation was performed in the absence of K+, the residues of the K+ binding site relaxed into an alpha helix, creating a barrier to the conformational change. The presence of K+ provides alternate interactions for the carbonyl oxygens of these residues, mobilizing this segment. A similar mechanism has been proposed for the activation of enzymes by salt in organic solvents (Eppler et al., 2006), and water molecules have been proposed to serve a similar role in ribozymes (Rhodes et al., 2006). K+ also changes the shape of the energy landscape. The energy well is narrow and steep in the absence of K+, but becomes broad in the presence of K+, and the minimum is in a more “open” position (Figure 8). These simulations suggest that K+ acts as a ball and socket joint, organizing the Cys319 loop while promoting the enslaved conformational transitions. Since the conformation of the Cys319 loop gates flap closure, K+ also influences this conformational change.
Figure 8.

Shape of the energy well for the closed conformation of the flap in the absence (A) and presence of K+ (B). Figure from (Riera et al., 2011) with permission. Potential of mean force calculations were used to probe the effects of K+ on the closed conformation (Riera et al., 2011). A model of the E-XMP*closed complex of C. parvum IMPDH was constructed based on 1pvn. The collective variable is comprised of the average of the distance of each Cα in the flap to the center of the purine ring of E-XMP*, so the smaller the collective variable, the more “closed” the conformation.
Monovalent cation specificity varies significantly among IMPDHs from different sources- for example, Na+ activates some IMPDHs, but has no effect, or inhibits, others (Hedstrom, 2009). This observation suggests that there are significant differences in the structure of the monovalent cation binding sites, which will translate into differences in the conformational dynamics of the Cys319 loop and the flap. Understanding, and predicting, how such functional distinctions arise from rather subtle changes in sequence will require even more detailed characterization of additional IMPDHs. In particular, direct methods for monitoring the conformational changes are needed to calibrate the computational approaches. This is no small task. The open-to-closed conformational change of the flap may be more analogous to an disorder-order transition than the rigid body lid closures that have been monitored to date.
The importance of the K+ site is dramatically illustrated with the MPA resistant IMPDHs found in Penicillium brevicompactum, the fungus that produces MPA (Hansen et al., 2011). MPA binds to E-XMP*, competing with the flap for the vacant cofactor binding site (Link and Straub, 1996, Sintchak et al., 1996). Thus the equilibrium between the open and closed conformation is an important determinant of drug sensitivity (Digits and Hedstrom, 2000, Kohler et al., 2005, Riera et al., 2008). P. brevicompactum contains two IMPDHs, PbIMPDH-A and PbIMPDH-B; these enzymes are 20- and 1000-fold more resistant to MPA, respectively, than a typical eukaryotic IMPDH (Hansen et al., 2011). Curiously, the active site and MPA binding site are completely conserved. However, the C-terminal helix that forms part of the K+ binding site has diverged; swapping this region between MPA-sensitive and resistant IMPDHs reveals that this region can account for 7-fold of the resistance. How these changes influence MPA inhibition is currently under investigation. More importantly, these observations indicate that additional “long-range” determinants of drug selectivity, and reaction properties, remain to be identified.
The GMPR reaction
Characterization of the GMPR reaction is rudimentary in comparison to IMPDH. The enzyme has been purified from a handful of sources: Aerobacter aerogenes (Mager and Magasanik, 1960, Brox and Hampton, 1968), human erythrocytes (Mackenzie and Sorensen, 1973, Spector, 1979 #838), calf thymus (Stephens and Whittaker, 1973), Leishmania donvani (Spector and Jones, 1982, Spector et al., 1984), Artemia salina (Renart et al., 1976a, Renart et al., 1976b), though its presence has been inferred in several other cases by the conversion of labeled guanine into adenine nucleotides. Only the human and Escherichia coli genes have been cloned and expressed to verify activity (Andrews and Guest, 1988, Moffat and Mackinnon, 1985, Martinelli et al., 2011, Patton et al., 2011, Li et al., 2006, Zhang et al., 2003, Deng et al., 2002). X-ray crystal structures are available for human GMPR1 and GMPR2 in several complexes: E•GMP (2ble, 2bwg, 2a7r, (Li et al., 2006)), E•IMP (2bzn, (Patton et al., 2011)) and E•IMP•NADH (2c6q, (Patton et al., 2011)). Two structures of B. anthracis GMPR can be found in the PDB (1ypf and 2a1y), but there is no record that this protein has actually been assayed for GMPR activity.
Using IMPDH as a template for GMPR reaction, the following mechanism would be expected: GMP binds, the deamination reaction occurs with formation of E-XMP*, ammonia departs and NADPH binds, E-XMP* is reduced to IMP. This mechanism would be described by ping-pong kinetics, which are most definitely not observed in GMPRs. Instead, an intersecting line pattern is found in Lineweaver-Burk plots (Spector et al., 1979, Deng et al., 2002, Martinelli et al., 2011, Patton et al., 2011), indicating that a ternary E•GMP•NADPH complex must form before deamination can proceed. Further, again by analogy to the IMPDH reaction, the deamination reaction would be expected to be rate-limiting and hydride transfer is expected to be fast. However, two investigations of the mechanism of E. coli GMPR report that an isotope effect is observed on hydride transfer, indicating that in fact the deamination step is fast and hydride transfer is rate-limiting (Martinelli et al., 2011, Patton et al., 2011). These findings create a conundrum: how can the deamination reaction occur in the presence of cofactor?
The above mechanism presumes that E-XMP* is an intermediate in the GMPR reaction as in IMPDH. However, an alternative concerted deamination/hydride transfer mechanism is also plausible, wherein Cys acts as a general acid to protonate the leaving group as hydride is transferred (Martinelli et al., 2011). Since hydride transfer is rate-limiting, if E-XMP* forms, it should accumulate in the steady state, and in fact the presence of E-XMP* has been demonstrated by mass spectroscopy (Patton et al., 2011). E-XMP* also forms in the presence of NADP+, but not when the cofactor is absent. Thus the presence of cofactor induces a conformational change required for the deamination of GMP and formation of E-XMP*. The nature of this conformational change can be deduced from the structures of E•GMP (1ble) and E•IMP•NADPH (2c6q). In the absence of cofactor, the catalytic Cys186 points away from GMP, as does the catalytic Glu289 and the conserved Tyr285-Arg286 dyad in the flap folds into the active site, with Arg286 protecting GMP from water (Figure 9A; (Li et al., 2006)). When the cofactor binds, this segment forms part of the 2′-phosphate binding site and the Cys186, Thr188 and Glu289 residues align for catalysis, as shown in the structure of the E•IMP•NAPDH complex (Figure 9B; 1c6q, (Patton et al., 2011)). While these structures explain how cofactor binding promotes the deamination reaction, they also provoke a new question: how is the amine is activated if the cofactor is where the acid/base catalyst should be?
Figure 9.

Conformational changes during the GMPR catalytic cycle. A. Structure of E•GMP (2a7r; (Li et al., 2006)). B. Structure of E&•IMP•NADPH in the “in” position for the hydride transfer reaction (1c6q; (Patton et al., 2011)). C. Structure of E•IMP•NADPH in the “out” position for the deamination reaction (1c6q; (Patton et al., 2011)). A color version of this figure is available online.
The answer to this question was also revealed in the structure of E•IMP•NADPH (Figure 9C). This structure has two GMPR tetramers in the asymmetric unit, and three different ligand states are found within the eight active sites: E•IMP alone, E•IMP•NADPHin, where the cofactor is optimally aligned for hydride transfer (Figure 9B), and E•IMP•NADPHout, where the cofactor is found in a new conformation that is too far away from the substrate to permit hydride transfer (Figure 9C). An electron density, modeled as water, is observed in the out conformation in the position that would be occupied by the ammonia product. This density is within hydrogen bonding distance of Thr188 and the carboxamide of the cofactor. These observations suggest that the deamination reaction occurs when the cofactor is in the “out” conformation, and that the Thr188-Glu289 dyad and cofactor amide together form the machinery that activates the amine.
Mutagenesis experiments were combined with substrate/cofactor analogs to investigate the role of Thr188-Glu289 in the deamination reaction (Table 2). 2-Cl-IMP is also a good substrate for GMP, with a relative kcat of 0.9 versus GMP. However, while the deamination of GMP requires the presence of a general acid catalyst, chloride is a much better leaving group, so the 2-Cl-IMP reaction should proceed even if the general acid catalyst has been removed. As expected, no reaction is observed with either GMP or 2-Cl-IMP when Cys186 was replaced with Ala (Patton et al., 2011). The substitution of Thr186 with Ala reduced the value of kcat by a factor of 500. A similar reduction was observed with the substitution of Glu289, demonstrating that this residue is also required for the GMPR reaction, unlike the IMPDH (Table 2). However, much smaller reductions were observed in the reactions of Thr186Ala and Glu289Ala with 2-Cl-IMP (factors of 14 and 18, respectively), as expected if Thr188 and Glu289 are involved in the activation of the amine.
A similar set of experiments was used to probe the role of the cofactor amide in the deamination reaction (Patton et al., 2011). Acetylpyridine adenine dinucleotide phosphate (APADP) contains a methylketone in place of the amide group of NADP. No reaction is observed between GMP and APADPH in the presence of GMPR (the relative value of kcat <0.006). In contrast, GMPR catalyzes the reaction of 2-Cl-IMP and APADPH with a relative kcat = 0.2. These observations indicate that the cofactor amide is part of the catalytic machinery activating ammonia. To the best of our knowledge, this is an unprecedented role for a nicotinamide cofactor.
Reaction specificity hinges on the ability to distinguish water and ammonia
The GMPR and IMPDH reactions involve the same covalent intermediate, E-XMP*, but this intermediate reacts with ammonia in GMPR and with water in IMPDH (considering the GMPR reaction from the reverse). GMPR does not catalyze the reacton of IMP and NADP+ to XMP and NADPH. However, GMPR does catalyze the formation of E-XMP* from IMP and NADP+. This complex is stable for days in the absence of ammonia, but forms GMP when NH4Cl is supplied (Patton et al., 2011). These observations indicate that GMPR has a >105 preference for ammonia/ammonium over water. If one assumes that ammonia is the actual substrate, this preference increases to >106 at pH 8, the conditions of these experiments. XMP is the more stable than IMP and GMP, in part because it is ionized at physiological pH, so XMP would form if GMPR catalyzed the reaction with water Likewise, IMPDH does not catalyze the hydrolysis of GMP (<0.1% the activity of GMPR; (Patton et al., 2011)). Therefore kinetic barriers must exist to prevent GMPR from reacting with water and IMPDH from reacting with GMP.
The ammonia selectivity of GMPR most likely results from a combination of intrinsic reactivity and specific binding. Ammonia is a much better nucleophile than water, by a factor of ~104 (Minegishi and Mayr, 2003). However, this leaves a selectivity factor of at least 100 that must derive from a specific binding site. Ammonia has one more hydrogen bond donor than water, which suggests that ~3 kcal/mol of specific binding energy could be available, providing approximately a factor of 500 in selectivity. Precedence for such selective interactions can be found in the ammonia-specific channels AmtB and RhAG (Musa-Aziz et al., 2009).
The nature of the kinetic barrier in IMPDH is more difficult to understand, because any mechanism that activates water would be expected to also activate ammonia. In IMPDH, water is activated by Arg418 with the assistance of Thr321/Glu289. One might reasonably expect that this constellation of active site residues could also activate the amine leaving group of GMP. However, this would require the flap to close in the E•GMP complex. As noted above, the conformation of the Cys319 loop gates the closure of the flap. Perhaps the proper Cys319 conformation cannot form in the presence of GMP. Another possibility is that GMP induces a new conformation of the flap that is not catalytically competent, as is observed in the E•GMP complex of GMPR. It’s worth noting that Glu289 is substituted with Gln in eukaryotic IMPDHs, so these enzymes would never be expected to react with GMP.
The cofactor amide participates in the activation of the leaving group in the GMPR reaction-could the cofactor perform a similar role in IMPDH? Cofactor participation requires that the adoption of the “out” position- this conformational change involves a rotation about the 5′ carbon of the cofactor adenosine. Importantly, the cofactor binding site, as defined by structures of cofactor complexes of human IMPDH1, human IMPDH2 and T. foetus IMPDH, occupies a different portion of the barrel domain than in GMPR, so a similar movement of the cofactor is not possible (Figure 10A and B). Therefore it seems unlikely that the cofactor could assist in the deamination of GMP in IMPDH, at least in these enzymes.
Figure 10.

Cofactor binding sites in IMPDH and GMPR. A. Surface of cofactor binding site in T. foetus IMPDH (1lrt). The complex of IMP (sticks) and an NADH analog, tiazofurin adenine dinucleotide (TAD; ball and stick), is shown. B. Surface of the cofactor binding site in human GMPR2 (1c6q). IMP (sticks) and NADPH (ball and stick) are shown. C. Structure of the inhibitor binding site of C. parvum IMPDH (3khj). IMP (sticks) and inhibitor C64 (ball and stick) are shown. Note that C. parvum IMPDH lacks the pocket that interacts with the cofactor adenine in T. foetus IMPDH, but does have a similar pocket to the one that binds the adenine of NAPDH in human GMPR2. D. Structure of the adenine binding site is human GMPR2 (1c6q) in dark, overlayed with the analogous region of C. parvum IMPDH in light (3khj). The nicotinamide riboside portion of NADPH is omitted for clarity, as are the residues that interact with the 2′-phosphate. Light lines depict hydrogen bonds between NADPH and human GMPR2. A color version of this figure is available online.
Curiously, some prokaryotic IMPDHs appear to contain a site very similar to the cofactor binding site of GMPR. The NADP binding site extends into the neighboring subunit where the adenine group interacts with Tyr318′ (the ‘ denotes the second subunit; Figure 10). The analogous region of IMPDH was previously identified as a potential binding site for selective inhibitors of prokaryotic IMPDHs (Zhang et al., 1999). Such inhibitors have recently been identified (MacPherson et al., 2010, Gollapalli et al., 2010). The structure of the C. parvum IMPDH complex with one of these inhibitors, C64, has recently been solved (MacPherson et al., 2010). The inhibitor also crosses the subunit interface, where it interacts with a Tyr358. Many of the residues in the inhibitor binding site are shared with the cofactor binding site of GMPR (Figure 10C and D). These observations suggest that such bacterial IMPDHs may bind the NAD+ in the same manner that GMPR binds NADPH. If this is true, these enzymes may be able to catalyze both reactions.
Implications for metabolism and evolution
The physiological role of GMPR should be reconsidered in light of the observation that the reverse reaction producing GMP from IMP and ammonia is reasonably facile (contrary to some previous reports). The over-expression of E. coli GMPR can complement bacteria lacking IMPDH and attenuated in GMPS, demonstrating that GMPR is sufficient to support life in ammonia/ammonium rich environments such as the mammalian gut (Patton et al., 2011). Curiously, the insect gut contains ammonia/ammonium concentrations as high as 130 mM (Ji and Brune, 2006). Ammonia is membrane permeable, so intracellular concentrations are likely to be correspondingly high. Indeed, the synthesis of GMP appears to occur via GMPR in this environment. The aphid symbiont Buchnera, a close relative of E. coli, lacks both IMPDH and GMPS but contains a GMPR (van Ham et al., 2003). Buchnera uses ammonia to synthesize amino acids (Hansen and Moran, 2011), so it is reasonable to propose that ammonia is also used to synthesize guanine nucleotides. Approximately 20 bacteria/archaea appear to be missing GMPS, and may therefore also synthesize GMP directly from ammonia and IMP (The SEED, http://theseed.uchicago.edu/FIG/index.cgi accessed June 11, 2011).
The evolution of IMPDH/GMPR
The implications for the evolution of IMPDH and GMPR are also intriguing (Figure 11). Life is believed to have emerged in an ammonia-rich reductive environment (Zahnle et al., 2010), so the founding IMPDH/GMPR enzyme would likely have been a GMPR operating to produce GMP. This hypothesis is supported by the suggestion that GMPS arose after IMPDH (Kim et al., 2006). The installation of the Arg419 pathway for water activation would allow the emergence/specialization of IMPDH. Since cofactor conformational changes would no longer be required, the hydride transfer step could be optimized, and since the Glu is not required for the activation of water, this residue could be substituted with Gln. Migration of the cofactor binding site may represent a further specialization to the IMPDH reaction. It may be possible to test the feasibility of this pathway.
Figure 11.

Proposed pathway for the evolution of IMPDH and GMPR.
Challenges and opportunities
The above discussion spotlights several gaping holes in our understanding of the mechanism of IMPDH and GMPR. For IMPDH, ammonia/water selectivity, monovalent cation activation, protein conformation in ternary cofactor complexes and the pathway of protein conformational exchange are all open questions. More x-ray crystal structures are clearly needed. The E•GMP complex of IMPDH would provide important insights into reaction specificity. Cofactor complexes with monovalent cations are needed to construct a structural model of the catalytic cycle. More complexes of bacterial IMPDHs are necessary to determine if the cofactor site varies among IMPDHs from different sources. Methods to directly interrogate the conformational changes are also sorely needed. This wish list also applies to GMPR, with the addition that more GMPRs must be characterized.
The reaction-defining features of IMPDH and GMPR are dynamical. In IMPDH, the cofactor departs after the hydride transfer step and the flap moves, bringing the catalytic base Arg418 into the active site for the hydrolysis reaction. In contrast, in GMPR, the cofactor assumes different conformations during the deamination and hydride transfer reactions. These motions are influenced by residues acting at long distances from the active site, as vividly illustrated in the MPA-resistant IMPDHs from P. brevicompactum. These observations suggest that structural determinants of reaction specificity may lie far from the active site.
Gene annotation in the IMPDH/GMPR family
The difficulty of distinguishing IMPDH and GMPR can be illustrated with putative GMPR from Bacillus anthracis, which is 28% identical to E. coli GMPR, but 30% identical to E. coli IMPDH. Thus the potential for misannotation is very high. The annotation problem is compounded by an unfortunate typographical error that designated a likely IMPDH as inositol monophosphate dehydrogenase (while an inositol dehydrogenase has been identified, no actual inositol monophosphate dehydrogenase has been reported). This error has propagated- some genes have even been given dual annotations as both inosine and inositol monophosphate dehydrogenase.
Given the paucity of structural and functional characterization of GMPR, IMPDH/GMPR annotations should be viewed with caution. Nonetheless, there may be sequence motifs that can reliably distinguish IMPDH from GMPR. To date, only IMPDHs contain CBS domains. However, some IMPDH lack CBS domains, so absence of these domains is not sufficient to identify a GMPR. The Arg418-Tyr419 dyad in the flap also appears to be a unique feature of IMPDH, but the Tyr can be substituted with relatively little effect on enzymatic activity (Table 2; (Guillen Schlippe et al., 2004)), so the absence of this motif is probably not sufficient to identify a GMPR. The Tyr285-Arg286 dyad of GMPR, which interacts with the 2′-phosphate of NADPH, may prove to be a distinguishing feature. Lastly, GMPRs require the presence of the Glu289, so if Gln is in this position, the enzyme is an IMPDH. All of these potentially distinguishing features are derived from the characterization of rather few enzymes; more work is needed to demonstrating the generality of these findings so that these assignments can be made in confidence. Given the central connection of nucleotide metabolism and proliferation and the importance of IMPDH as a drug target, this is a goal worth pursuing.
Acknowledgments
The author thanks Jeff Boucher for the alignment of IMPDH and GMPR sequences.
Footnotes
Declaration of Interest
This work was supported by NIH grant GM054403 (LH). Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR001081).
References
- ANANTHARAMAN V, ARAVIND L, KOONIN EV. Emergence of diverse biochemical activities in evolutionarily conserved structural scaffolds of proteins. Curr Opin Chem Biol. 2003;7:12–20. doi: 10.1016/s1367-5931(02)00018-2. [DOI] [PubMed] [Google Scholar]
- ANDREWS SC, GUEST JR. Nucleotide sequence of the gene encoding the GMP reductase of Escherichia coli K12. Biochem J. 1988;255:35–43. doi: 10.1042/bj2550035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BATEMAN A. The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem Sci. 1997;22:12–13. doi: 10.1016/s0968-0004(96)30046-7. [DOI] [PubMed] [Google Scholar]
- BOWNE SJ, SULLIVAN LS, MORTIMER SE, HEDSTROM L, ZHU J, SPELLICY CJ, GIRE AI, HUGHBANKS-WHEATON D, BIRCH DG, LEWIS RA, HECKENLIVELY JR, DAIGER SP. Spectrum and frequency of mutations in IMPDH1 associated with autosomal dominant retinitis pigmentosa and leber congenital amaurosis. Invest Ophthalmol Vis Sci. 2006;47:34–42. doi: 10.1167/iovs.05-0868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BROX LW, HAMPTON A. Inactivation of guanosine 5′-monphosphate reductase by 6-chloro-, 6-mercapto-, and 2-amino-6-mercapto-9-b-D-ribofuranosyl purine 5′-nucleotides. Biochemistry. 1968;7:398–406. doi: 10.1021/bi00841a050. [DOI] [PubMed] [Google Scholar]
- CARLTON JM, HIRT RP, SILVA JC, DELCHER AL, SCHATZ M, ZHAO Q, WORTMAN JR, BIDWELL SL, ALSMARK UC, BESTEIRO S, SICHERITZ-PONTEN T, NOEL CJ, DACKS JB, FOSTER PG, SIMILLION C, VAN DE PEER Y, MIRANDA-SAAVEDRA D, BARTON GJ, WESTROP GD, MULLER S, DESSI D, FIORI PL, REN Q, PAULSEN I, ZHANG H, BASTIDA-CORCUERA FD, SIMOES-BARBOSA A, BROWN MT, HAYES RD, MUKHERJEE M, OKUMURA CY, SCHNEIDER R, SMITH AJ, VANACOVA S, VILLALVAZO M, HAAS BJ, PERTEA M, FELDBLYUM TV, UTTERBACK TR, SHU CL, OSOEGAWA K, DE JONG PJ, HRDY I, HORVATHOVA L, ZUBACOVA Z, DOLEZAL P, MALIK SB, LOGSDON JM, JR, HENZE K, GUPTA A, WANG CC, DUNNE RL, UPCROFT JA, UPCROFT P, WHITE O, SALZBERG SL, TANG P, CHIU CH, LEE YS, EMBLEY TM, COOMBS GH, MOTTRAM JC, TACHEZY J, FRASER-LIGGETT CM, JOHNSON PJ. Draft genome sequence of the sexually transmitted pathogen Trichomonas vaginalis. Science. 2007;315:207–212. doi: 10.1126/science.1132894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARR SF, PAPP E, WU JC, NATSUMEDA Y. Characterization of human type I and type II IMP dehydrogenases. J Biol Chem. 1993;268:27286–27290. [PubMed] [Google Scholar]
- CHEN L, PANKIEWICZ KW. Recent development of IMP dehydrogenase inhibitors for the treatment of cancer. Curr Opin Drug Discov Devel. 2007;10:403–12. [PubMed] [Google Scholar]
- CORNUEL JF, MORAILLON A, GUERON M. Participation of yeast inosine 5′-monophosphate dehydrogenase in an in vitro complex with a fragment of the C-rich telomeric strand. Biochimie. 2002;84:279–89. doi: 10.1016/s0300-9084(02)01400-1. [DOI] [PubMed] [Google Scholar]
- DENG Y, WANG Z, YING K, GU S, JI C, HUANG Y, GU X, WANG Y, XU Y, LI Y, XIE Y, MAO Y. NADPH-dependent GMP reductase isoenzyme of human (GMPR2). Expression, purification, and kinetic properties. Int J Biochem Cell Biol. 2002;34:1035–50. doi: 10.1016/s1357-2725(02)00024-9. [DOI] [PubMed] [Google Scholar]
- DIGITS JA, HEDSTROM L. Kinetic mechanism of Tritrichomonas foetus inosine-5′-monophosphate dehydrogenase. Biochemistry. 1999;38:2295–2306. doi: 10.1021/bi982305k. [DOI] [PubMed] [Google Scholar]
- DIGITS JA, HEDSTROM L. Drug selectivity is determined by coupling across the NAD+ site of IMP dehydrogenase. Biochemistry. 2000;39:1771–1777. doi: 10.1021/bi992288e. [DOI] [PubMed] [Google Scholar]
- EPPLER RK, KOMOR RS, HUYNH J, DORDICK JS, REIMER JA, CLARK DS. Water dynamics and salt-activation of enzymes in organic media: mechanistic implications revealed by NMR spectroscopy. Proc Natl Acad Sci U S A. 2006;103:5706–10. doi: 10.1073/pnas.0601113103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAN L, PETSKO GA, HEDSTROM L. Crystal structure of a ternary complex of Tritrichomonas foetus inosine 5′-monophosphate dehydrogenase: NAD+ orients the active site loop for catalysis. Biochemistry. 2002;41:13309–17. doi: 10.1021/bi0203785. [DOI] [PubMed] [Google Scholar]
- GAN L, SEYEDSAYAMDOST MR, SHUTO S, MATSUDA A, PETSKO GA, HEDSTROM L. The immunosuppressive agent mizoribine monophosphate forms a transition state analog complex with IMP dehydrogenase. Biochemistry. 2003;42:857–63. doi: 10.1021/bi0271401. [DOI] [PubMed] [Google Scholar]
- GERLT JA, RAUSHEL FM. Evolution of function in (beta/alpha)8-barrel enzymes. Curr Opin Chem Biol. 2003;7:252–64. doi: 10.1016/s1367-5931(03)00019-x. [DOI] [PubMed] [Google Scholar]
- GLASNER ME, GERLT JA, BABBITT PC. Evolution of enzyme superfamilies. Curr Opin Chem Biol. 2006;10:492–7. doi: 10.1016/j.cbpa.2006.08.012. [DOI] [PubMed] [Google Scholar]
- GOLLAPALLI DR, MACPHERSON IS, LIECHTI G, GORLA SK, GOLDBERG JB, HEDSTROM L. Structural determinants of inhibitor selectivity in prokaryotic IMP dehydrogenases. Chem Biol. 2010;17:1084–91. doi: 10.1016/j.chembiol.2010.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUILLEN SCHLIPPE YV, HEDSTROM L. Is Arg418 the catalytic base required for the hydrolysis step of the IMP dehydrogenase reaction? Biochemistry. 2005;44:11700–7. doi: 10.1021/bi048342v. [DOI] [PubMed] [Google Scholar]
- GUILLÉN SCHLIPPE YV, HEDSTROM L. Guanidine derivatives rescue the Arg418Ala mutation of Tritrichomonas foetus IMP dehydrogenase. Biochemistry. 2005a;44:16695–700. doi: 10.1021/bi051603w. [DOI] [PubMed] [Google Scholar]
- GUILLÉN SCHLIPPE YV, HEDSTROM L. A twisted base? The role of arginine in enzyme-catalyzed proton abstractions. Arch Biochem Biophys. 2005b;433:266–78. doi: 10.1016/j.abb.2004.09.018. [DOI] [PubMed] [Google Scholar]
- GUILLEN SCHLIPPE YV, RIERA TV, SEYEDSAYAMDOST MR, HEDSTROM L. Substitution of the conserved Arg-Tyr dyad selectively disrupts the hydrolysis phase of the IMP dehydrogenase reaction. Biochemistry. 2004;43:4511–21. doi: 10.1021/bi035823q. [DOI] [PubMed] [Google Scholar]
- GUNTER JH, THOMAS EC, LENGEFELD N, KRUGER SJ, WORTON L, GARDINER EM, JONES A, BARNETT NL, WHITEHEAD JP. Characterisation of inosine monophosphate dehydrogenase expression during retinal development: Differences between variants and isoforms. Int J Biochem Cell Biol. 2008;40:1716–1728. doi: 10.1016/j.biocel.2007.12.018. [DOI] [PubMed] [Google Scholar]
- HANSEN AK, MORAN NA. Aphid genome expression reveals host-symbiont cooperation in the production of amino acids. Proc Natl Acad Sci U S A. 2011;108:2849–54. doi: 10.1073/pnas.1013465108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HANSEN BG, SUN XE, GENEE HJ, KAAS CS, NIELSEN JB, MORTENSEN UH, FRISVAD JC, HEDSTROM L. Adaptive evolution of drug targets in producer and non-producer organisms. The Biochemical journal. 2011;441:219–26. doi: 10.1042/BJ20111278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEDSTROM L. IMP Dehydrogenase: structure, mechanism and inhibition. Chem Rev. 2009;109:2903– 2928. doi: 10.1021/cr900021w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEDSTROM L, GAN L. IMP dehydrogenase: structural schizophrenia and an unusual base. Curr Opin Chem Biol. 2006;10:520–5. doi: 10.1016/j.cbpa.2006.08.005. [DOI] [PubMed] [Google Scholar]
- HEDSTROM L, LIECHTI G, GOLDBERG JB, GOLLAPALLI DR. The antibiotic potential of prokaryotic IMP dehydrogenase inhibitors. Current medicinal chemistry. 2011;18:1909–18. doi: 10.2174/092986711795590129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEYDE E, NAGABHUSHANAM A, VONARX M, MORRISON J. Studies on inosine monophosphate dehydrogenase. Steady state kinetics. Biochim Biophys Acta. 1976;429:645–660. doi: 10.1016/0005-2744(76)90314-4. [DOI] [PubMed] [Google Scholar]
- HOLMES E, PEHLKE D, KELLEY W. Human IMP dehydrogenase. Kinetics and regulatory properties. Biochim Biophys Acta. 1974;364:209–217. doi: 10.1016/0005-2744(74)90006-0. [DOI] [PubMed] [Google Scholar]
- IGNOUL S, EGGERMONT J. CBS domains: structure, function, and pathology in human proteins. Am J Physiol Cell Physiol. 2005;289:C1369–78. doi: 10.1152/ajpcell.00282.2005. [DOI] [PubMed] [Google Scholar]
- JACKSON R, WEBER G, MORRIS HP. IMP dehydrogenase, an enzyme linked with proliferation and malignancy. Nature. 1975;256:331–333. doi: 10.1038/256331a0. [DOI] [PubMed] [Google Scholar]
- JAMSEN J, TUOMUNEN H, SALMINEN A, BELOGUROV GA, MAGRETOVA NN, BAYKOV AA, LAHTI R. A CBS domain-containing pyrophosphatase of Moorella thermoacetica is regulated by adenine nucleotides. Biochem J. 2007;408:327–333. doi: 10.1042/BJ20071017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JANOSIK M, KERY V, GAUSTADNES M, MACLEAN KN, KRAUS JP. Regulation of human cystathionine beta-synthase by S-adenosyl-L-methionine: evidence for two catalytically active conformations involving an autoinhibitory domain in the C-terminal region. Biochemistry. 2001;40:10625–33. doi: 10.1021/bi010711p. [DOI] [PubMed] [Google Scholar]
- JENTSCH TJ. CLC chloride channels and transporters: from genes to protein structure, pathology and physiology. Critical reviews in biochemistry and molecular biology. 2008;43:3–36. doi: 10.1080/10409230701829110. [DOI] [PubMed] [Google Scholar]
- JI R, BRUNE A. Nitrogen mineralization, ammonia accumulation, and emission of gaseous NH3 by soil-feeding termites. Biogeochemistry. 2006;78:267–283. [Google Scholar]
- JOSEPHINE HR, RAVICHANDRAN KR, HEDSTROM L. The Cys319 loop modulates the transition between dehydrogenase and hydrolase conformations in IMP dehydrogenase. Biochemistry. 2010;49:10674–81. doi: 10.1021/bi101590c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KERR KM, CAHOON MC, BOSCO DA, HEDSTROM L. Monovalent cation activation in Escherichia coli IMP dehydrogenase. Arch Biochem Biophys. 2000;375:131–137. doi: 10.1006/abbi.1999.1644. [DOI] [PubMed] [Google Scholar]
- KESSLER AI, GOTS JS. Regulation of guaC expression in Escherichia coli. Journal of bacteriology. 1985;164:1288–93. doi: 10.1128/jb.164.3.1288-1293.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIM HS, MITTENTHAL JE, CAETANO-ANOLLES G. MANET: tracing evolution of protein architecture in metabolic networks. BMC Bioinformatics. 2006;7:351. doi: 10.1186/1471-2105-7-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOHLER GA, GONG X, BENTINK S, THEISS S, PAGANI GM, AGABIAN N, HEDSTROM L. The functional basis of mycophenolic acid resistance in Candida albicans IMP dehydrogenase. J Biol Chem. 2005;280:11295–302. doi: 10.1074/jbc.M409847200. [DOI] [PubMed] [Google Scholar]
- KUEHNER JN, BROW DA. Regulation of a eukaryotic gene by GTP-dependent start site selection and transcription attenuation. Molecular cell. 2008;31:201–11. doi: 10.1016/j.molcel.2008.05.018. [DOI] [PubMed] [Google Scholar]
- LI J, WEI Z, ZHENG M, GU X, DENG Y, QIU R, CHEN F, JI C, GONG W, XIE Y, MAO Y. Crystal structure of human guanosine monophosphate reductase 2 (GMPR2) in complex with GMP. J Mol Biol. 2006;355:980–8. doi: 10.1016/j.jmb.2005.11.047. [DOI] [PubMed] [Google Scholar]
- LINK JO, STRAUB K. Trapping of an IMP dehydrogenase-substrate covalent interemediate by mycophenolic acid. J Am Chem Soc. 1996;118:2091–2092. [Google Scholar]
- LIU YC, LI F, HANDLER J, HUANG CR, XIANG Y, NERETTI N, SEDIVY JM, ZELLER KI, DANG CV. Global regulation of nucleotide biosynthetic genes by c-Myc. PloS one. 2008;3:e2722. doi: 10.1371/journal.pone.0002722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LO CONTE L, BRENNER SE, HUBBARD TJ, CHOTHIA C, MURZIN AG. SCOP database in 2002: refinements accommodate structural genomics. Nucleic Acids Res. 2002;30:264–7. doi: 10.1093/nar/30.1.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOOKER DL, MARR JJ, BERENS RL. Mechanisms of action of pyrazolopyrimidines in Leishmania donvani. J Biol Chem. 1986;261:9412–9415. [PubMed] [Google Scholar]
- MACKENZIE J, SORENSEN L. Guanosine 5′-phosphate reductase of human erythrocytes. Biochim Biophys Acta. 1973;327:282–294. doi: 10.1016/0005-2744(73)90411-7. [DOI] [PubMed] [Google Scholar]
- MACPHERSON IS, KIRUBAKARAN S, GORLA SK, RIERA TV, D’AQUINO JA, ZHANG M, CUNY GD, HEDSTROM L. The structural basis of Cryptosporidium -specific IMP dehydrogenase inhibitor selectivity. J Am Chem Soc. 2010;132:1230–1. doi: 10.1021/ja909947a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAGER J, MAGASANIK B. Guanosine 5′-phosphate reductase and its role in the interconversion of purine nucleotides. The Journal of biological chemistry. 1960;235:1474–8. [PubMed] [Google Scholar]
- MARTINELLI LK, DUCATI RG, ROSADO LA, BREDA A, SELBACH BP, SANTOS DS, BASSO LA. Recombinant Escherichia coli GMP reductase: kinetic, catalytic and chemical mechanisms, and thermodynamics of enzyme-ligand binary complex formation. Molecular bioSystems. 2011;7:1289–305. doi: 10.1039/c0mb00245c. [DOI] [PubMed] [Google Scholar]
- MCLEAN JE, HAMAGUCHI N, BELENKY P, MORTIMER SE, STANTON M, HEDSTROM L. Inosine 5′-monophosphate dehydrogenase binds nucleic acids in vitro and in vivo. Biochem J. 2004;379:243–251. doi: 10.1042/BJ20031585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIN D, JOSEPHINE HR, LI H, LAKNER C, MACPHERSON IS, NAYLOR GJ, SWOFFORD D, HEDSTROM L, YANG W. An enzymatic atavist revealed in dual pathways for water activation. PLoS Biol. 2008;6:e206. doi: 10.1371/journal.pbio.0060206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MINEGISHI S, MAYR H. How constant are Ritchie’s “constant selectivity relationships”? A general reactivity scale for n-, pi-, and sigma-nucleophiles. Journal of the American Chemical Society. 2003;125:286–295. doi: 10.1021/ja021010y. [DOI] [PubMed] [Google Scholar]
- MOFFAT K, MACKINNON G. Cloning of the Escherichia coli K-12 guaC gene following its transposition into the RP4::Mu cointegrate. Gene. 1985;40:141–143. doi: 10.1016/0378-1119(85)90034-4. [DOI] [PubMed] [Google Scholar]
- MORRISON HG, MCARTHUR AG, GILLIN FD, ALEY SB, ADAM RD, OLSEN GJ, BEST AA, CANDE WZ, CHEN F, CIPRIANO MJ, DAVIDS BJ, DAWSON SC, ELMENDORF HG, HEHL AB, HOLDER ME, HUSE SM, KIM UU, LASEK-NESSELQUIST E, MANNING G, NIGAM A, NIXON JE, PALM D, PASSAMANECK NE, PRABHU A, REICH CI, REINER DS, SAMUELSON J, SVARD SG, SOGIN ML. Genomic minimalism in the early diverging intestinal parasite Giardia lamblia. Science. 2007;317:1921–1926. doi: 10.1126/science.1143837. [DOI] [PubMed] [Google Scholar]
- MORTIMER SE, HEDSTROM L. Autosomal dominant retinitis pigmentosa mutations in inosine 5′-monophosphate dehydrogenase type I disrupt nucleic acid binding. Biochem J. 2005;390:41–7. doi: 10.1042/BJ20042051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MORTIMER SE, XU D, MCGREW D, HAMAGUCHI N, LIM HC, BOWNE SJ, DAIGER SP, HEDSTROM L. IMP Dehydrogenase Type 1 Associates with Polyribosomes Translating Rhodopsin mRNA. J Biol Chem. 2008;283:36354–60. doi: 10.1074/jbc.M806143200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MUSA-AZIZ R, CHEN LM, PELLETIER MF, BORON WF. Relative CO2/NH3 selectivities of AQP1, AQP4, AQP5, AmtB, and RhAG. Proc Natl Acad Sci U S A. 2009;106:5406–11. doi: 10.1073/pnas.0813231106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAGANO N, ORENGO CA, THORNTON JM. One fold with many functions: the evolutionary relationships between TIM barrel families based on their sequences, structures and functions. J Mol Biol. 2002;321:741–65. doi: 10.1016/s0022-2836(02)00649-6. [DOI] [PubMed] [Google Scholar]
- NAIR V, SHU Q. Inosine monophosphate dehydrogenase as a probe in antiviral drug discovery. Antivir Chem Chemother. 2007;18:245–258. doi: 10.1177/095632020701800501. [DOI] [PubMed] [Google Scholar]
- NIMMESGERN E, BLACK J, FUTER O, FULGHUM JR, CHAMBERS SP, BRUMMEL CL, RAYBUCK SA, SINTCHAK MD. Biochemical analysis of the modular enzyme inosine monophosphate dehydrogenase. Protein Expression and Purification. 1999;17:282–289. doi: 10.1006/prep.1999.1136. [DOI] [PubMed] [Google Scholar]
- OLAH E, KOKENY S, PAPP J, BOZSIK A, KESZEI M. Modulation of cancer pathways by inhibitors of guanylate metabolism. Adv Enzyme Regul. 2006;46:176–90. doi: 10.1016/j.advenzreg.2006.01.002. [DOI] [PubMed] [Google Scholar]
- ORENGO CA, MICHIE AD, JONES S, JONES DT, SWINDELLS MB, THORNTON JM. CATH--a hierarchic classification of protein domain structures. Structure. 1997;5:1093–108. doi: 10.1016/s0969-2126(97)00260-8. [DOI] [PubMed] [Google Scholar]
- PANKIEWICZ KW, PATTERSON SE, BLACK PL, JAYARAM HN, RISAL D, GOLDSTEIN BM, STUYVER LJ, SCHINAZI RF. Cofactor mimics as selective inhibitors of NAD-dependent inosine monophosphate dehydrogenase (IMPDH)--the major therapeutic target. Curr Med Chem. 2004;11:887–900. doi: 10.2174/0929867043455648. [DOI] [PubMed] [Google Scholar]
- PARK JH, AHN SH. IMP dehydrogenase is recruited to the transcription complex through serine 2 phosphorylation of RNA polymerase II. Biochem Biophys Res Commun. 2010;392:588–92. doi: 10.1016/j.bbrc.2010.01.079. [DOI] [PubMed] [Google Scholar]
- PATTON GC, STENMARK P, GOLLAPALLI DR, SEVASTIK R, KURSULA P, FLODIN S, SCHULER H, SWALES CT, EKLUND H, HIMO F, NORDLUND P, HEDSTROM L. Cofactor mobility determines reaction outcome in the IMPDH/GMPR (β/α)8 barrel enzymes. Nature Chem Biol. 2011;7:950–8. doi: 10.1038/nchembio.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PIMKIN M, MARKHAM GD. The CBS subdomain of inosine 5′-monophosphate dehydrogenase regulates purine nucleotide turnover. Molecular Microbiology. 2008;69:342–359. doi: 10.1111/j.1365-2958.2008.06153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PIMKIN M, PIMKINA J, MARKHAM GD. A regulatory role of the Bateman domain of IMP dehydrogenase in adenylate nucleotide biosynthesis. J Biol Chem. 2009;284:7960–9. doi: 10.1074/jbc.M808541200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PROSISE GL, LUECKE H. Crystal Structures of Tritrichomonas foetus Inosine Monophosphate Dehydrogenase in Complex with Substrate, Cofactor and Analogs: A Structural Basis for the Random-in Ordered-out Kinetic Mechanism. J Mol Biol. 2003;326:517–27. doi: 10.1016/s0022-2836(02)01383-9. [DOI] [PubMed] [Google Scholar]
- PROSISE GL, WU JZ, LUECKE H. Crystal Structure of Tritrichomonas foetus Inosine Monophosphate Dehydrogenase in Complex with the Inhibitor Ribavirin Monophosphate Reveals a Catalysis-dependent Ion-binding Site. J Biol Chem. 2002;277:50654–9. doi: 10.1074/jbc.M208330200. [DOI] [PubMed] [Google Scholar]
- RATCLIFFE AJ. Inosine 5′-monophosphate dehydrogenase inhibitors for the treatment of autoimmune diseases. Curr Opin Drug Discov Devel. 2006;9:595–605. [PubMed] [Google Scholar]
- RENART MF, RENART J, SILLERO MA, SILLERO A. Guanosine monophosphate reductase from Artemia salina: Inhibition by xanthosine monophosphate and activation by diguanosine tetraphosphate. Biochemistry. 1976a;15:4962–6. doi: 10.1021/bi00668a003. [DOI] [PubMed] [Google Scholar]
- RENART MF, RENART J, SILLERO MAG, SILLERO A. Guanosine monphosphate dehydrogenase from Artemia salina: inhibition by xanthosine monophosphate and activation by diguanosine tetraphosphate. Biochemistry. 1976b;15:4962–4966. doi: 10.1021/bi00668a003. [DOI] [PubMed] [Google Scholar]
- RHODES MM, REBLOVA K, SPONER J, WALTER NG. Trapped water molecules are essential to structural dynamics and function of a ribozyme. Proc Natl Acad Sci U S A. 2006;103:13380–5. doi: 10.1073/pnas.0605090103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RIERA TV, WANG W, JOSEPHINE HR, HEDSTROM L. A kinetic alignment of orthologous inosine-5′-monophosphate dehydrogenases. Biochemistry. 2008;47:8689–96. doi: 10.1021/bi800674a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RIERA TV, ZHENG L, JOSEPHINE HR, MIN D, YANG W, HEDSTROM L. Allosteric Activation via Kinetic Control: Potassium Accelerates a Conformational Change in IMP Dehydrogenase. Biochemistry. 2011;50:8508–18. doi: 10.1021/bi200785s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SALVATORE D, BARTHA T, LARSEN PR. The guanosine monophosphate reductase gene is conserved in rats and its expression increases rapidly in brown adipose tissue during cold exposure. J Biol Chem. 1998;273:31092–6. doi: 10.1074/jbc.273.47.31092. [DOI] [PubMed] [Google Scholar]
- SCOTT JW, HAWLEY SA, GREEN KA, ANIS M, STEWART G, SCULLION GA, NORMAN DG, HARDIE DG. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J Clin Invest. 2004;113:274–84. doi: 10.1172/JCI19874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SINTCHAK MD, FLEMING MA, FUTER O, RAYBUCK SA, CHAMBERS SP, CARON PR, MURCKO M, WILSON KP. Structure and mechanism of inosine monophosphate dehydrogenase in complex with the immunosuppressant mycophenolic acid. Cell. 1996;85:921–930. doi: 10.1016/s0092-8674(00)81275-1. [DOI] [PubMed] [Google Scholar]
- SNYDER F, HENDERSON J, KIM S, PATERSON A, BROX L. Purine nucleotide metabolism and nucleotide pool sizes in synchronized lymphoma L5178Y cells. Cancer Res. 1973;33:2425–2430. [PubMed] [Google Scholar]
- SOSKINE M, TAWFIK DS. Mutational effects and the evolution of new protein functions. Nat Rev Genet. 2010;11:572–82. doi: 10.1038/nrg2808. [DOI] [PubMed] [Google Scholar]
- SPECTOR T, JONES TE. Guanosine 5′-monophosphate reductase from Leishmania donovani. A possible chemotherapeutic target. Biochem Pharmacol. 1982;31:3891–7. doi: 10.1016/0006-2952(82)90307-0. [DOI] [PubMed] [Google Scholar]
- SPECTOR T, JONES TE, LAFON SW, NELSON DJ, BERENS RL, MARR JJ. Monophosphates of formycin B and allopurinol riboside. Interactions with leishmanial and mammalian succino-AMP synthetase and GMP reductase. Biochem Pharmacol. 1984;33:1611–7. doi: 10.1016/0006-2952(84)90282-x. [DOI] [PubMed] [Google Scholar]
- SPECTOR T, JONES TE, MILLER RL. Reaction mechanism and specificity of human GMP reductase. Substrates, inhibitors, activators, and inactivators. J Biol Chem. 1979;254:2308–15. [PubMed] [Google Scholar]
- SPELLICY CJ, DAIGER SP, SULLIVAN LS, ZHU J, LIU Q, PIERCE EA, BOWNE SJ. Characterization of retinal inosine monophosphate dehydrogenase 1 in several mammalian species. Mol Vis. 2007;13:1866–72. [PubMed] [Google Scholar]
- STEPHENS RW, WHITTAKER VK. Calf thymus GMP reductase: control by XMP. Biochem Biophys Res Commun. 1973;53:975–81. doi: 10.1016/0006-291x(73)90187-3. [DOI] [PubMed] [Google Scholar]
- THOMAS M, DRABBLE W. Molecular cloning and characterisation of the gua regulatory region of Escherichia coli K12. Mol Gen Genet. 1984;195:238–245. doi: 10.1007/BF00332753. [DOI] [PubMed] [Google Scholar]
- VAN HAM RC, KAMERBEEK J, PALACIOS C, RAUSELL C, ABASCAL F, BASTOLLA U, FERNANDEZ JM, JIMENEZ L, POSTIGO M, SILVA FJ, TAMAMES J, VIGUERA E, LATORRE A, VALENCIA A, MORAN F, MOYA A. Reductive genome evolution in Buchnera aphidicola. Proc Natl Acad Sci U S A. 2003;100:581–6. doi: 10.1073/pnas.0235981100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG W, HEDSTROM L. The kinetic mechanism of human inosine 5′-monophosphate type II: random addition of substrates, ordered release of products. Biochemistry. 1997;36:8479–8483. doi: 10.1021/bi970226n. [DOI] [PubMed] [Google Scholar]
- WEBER G, NAKAMURA H, NATSUMEDA Y, SZEKERES T, NAGAI M. Regulation of GTP biosynthesis. Adv Enzyme Regul. 1992;32:57–69. doi: 10.1016/0065-2571(92)90008-n. [DOI] [PubMed] [Google Scholar]
- WISE EL, RAYMENT I. Understanding the importance of protein structure to nature’s routes for divergent evolution in TIM barrel enzymes. Acc Chem Res. 2004;37:149–58. doi: 10.1021/ar030250v. [DOI] [PubMed] [Google Scholar]
- XIANG B, TAYLOR JC, MARKHAM GD. Monovalent cation activation and kinetic mechanism of inosine 5′-monophosphate dehydrogenase. J Biol Chem. 1996;271:1435–1440. doi: 10.1074/jbc.271.3.1435. [DOI] [PubMed] [Google Scholar]
- ZAHNLE K, SCHAEFER L, FEGLEY B. Earth’s earliest atmospheres. Cold Spring Harb Perspect Biol. 2010;2:a004895. doi: 10.1101/cshperspect.a004895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZALATAN JG, HERSCHLAG D. The far reaches of enzymology. Nat Chem Biol. 2009;5:516–20. doi: 10.1038/nchembio0809-516. [DOI] [PubMed] [Google Scholar]
- ZHANG J, ZHANG W, ZOU D, CHEN G, WAN T, ZHANG M, CAO X. Cloning and functional characterization of GMPR2, a novel human guanosine monophosphate reductase, which promotes the monocytic differentiation of HL-60 leukemia cells. J Cancer Res Clin Oncol. 2003;129:76–83. doi: 10.1007/s00432-002-0413-7. [DOI] [PubMed] [Google Scholar]
- ZHANG R, GE, ROTELLA F, WESTBROOK E, HUBERMAN E, JOACHIMIAK A, COLLART FR. Differential signatures of bacterial and mammalian IMP dehydrogenase enzymes. Curr Med Chem. 1999;6:537–43. [PubMed] [Google Scholar]