

Abstract
There remains an ongoing need for fast, highly sensitive, and quantitative technologies that can detect and profile rare cells in freshly harvested samples. Recent developments in nanomaterial-based detection platforms provide advantages over traditional approaches in terms of signal sensitivity, stability, and the possibility for performing multiplexed measurements. Here, we describe a bioorthogonal, nanoparticle amplification technique capable of rapid augmentation of detection sensitivities by up to 1-2 orders of magnitude over current methods. This improvement in sensitivity was achieved by i) significantly reducing background noise arising from non-specific nanoparticle binding, ii) increasing nanomaterial binding through orthogonal rounds of amplification, and iii) implementing a cleavage step to improve assay robustness. The developed method allowed sensitive detection and molecular profiling of scant tumor cells directly in unpurified human clinical samples such as ascites. With its high sensitivity and simplified assay steps, this technique will likely have broad utility in nanomaterial-based diagnostics.
Keywords: nanoparticles, targeting, orthogonal chemistry, tetrazine, NMR, diagnostics, cancer
Nanoparticles (NPs) of different sizes, shapes and compositions have been increasingly employed for in vitro diagnostics.1,2 NP-based sensing technologies are often more sensitive than small molecule sensors, due to their multivalency,3 exploitation of novel physical effects,4,5 simplified purification and analysis, and because assays can be multiplexed. Recently, enormous progress has been made in developing NPs with unique optical or magnetic signatures. For example, advanced gold/silver clusters1,6 and newer doped ferrites with high magnetization can detect analytes within the femtomolar (fM) range.7,8 However, there is still a gap between current detection limits and the abundance of biological targets, which requires either purification and concentration, or amplification. This is especially the case in clinical diagnostic settings, such as cancer9-11 or infectious diseases,12-14 where detection of rare targets (e.g., cells or bacteria) in clinical samples is necessary.
A variety of amplification methods have been previously described; these include two-step methods (avidin-biotin, click chemistry15,16), DNA-templated amplification17 and supramolecular host chemistry.18 Based on newer cycloaddition chemistries for rapid conjugation, we hypothesized that multiple steps of alternating orthogonal chemistries could be used as an alternative amplification method with higher sensitivity. Of particular interest are {4+2} cycloaddition reactions, which are extremely fast and selective, and for which a number of orthogonal reaction partners have already been described.19,20 Unlike DNA methods, these cycloadditions do not require sample heating (annealing) nor do they require sensitive polymerases or catalysts. Here, we describe the systematic exploration of one such method. Specifically, we investigated the effect of repeated rounds of orthogonal NP labeling on amplification (signal over noise), and the implementation of an additional cleavage modification that would confer synergistic improvements to the assay's performance. We show that this optimized labeling method significantly improves detection sensitivities of nuclear magnetic resonance (NMR)-based sensing (diagnostic magnetic resonance, DMR). Unlike conventional methods such as flow cytometry, whose uses are often limited due to time-consuming sample-purification and accompanying cell loss, this new labeling strategy allowed cancer cells to be detected and molecularly profiled in unpurified clinical samples. We expect that this new technique will have broad applications in future nanomaterial-based diagnostics.
Results
New labeling strategy for high detection sensitivity
Figure 1 summarizes the scheme of the developed labeling method. We hypothesized that the cellular loading of nanoagents, specifically magneto-fluorescent nanoparticles (MFNPs), could be maximized via the sequential application of MFNPs conjugated with orthogonal binding partners. Specifically, to form an initial MFNP layer, cellular targets were first labeled with antibodies modified with trans-cyclooctene (TCO) before being coupled with MFNPs derivatized with tetrazine (Tz).15 This primary labeling can then be amplified through alternating applications of MFNP-TCO (Amplification 1; AMP1) and MFNP-Tz (AMP2) to form multiple MFNP layers. In addition to amplification, bound MFNPs could also be released from cells, collected and resuspended in buffer prior to performing analytical measurements. In so doing, it is theoretically possible to confer improved detection sensitivity and reliability by 1) eliminating biological contaminants (e.g., cellular debris, components of extracellular matrix, non-targeted cells), and 2) reducing measurement artifacts caused by the sedimentation of labeled cells. In order to provide such functionality, the orthogonal reactants (TCO and Tz) were immobilized onto the MFNP surface through a cleavable linker (e.g., disulfide bond; see Methods for details).
Figure 1. Schematic of the labeling strategy used to amplify biomarker signals.
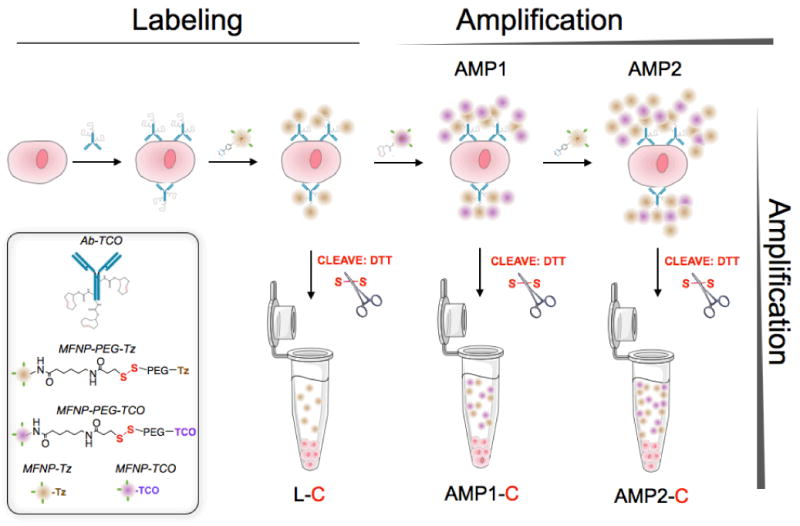
The labeling step (L) refers to the initial antibody-trans-cyclooctene (TCO) conjugate binding to the target followed by the addition of magneto-fluorescent nanoparticles (MFNPs) conjugated to the orthogonal reactant, tetrazine (Tz). The signal can be subsequently amplified through additional rounds of complementary orthogonal MFNP conjugates (AMP1, AMP2) and through cleavage/purification using dithiothreitol (DTT; AMP1-C, AMP2-C).
Our first goal was to optimize both the NPs as well as the labeling protocols. Cancer cells (SK-OV-3, human ovarian carcinoma) overexpressing HER2 (human epidermal growth factor receptor 2; ∼1×106 receptors per cell) were used as a model cell line. Anti-HER2 antibodies (trastuzumab) were first modified with TCO (HER2-TCO; with each antibody bearing ∼ 20 TCO 15). Two types of orthogonal MFNP were then prepared: one with Tz directly conjugated to the particles (MFNP-Tz) and the other with a polyethylene glycol (PEG) spacer between the particle and the orthogonal reactant (MFNP-PEG-Tz). The PEGylation was expected to minimize non-specific MFNP binding to cells. Indeed, when SK-OV-3 cells (in the absence of the primary antibody labeling step) were incubated with MFNP-PEG-Tz, the background signal from nonspecific binding was significantly smaller (>20-fold) than that of MFNP-Tz (Figure 2A). SK-OV-3 cells targeted with HER2-TCO followed by the application of either MFNP-Tz or MFNP-PEG-Tz showed similar dose-dependent responses (Figure 2B). With MFNP-PEG-Tz, however, the background signal remained significantly low (Figure 2B), which in turn increased the achievable signal-to-noise ratio (SNR; Figure 2C). Note that keeping the background signal low is critical to the amplification strategy, as it prevents SNR degeneration during multiple rounds of MFNP-Tz / MFNP-TCO layering.
Figure 2. Effect of nanoparticle (NP) PEGylation on signal-to-noise ratio (SNR).
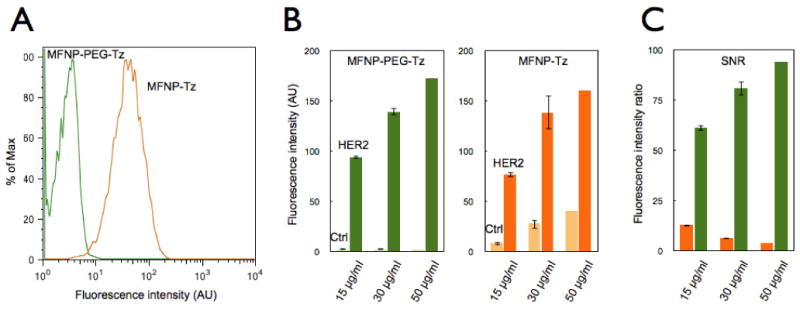
(A) Flow cytometry experiments comparing SK-OV-3 cells (in the absence of the primary antibody labeling step) incubated with either MFNP-PEG-Tz (green) or MFNP-Tz (orange) for 15 minutes to determine nonspecific cellular binding. PEGylated particles displayed significantly reduced nonspecific cellular binding. (B) Dose response graphs of specific cellular (i.e. HER2 targeting) versus nonspecific cellular binding with different NPs. With PEGylated MFNPs (left panel), non-specific binding is kept at a minimum non-significant level, whereas the non-specific binding increases in a dose-dependent manner with non-PEGylated MFNPs (right panel). (C) The overall signal-to-noise ratio (SNR) with PEGylated (green) and non-PEGylated (orange) MFNPs.
Improved robustness and sensitivity through cleavage
After labeling, we next quantitated the effects of MFNP-cleavage on detection sensitivity. Following primary cell labeling with HER2-TCO and MFNP-PEG-Tz, SK-OV-3 cells were further treated with MFNP-PEG-TCO (AMP1). Cell-bound MFNPs were then released by cleaving disulfide linkers (AMP1-C), and separated from cellular contents via centrifugation. The transverse relaxation rate (R2) of samples was subsequently measured by DMR. The cell-number matched comparisonshowed a significantly higher R2 following the cleaving method (Figure 3A; >200% enhancement in SNR). The observed high R2 is presumably due to an increase in particle size, as a result of inter-particle clustering between MFNP-PEG-Tz and MFNP-PEG-TCO. It has previously been shown that clusters of small magnetic NPs are more efficient at accelerating NMR signal decay and thereby result in higher R2.5,21-23 Further measurement of particle size by dynamic light scattering supported this hypothesis. The cleaved materials (AMP1-C) had a monodisperse hydrodynamic diameter of ∼100 nm, whereas the size of the original particle was ∼30 nm. Note that the effect of clustering is more pronounced in the AMP1-C stage as particles are free in suspension, and thus able to further interact with surrounding water molecules. The higher R2 and homogeneous dispersion of MFNPs in solution rendered the cleaving method more sensitive (>10 times) and robust than direct cellular detection (Figure 3B). When subsequently compared with conventional flow cytometry, the gold standard for cellular detection, the cleave-based DMR technique showed an excellent correlation (R2 = 0.99), a finding that validated its analytical capacity (Figure 3C).
Figure 3. Comparison of the cleavage method to whole cell detection.
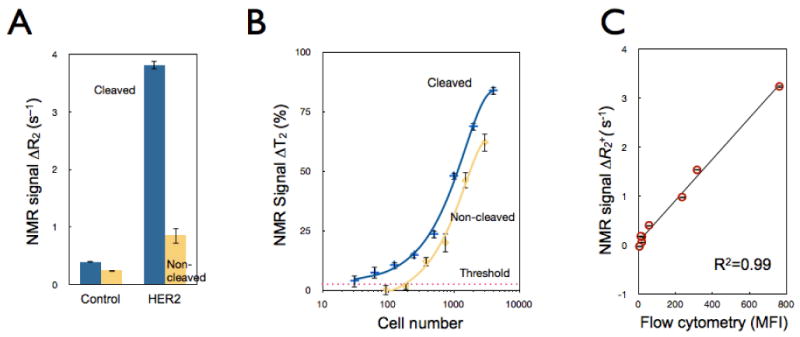
(A) Comparative NMR signals for HER2-targeted SK-OV-3 cells using the cleave (blue) versus the non-cleave (yellow) method (∼3,500 cells); control samples were incubated with NPs alone. (B) Detection sensitivity of SK-OV-3 cancer cells using the AMP1 and AMP1-C methods (See Figure 1). Note the ∼10-fold increase in detection sensitivity following the cleavage method. Data are expressed as a mean ± standard deviation. (C) Comparative detection between the NMR-based cleavage method and flow cytometry demonstrated an excellent correlation (R2=0.99). Each data point represents different target expression levels (EGFR, EpCAM, HER2, MUC1) across two model cell lines (SK-OV-3, SK-BR-3). NMR and flow cytometry detection volumes contained 3,500 and 35,000 cells, respectively. MFI: mean fluorescent intensity.
Multiple amplification rounds yield higher SNRs
We next characterized the signal amplification strategy through multiple applications of MFNPs. Cancer cells (SK-BR-3), primarily labeled with HER2-TCO and MFNP-PEG-Tz, were incubated with alternating applications of MFNPs and their orthogonal binding partners: MFNP-PEG-TCO (AMP1), MFNP-PEG-Tz (AMP2). Figure 4A shows fluorescent micrographs of labeled cells, where the labeling and amplification steps were made distinguishable by conjugating different fluorescent dyes to the MFNPs. The images show excellent co-localization between these steps, and thereby confirm that the layering indeed amplifies the primary target and not other cellular structures/processes. Equally important was the finding that the MFNP-cleaved cells display negligible fluorescent signals, suggesting that there is maximal MFNP release into suspension. The DMR assays were performed using MFNP samples that had been cleaved from SK-OV-3 cells after each labeling and amplification step. Successive increases in R2 were observed with each round of amplification (Figure 4B); there was likewise a corresponding improvement in detection sensitivities (Figure 4C).
Figure 4. Comparison of successive amplification rounds.
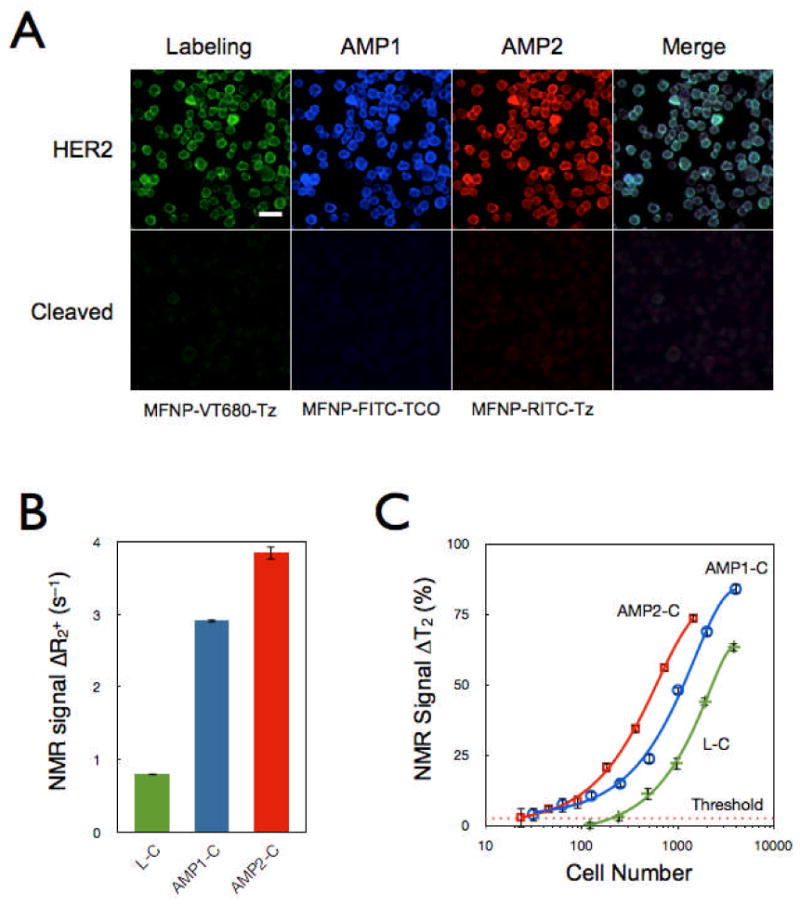
(A) Fluorescent signals detected from the initial labeling step and for each subsequent amplification step showed excellent co-localization, confirming that multiple MFNP layers can be applied to cellular targets for signal amplification. The MFNP-cleaved cells displayed negligible fluorescent signal, suggesting maximal MFNP release into suspension. The scale bar represents 30 μm. (B) Comparative NMR signal for HER2-targeted SK-OV-3 cells (3,500 cells). AMP2-C conferred the largest increase in SNR but AMP1-C had the highest SNR/unit time. (C) Cellular detection threshold for different cleavage methods (Labeling, AMP1, and AMP2; see Fig. 1) based on HER2 targeting of SK-OV-3 cells.
Application to Clinical Samples
The overall goal of the study was to improve assay sensitivity and robustness in native clinical samples (e.g., fine needle aspirates, biopsies, ascites, blood, sputum), which are inherently complex in composition, as well as heterogeneous and variable in cell number.24 We therefore tested our new method for cancer cell detection in malignant human ascites from patients with pancreatic cancer. Samples were divided into two sets, one for the non-cleaving (AMP1) and the other for the cleaving method (AMP1-C). For each set, samples were screened for EGFR (epidermal growth factor receptor), EpCAM (epithelial cell adhesion molecule), HER2, and MUC1 (mucin-1) biomarkers. The cleaving approach was found to produce superior results, revealing otherwise barely or undetectable markers (e.g., MUC1 and EpCAM; Figure 5A). Integral to this method's successful detection of low levels of biomarkers is the preferential amplification of signals emanating from target rather than from background, which effectively maximizes the SNRs.
Figure 5. Processing of primary clinical samples.
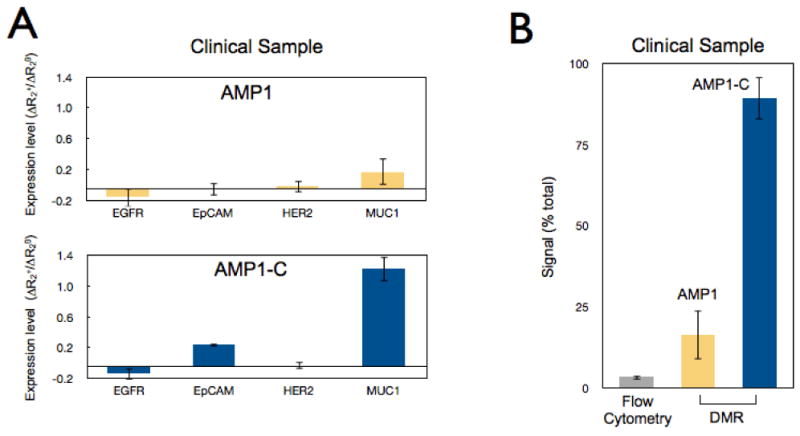
(A) Human clinical ascites from pancreatic cancer was profiled for 4 different biomarkers: EGFR, EpCAM, HER2, and MUC1. The AMP1 labeling method was then assessed with and without the subsequent cleavage step. The cleavage step generated higher signal levels. (B) Comparison of different analytical methods (flow cytometry, diagnostic magnetic resonance) for MUC1 detection in clinical samples. Error bars represent the standard error from at least three NMR measurements.
For this study, we tested both purified and non-purified samples to reflect the clinical reality and clinical need, respectively. Purification in our study was achieved by negatively selecting CD45+ cells, which comprise ∼90% of the total cell concentration in the samples. Due to significant and inevitable cell losses (often >40% of the initial cell number), it is always advantageous to avoid or minimize sample purification steps, especially when dealing with complex clinical samples. In both purified and non-purified specimens, MUC1 was labeled using non-cleaving (AMP1) and cleaving (AMP1-C) techniques. DMR measurements of AMP1-C generated the least signal variation between the non-purified and purified sets. Importantly, the non-purified signal was ∼90% of the purified signal. Eliminating the cleaving steps (AMP1) reduced the non-purified signal to 16% of the purified signal, as measured by DMR, and to 3% of the purified signal, as measured by flow cytometry (Figure 5B). In summary, the cleaving method enhanced detection in unaltered samples, and thus obviated the need for purification steps. Flow cytometry, however, clearly benefits from the inclusion of a purification step when detecting and profiling scarce cells in heterogeneous biological samples such as human derived specimens.
Discussion
Utilizing the cycloaddition chemistry for signal amplification, we have developed a new NP-based diagnostic strategy for higher detection sensitivity and robustness. The method relies on i) increasing the number of NPs bound to the target for signal amplification and ii) cleavage of the NP from its target prior to measurement. Amplification is achieved by labeling with successive rounds of complementary TCO and Tz NPs. However, we found that this strategy only works well when background noise remains low. We achieved this by using PEG spacers on the NP surface to minimize non-specific binding of the NP. The cleavage of NPs from labeled cells further increased the detection sensitivity by over an order of magnitude. This was likely as a result of a) the MFNPs being surrounded by large numbers of water molecules, which could increase the R2; and b) cleaved MFNP-Tz and MFNP-TCO forming clusters, which could also increase the R2. In addition to enhancing sensitivity, the cleavage method also improves the detection reliability; the measurement is free from artifacts caused either by cell sedimentation or by the presence of clumps/extraneous matter often found in clinical samples. Finally, the cleavage method simplifies operation. Results are highly reproducible and longitudinal samples do not have to be analyzed in real time (unless desired). Rather, measurements can be done at the investigator's convenience since, unlike labeled cells where variations can occur due to dissociation of the MFNPs from cells, cleaved MFNPs are stable and do not vary over time. Lastly, because the analytical measurement does not require the cells after the cleavage step, it is possible that rare cells could be relabeled for other biomarkers. For instance, samples could be first profiled for a less abundant marker (MFNP labeling and amplification followed by cleavage); the same sample then can be screened for more abundant markers using the same MFNP labeling and cleavage strategy.17
Conclusion
We envision a variety of applications for this technology. While originally developed and optimized for robust cellular analyses and measurements in ascitic fluid, we anticipate that this method could likewise be applied to fine needle aspirates, blood, biopsy specimens, sputum and other biological sources. A particularly interesting application is the possibility of performing multiplexed measurements of rare cells such as circulating cancer cells, immune cell subpopulations or stem cells. Finally, we envision that this novel method could be applied to other profiling methods and nanomaterials. For example, the method could be adapted to ELISA-based MFNP approaches to enable detection of soluble markers in blood or urine. In such assays, the soluble marker would be first captured on micron sized polystyrene beads or microtiter plates before undergoing an AMP2-C procedure to augment sensitivities. It is also possible that this method could be adapted to non-magnetic NPs, i.e. using particles detectable by light sensing25 or by plasmon resonance techniques.26-28
Materials and Methods
Preparation of cleavable pegylated Tz and TCO nanoparticles (NPs)
Magnetofluorescent nanoparticles (MFNP) were synthesized by reacting cross-linked iron oxide (CLIO) NPs with amine reactive cyanine dyes (VT-680xl, Perkin Elmer), as previously described.15 The amino-MFNP contained approximately 62 primary amine groups and ∼7 VT-680 molecules conjugated to the surface. The hydrodynamic diameter was 27 nm, as determined by dynamic light scattering (Zetasizer 1000HS; Malvern Instruments), and the r1 and r2 relaxivities were 26.3 and 52.3 mM-1 s-1, respectively, at 40°C and 0.47 T (Minispec MQ20; Bruker). MFNP molar concentration was determined based on an estimated molecular mass of 447,000 daltons (8000 Fe atoms per core crystal, 55.85 daltons per Fe atom29,30).
MFNPs with a polyethylene glycol (PEG) spacer between the particle and the orthogonal reactant were prepared in a three-step process. First, the MFNPs were reacted with 2000 molar equivalents (relative to the MFNPs) of sulfosuccinimidyl 6-[3′(2-pyridyldithio)-propionamido] hexanoate (sulfo- LC-SPDP, Thermo Scientific) in phosphate buffered saline (PBS) for 1.5 hours at room temperature. Excess sulfo-LC-SPDP was removed using a 100 kD ultracentrifugation unit (Amicon), and washed three times with PBS at 1800 rcf for 15 minutes. In the second step, 2000 molar equivalents of thiol-PEG-amine (3.4 kDA, Creative Pegworks) relative to the MFNPs, were reacted in PBS and aged overnight at 4°C on a shaker. Excess thiol-PEG-amine was removed using a 100 kDa ultracentrifugation unit (Amicon), and washed three times with PBS at 1800 rcf for 15 minutes. In the third step, amine-PEG terminated MFNPs were modified with either 2,5-dioxo-pyrrolidin-1-yl 5-(4-(1,2,4,5-tetrazin-3-yl)benzylamino)-5-oxo-pentanoate (Tz-NHS) or (E)-Cyclooct-4-enyl 2,5-dioxopyrro-lidin-1-yl carbonate (trans-cyclooctene N-hydroxy-succinimidyl ester; TCO-NHS), synthesized as previously reported.31 This reaction was performed using 250 molar equivalents of Tz-NHS or 2000 molar equivalents of TCO-NHS (relative to the MFNPs), and proceeded in PBS containing 10% dimethylformamide (DMF) and 10 mM of sodium bicarbonate at room temperature for 4 hours. Excess orthogonal reactant was first removed using a 100 kDA ultracentrifugation unit (Amicon), which concentrated the sample down to ∼0.25 ml for the final purification step using gel filtration (Sephadex G-50, GE Healthcare). For MFNPs conjugated to Tz without the PEG spacer, the first two steps were omitted (Figure 2).
Preparation of TCO-modified antibodies
Monoclonal antibodies: trastuzumab (Genentech), Cetuximab (Bristo Myers Squibb), anti-EpCAM (clone 158206, R&D systems) and anti-MUC1 (clone M01102909, Fitzgerald Industries) were modified with TCO-NHS. If sodium azide was present, it was removed using a 2 ml Zeba desalting column (Thermo Fisher). The reaction was performed using 1000 molar equivalents of TCO-NHS and 0.5 mg of antibody in PBS containing 10% (v/v) DMF and 10 mM sodium bicarbonate for 4 hours at room temperature. Samples were then purified using Zeba columns and the antibody concentration was determined by absorbance measurement (NanoDrop 1000 Spectrophotometer, Thermo Scientific). On average, antibodies bore ∼15 TCO molecules. The TCO-modified antibodies retained their affinity as previously confirmed.15 In addition, this conjugation can be further improved by directing the chemical modification to the FC portion of antibody (e.g., via oxidation of its glycosidic chains).32
Nanoparticle labeling
The human cancer cell lines SK-OV-3 and SK-BR-3 were obtained from ATCC and maintained in McCoy's 5A with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, 3% sodium bicarbonate, and 1% L-glutamine. Prior to experiments, cells were grown to ∼90% confluency, released using 0.05% trypsin/0.53 mM ethylenediaminetetraacetic acid (EDTA), and washed once with PBS containing 2% bovine serum albumin (PBS+). Cells were then fixed with Lyse/Fix buffer (BD Biosciences 558049) for 10 minutes at 37 °C and washed twice with PBS+. The fixed cells were then either analyzed real-time or frozen down at -20°C for subsequent labeling. In the next step, cells were labeled with TCO modified monoclonal antibodies (10 μg ml-1) in 0.15 ml of PBS+ for 30 minutes at room temperature; antibodies were omitted in control samples. Cells were washed once with PBS to remove the excess antibody. For the initial labeling with MFNPs, cells were resuspended in 0.4 ml MFNP-PEG-Tz (40 μg Fe/ml) for 15 minutes at room temperature. The NP concentration was then determined by measuring the iron (Fe) content through absorbance, at a characteristic wavelength of 400 nm (NanoDrop 1000 Spectrophotometer, Thermo Scientific) and with a known standard for calibration. For the initial amplification (AMP1), cells were washed once and resuspended in 0.4 ml of 40 μg Fe/ml MFNP-PEG-TCO (15 minutes, room temperature). Likewise, cells from the AMP1 step were washed once and resuspended in 0.4 ml of 40 μg Fe/ml of MFNP-PEG-Tz (15 minutes, room temperature) for the second amplification (AMP2). After the last round of labeling, cells were washed once with PBS+ and this was followed by a final wash with PBS. For the cleaving step, samples were mixed with dithiothreitol (DTT; 100 mM) and kept at 37°C for 15 minutes. Finally, cells were centrifuged down and the supernatant (containing the MFNPs) was removed for magnetic resonance measurements. The cleaved MFNPs in DTT (AMP1-C) had a monodisperse mean diameter of ∼100 nm (Malvern).
DMR measurements
Magnetic resonance measurements were performed using the miniature NMR system developed for point-of-care diagnostics.33 The miniaturized NMR device was used to measure the transverse relaxation rate on 1-2 μl sample volumes, using Carr-Purcell-Meiboom-Gill pulse sequences with the following parameters: echo time, 3 ms; repetition time, 4 s; number of 180° pulses per scan, 900; number of scans, 7. A detection threshold of ΔT2 ≥ 2.5% was used to rule out instrumental errors.33 All measurements were performed in triplicate, and the data are presented as the mean ± standard error of the mean. The measured T2 values were then converted to ΔR2+, (R2 = 1/T2, ΔR2+ = R2sig - R2θ), where R2sig and R2θ are the transverse relaxation rates for targeted and control samples, respectively. For the same cell concentration the measured ΔR2+ value is proportional to the amount of MFNP loaded onto each cancer cell.17,24 A negative or zero signal signifies that there is no significant difference in DMR signal between biomarker labeled cells and non-specific binding of MNP to cells within the detection limit. To determine the absolute number of biomarkers, polymer microspheres (Bangs Laboratories) with a known amount of binding sites can be used to create calibration curves to translate DMR levels to the number of biomarkers present.
Flow cytometry
At the conclusion of the MFNP amplification step, but before the cleavage step, samples were measured for VT-680 fluorescence using an LSRII flow cytometer (Becton Dickinson). Mean fluorescence intensity (MFI) was determined using FlowJo software to quantitate the amount of NP present. Two standard deviations above the non-labeled cell measurement was used as the lower limit of detection.
Microscopy
SK-BR-3 cells were magnetically labeled using the method described above. MFNPs with different fluorophores were employed: VT-680-MNP-PEG-Tz for labeling, FITC-MNP-PEG-TCO for AMP1, and RITC-MNP-PEG-Tz for AMP2. Samples were transferred to a 96-well plate at the end of labeling, and again after the cleavage step. Images were acquired at 20× with a DeltaVision screening system (Applied Precision Instruments) and images were analyzed using FIJI software (version 1.45).
Clinical samples
Human clinical ascites from pancreatic cancer was profiled. To compare the cleaving and non-cleaving methods, non-purified samples were divided and screened for EGFR, EpCAM, HER2, and MUC1. For each marker, both AMP1 and AMP1-C were employed as described above. Marker expression levels were determined based on the ratio of positive marker (ΔR2+) and control (ΔR2θ=R2θ - R2PBS). Purified clinical samples were prepared through CD45 negative selection using CD45 magnetic beads and LS columns (Miltenyi Biotec). Both purified and non-purified samples were then targeted for MUC1, and their magnetic labeling was amplified via AMP1 and AMP1-C. Samples were analyzed using either DMR or flow cytometry, and the percent ratio of the non-purified signal to the purified signal was determined (Figure 5B).
Acknowledgments
The authors thank T. Reiner and J. Klubnik for synthesizing the TCO precursor, S. Hilderbrand, M. Karver, and S. Agasti for synthesizing the tetrazine precursor, N. Sergeyev for synthesizing cross-linked dextran iron oxide nanoparticles, J. Dunham with imaging, and C. Min, B. Marinelli, and R. Gorbatov for help with NMR measurements. We especially thank J. Haun, G. Thurber, M. Liong, K. Prather and R. Langer for many helpful discussions and Y. Fisher-Jeffes for review of the manuscript. VP was supported in part by a Merck Educational Assistance Fellowship. This work was funded in part by U54CA151884, R01EB010011, 2R01EB004626, HHSN268201000044C, 2-P50-CA86355, K12CA087723, and DOD W81XWH-11-1-0706.
References and Notes
- 1.Giljohann DA, Mirkin CA. Drivers of Biodiagnostic Development. Nature. 2009;462:461–464. doi: 10.1038/nature08605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haun JB, Yoon TJ, Lee H, Weissleder R. Molecular Detection of Biomarkers and Cells Using Magnetic Nanoparticles and Diagnostic Magnetic Resonance. Methods Mol Biol. 2011;726:33–49. doi: 10.1007/978-1-61779-052-2_3. [DOI] [PubMed] [Google Scholar]
- 3.Lees WJ, Spaltenstein A, Kingery-Wood JE, Whitesides GM. Polyacrylamides Bearing Pendant Alpha-Sialoside Groups Strongly Inhibit Agglutination of Erythrocytes By Influenza a Virus: Multivalency and Steric Stabilization of Particulate Biological Systems. J Med Chem. 1994;37:3419–3433. doi: 10.1021/jm00046a027. [DOI] [PubMed] [Google Scholar]
- 4.Jones MR, Osberg KD, Macfarlane RJ, Langille MR, Mirkin CA. Templated Techniques for the Synthesis and Assembly of Plasmonic Nanostructures. Chem Rev. 2011;111:3736–3827. doi: 10.1021/cr1004452. [DOI] [PubMed] [Google Scholar]
- 5.Perez JM, Josephson L, O'Loughlin T, Hogemann D, Weissleder R. Magnetic Relaxation Switches Capable of Sensing Molecular Interactions. Nat Biotechnol. 2002;20:816–820. doi: 10.1038/nbt720. [DOI] [PubMed] [Google Scholar]
- 6.Kim D, Daniel WL, Mirkin CA. Microarray-Based Multiplexed Scanometric Immunoassay for Protein Cancer Markers Using Gold Nanoparticle Probes. Anal Chem. 2009;81:9183–9187. doi: 10.1021/ac9018389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoon TJ, Lee H, Shao H, Weissleder R. Highly Magnetic Core-Shell Nanoparticles With a Unique Magnetization Mechanism. Angew Chem, Int Ed Engl. 2011;50:4663–4666. doi: 10.1002/anie.201100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoon TJ, Lee H, Shao H, Hilderbrand SA, Weissleder R. Multicore Assemblies Potentiate Magnetic Properties of Biomagnetic Nanoparticles. Adv Mater. 2011;23:4793–4797. doi: 10.1002/adma.201102948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aktas B, Kasimir-Bauer S, Heubner M, Kimmig R, Wimberger P. Molecular Profiling and Prognostic Relevance of Circulating Tumor Cells in the Blood of Ovarian Cancer Patients At Primary Diagnosis and After Platinum-Based Chemotherapy. Int J Gynecol Cancer. 2011;21:822–830. doi: 10.1097/IGC.0b013e318216cb91. [DOI] [PubMed] [Google Scholar]
- 10.Armstrong AJ, Marengo MS, Oltean S, Kemeny G, Bitting RL, Turnbull JD, Herold CI, Marcom PK, George DJ, Garcia-Blanco MA. Circulating Tumor Cells From Patients With Advanced Prostate and Breast Cancer Display Both Epithelial and Mesenchymal Markers. Mol Cancer Res. 2011;9:997–1007. doi: 10.1158/1541-7786.MCR-10-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Attard G, de Bono JS. Utilizing Circulating Tumor Cells: Challenges and Pitfalls. Curr Opin in Genet Dev. 2011;21:50–58. doi: 10.1016/j.gde.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 12.Chin CD, Linder V, Sia SK. Lab-on-a-Chip Devices for Global Health: Past Studies and Future Opportunities. Lab Chip. 2007;7:41–57. doi: 10.1039/b611455e. [DOI] [PubMed] [Google Scholar]
- 13.Struelens MJ, Denis O, Rodriguez-Villalobos H. Microbiology of Nosocomial Infections: Progress and Challenges. Microbes Infect. 2004;6:1043–1048. doi: 10.1016/j.micinf.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 14.Urdea M, Penny LA, Olmsted SS, Giovanni MY, Kaspar P, Shepherd A, Wilson P, Dahl CA, Buchsbaum S, Moeller G, et al. Requirements for High Impact Diagnostics in the Developing World. Nature. 2006;444(1):73–79. doi: 10.1038/nature05448. [DOI] [PubMed] [Google Scholar]
- 15.Haun JB, Devaraj NK, Hilderbrand SA, Lee H, Weissleder R. Bioorthogonal Chemistry Amplifies Nanoparticle Binding and Enhances the Sensitivity of Cell Detection. Nat Nanotechnol. 2010;5:660–665. doi: 10.1038/nnano.2010.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haun JB, Devaraj NK, Marinelli BS, Lee H, Weissleder R. Probing Intracellular Biomarkers and Mediators of Cell Activation Using Nanosensors and Bioorthogonal Chemistry. ACS Nano. 2011;5:3204–3213. doi: 10.1021/nn200333m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liong M, Tassa C, Shaw SY, Lee H, Weissleder R. Multiplexed Magnetic Labeling Amplification Using Oligonucleotide Hybridization. Adv Mater. 2011;23:H254–7. doi: 10.1002/adma.201101401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agasti SS, Liong M, Tassa C, Chung HJ, Shaw SY, Lee H, Weissleder R. Supramolecular Host-Guest Interaction for Labeling and Detection of Cellular Biomarkers. Angew Chem, Int Ed Engl. 2011;51:450–454. doi: 10.1002/anie.201105670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karver MR, Weissleder R, Hilderbrand SA. Bioorthogonal Reaction Pairs Enable Simultaneous, Selective, Multi-Target Imaging. Angew Chem, Int Ed Engl. 2011;51:920–922. doi: 10.1002/anie.201104389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karver MR, Weissleder R, Hilderbrand SA. Synthesis and Evaluation of a Series of 1,2,4,5-Tetrazines for Bioorthogonal Conjugation. Bioconjug Chem. 2011;22:2263–2270. doi: 10.1021/bc200295y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brooks RA. T(2)-Shortening By Strongly Magnetized Spheres: A Chemical Exchange Model. Magn Reson Med. 2002;47:388–391. doi: 10.1002/mrm.10064. [DOI] [PubMed] [Google Scholar]
- 22.Haun JB, Yoon TJ, Lee H, Weissleder R. Magnetic Nanoparticle Biosensors. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2010;2:291–304. doi: 10.1002/wnan.84. [DOI] [PubMed] [Google Scholar]
- 23.Shao H, Yoon TJ, Liong M, Weissleder R, Lee H. Magnetic Nanoparticles for Biomedical Nmr-Based Diagnostics. Beilstein J Nanotechnol. 2010;1:142–154. doi: 10.3762/bjnano.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee H, Yoon TJ, Figueiredo JL, Swirski FK, Weissleder R. Rapid Detection and Profiling of Cancer Cells in Fine-Needle Aspirates. Proc Natl Acad Sci U S A. 2009;106:12459–12464. doi: 10.1073/pnas.0902365106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han HS, Devaraj NK, Lee J, Hilderbrand SA, Weissleder R, Bawendi MG. Development of a Bioorthogonal and Highly Efficient Conjugation Method for Quantum Dots Using Tetrazine-Norbornene Cycloaddition. J Am Chem Soc. 2010;132:7838–7839. doi: 10.1021/ja101677r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Homola J. Surface Plasmon Resonance Sensors for Detection of Chemical and Biological Species. Chem Rev. 2008;108:462–493. doi: 10.1021/cr068107d. [DOI] [PubMed] [Google Scholar]
- 27.Sendroiu IE, Gifford LK, Luptak A, Corn RM. Ultrasensitive DNA Microarray Biosensing Via in Situ RNA Transcription-Based Amplification and Nanoparticle-Enhanced Spr Imaging. J Am Chem Soc. 2011;133:4271–4273. doi: 10.1021/ja2005576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee K, Drachev VP, Irudayaraj J. DNA– Gold Nanoparticle Reversible Networks Grown on Cell Surface Marker Sites: Application in Diagnostics. ACS Nano. 2011;5:2109–17. doi: 10.1021/nn1030862. [DOI] [PubMed] [Google Scholar]
- 29.Haun JB, Castro CM, Wang R, Peterson VM, Marinelli BS, Lee H, Weissleder R. Micro-Nmr for Rapid Molecular Analysis of Human Tumor Samples. Sci Transl Med. 2011;3:71ra16. doi: 10.1126/scitranslmed.3002048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reynolds F, O'loughlin T, Weissleder R, Josephson L. Method of Determining Nanoparticle Core Weight. Anal Chem. 2005;77:814–817. doi: 10.1021/ac049307x. [DOI] [PubMed] [Google Scholar]
- 31.Devaraj NK, Upadhyay R, Haun JB, Hilderbrand SA, Weissleder R. Fast and Sensitive Pretargeted Labeling of Cancer Cells Through a Tetrazine/Trans-Cyclooctene Cycloaddition. Angew Chem, Int Ed Engl. 2009;48:7013–7016. doi: 10.1002/anie.200903233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hermanson GT. Bioconjugate Techniques. 2nd. Academic Press; San Diego: 2008. pp. 784–804. [Google Scholar]
- 33.Issadore D, Min C, Liong M, Chung J, Weissleder R, Lee H. Miniature Magnetic Resonance System for Point-of-Care Diagnostics. Lab Chip. 2011;11:2282–2287. doi: 10.1039/c1lc20177h. [DOI] [PMC free article] [PubMed] [Google Scholar]