

Abstract
Cutaneous melanomas can be divided into three mutually exclusive genetic subsets: tumors with mutated BRAF, tumors with mutated NRAS, and tumors wild type at both loci (wt/wt). Targeted therapy for melanoma has been advancing with agents directed to mutated BRAF, accounting for 50% of melanoma patients. The c-Met pathway is known to play a role in melanoma tumorigenesis and preliminary data from our laboratory suggested that this pathway is preferentially activated in NRAS-mutated tumors. The objective of this study was to test the hypothesis that melanomas carrying the mutated NRAS genotype are uniquely sensitively to c-Met inhibition, thus providing rationale for therapeutic targeting of c-Met in this patient cohort. Using primary human melanomas with known BRAF/NRAS genotypes, we observed greater immunostaining for phosphorylated (activated) c-Met in NRAS-mutated and wt/wt tumors, compared to BRAF-mutated tumors. NRAS-mutated and wt/wt cell lines also demonstrated more robust c-Met activation in response to hepatocyte growth factor (HGF). Knock-down of mutated N-Ras, but not wild type N-Ras, by RNA interference resulted in decreased c-Met phosphorylation. Compared to BRAF mutants, NRAS-mutated melanoma cells were more sensitive to pharmacologic c-Met inhibition in terms of c-Met activation, Akt phosphorylation, tumor cell proliferation, migration, and apoptosis. This enhanced sensitivity was observed in wt/wt cells as well, but was a less consistent finding. Based on these experimental results, we propose that c-Met inhibition may be a useful therapeutic strategy for melanomas with NRAS mutations, as well as some tumors with a wt/wt genotype.
Keywords: melanoma, c-Met, HGF, NRAS mutation, PHA665752
Introduction
The c-Met/HGF pathway is known to stimulate cancer cell growth and metastasis.1 c-Met (cellular-mesenchymal to epithelial transition factor) is a plasma membrane tyrosine kinase activated by auto-phosphorylation after ligand binding. Hepatocyte growth factor (HGF), the only known ligand for c-Met, functions in a paracrine manner under normal physiologic conditions.2 In contrast, some cancer cells produce both HGF and c-Met, leading to autocrine activation of the receptor. c-Met can also be constitutively active in cancer cells due to expression of the fusion protein Tpr-met, the presence of a mutation in the c-Met kinase domain, or c-Met protein overexpression.3–5 Activation of c-Met through these various mechanisms drives multiple features of the malignant phenotype including cell proliferation, motility, and some aspects of differentiation. Molecular analysis of the c-Met pathway has identified a number of adaptor proteins that become phosphorylated and contribute to c-Met signaling, including components of the phosphatidylinositol-3 kinase (PI3K)/Akt and mitogen-activated protein kinase (MAPK) pathways. 6, 7 c-Met activation also induces the activation and nuclear translocation of beta catenin, a key component of the Wnt pathway.
Human cutaneous melanoma is one of the many malignancies that express activated c-Met protein. c-Met has been shown to be up-regulated in the invading front of the tumor, and over-expression of c-Met is associated with melanoma growth and metastatic spread.8–12 Furthermore, HGF transgenic mice spontaneously develop melanomas with a high invasive and metastatic potential.13 These findings strongly implicate the c-Met pathway in melanoma progression and suggest that c-Met inhibition might provide an effective therapeutic approach.
Cutaneous melanoma is understood to represent at least three patient subsets based on the presence or absence of two established somatic mutations.14 The major population with mutated BRAF (~50%) is mutually exclusive of those with mutated NRAS, representing 15–20% of tumors. A third patient subset carries neither mutation (hereafter referred to as “wt/wt”). These three unique genotypes provide the opportunity for personalized, targeted melanoma therapy. The past two decades have witnessed small improvements over conventional chemotherapeutics, such as DTIC, which are palliative at best. A portion of patients with advanced melanoma benefit from immune-stimulating drugs, such as Interleukin-2 and Ipilimumab, but responders cannot be identified prior to treatment by any molecular or genetic features of the tumor or the patient.15 More recently compounds directed to mutated B-Raf have entered clinical trials and at least one of these drugs has demonstrated improved rates of response and survival in patients whose tumors bear this genotype.16 Although such responses are generally not durable, these compounds have provided a basis for optimism in the treatment of this melanoma patient subset.17 In contrast, the means to target mutant NRAS and wt/wt melanomas lag behind in development. Adding to the critical nature of this issue is the general consensus that NRAS-mutated melanomas are more aggressive than tumors bearing either of the other two genotypes.18 In the course of preliminary immunohistochemistry (IHC) experiments examining c-Met phosphorylation in human primary melanomas, we detected a trend for enhanced phospho-c-Met staining in NRAS-mutated tumors. This finding was intriguing in view of the aggressive nature of NRAS-mutated melanomas as well as other types of solid tumors which express aberrant c-Met activation. This led us to form the hypothesis that melanomas with mutated NRAS carry a unique dependence on c-Met signaling, making them vulnerable to c-Met inhibition. We now report confirmatory data that c-Met is more likely to be activated in both NRAS-mutated and wt/wt melanomas, and that melanoma cells with these genotypes, particularly NRAS mutants, are more sensitive to pharmacologic c-Met inhibition.
Materials and Methods
Reagents
The small molecule c-Met inhibitor, PHA665752, (3Z)-5-[(2,6-dichlorobenzyl)sulfonyl]-3-[(3,5-dimethyl-4-1H-pyrrol-2-yl)methylene]-1,3-dihydro-2H-indol-2-one, was obtained under a material transfer agreement with Pfizer Inc. (La Jolla, CA). PHA665752 was dissolved in dimethyl sulfoxide (DMSO) to prepare a stock solution of 30 mM, and diluted in fresh medium. In all experiments, the final concentration of DMSO was < 0.1%. HGF was purchased from R& D Systems (Minneapolis, MN).
Cells and cell culture
Melanoma cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum. Primary cultured melanocytes (FMC15H) were derived and grown as previously described.19 The A375, MeWo and SK-Mel-2 cell lines were purchased from American Type Culture Collection (Manassas, VA). SB2 cells were provided by Dr. Michael Davies at the M. D. Anderson Cancer Center, Houston, TX (MDACC). The WM852, 451Lu, and WM1361A cell lines were obtained from Dr. Meenhard Herlyn (Wistar Institute, Philadelphia, PA). The WM35 and WM793 cell lines were provided by Dr Robert Kerbel (Sunnybrook Health Science Center, Toronto, ON, Canada).
Cell line validation was accomplished by short random repeat (STR) DNA fingerprinting techniques and mutational analysis by MDACC Cancer Center Support Grant (CCSG)–supported Characterized Cell Line Core. Cell lines were validated by STR DNA fingerprinting using the AmpF_STR Identifier Kit (Applied Biosystems, Foster City, CA) according to manufacturer’s instructions. The STR profiles were compared to known ATCC fingerprints (ATCC.org), and to the Cell Line Integrated Molecular Authentication database (CLIMA) version 0.1.200808 (http://bioinformatics.istge.it/clima/).20 The STR profiles matched known DNA fingerprints or were unique.
Tissue sections and immunohistochemical staining
Use of patient materials was approved by the MDACC Institutional Review Board and research was conducted in compliance with Health Insurance Portability and Accountability Act. The melanoma tumor samples used in this study were formalin-fixed, paraffin-embedded specimens of primary cutaneous melanomas provided by the MDACC Melanoma Program Informatics, Tissue Resource, and Pathology Core. The tumors examined were residual sections from a previous study of 223 consecutive tumor bank entries in which clinical data were correlated with BRAF and NRAS genotypes.21 Tumor samples were examined for c-Met and phospho-c-Met expression by IHC using anti-c-Met (Santa Cruz Biotechnology, Santa Cruz, CA) and anti-phospho c-Met (Invitrogen, Camarillo, CA) polyclonal antibodies. Pre-immune normal rabbit IgG (Vector Laboratories, Burlingame, CA) and anti-vimentin antibody (BioGenex Laboratories, San Ramon, CA) were used as negative and positive controls, respectively. The staining procedure has been previously described.22 Immunostaining was scored by two independent observers (SE and VRG) on the following scale: <5%, “0”; 5–25%, “1+”; 26–75%, “2+”; >75%,“3+”;. Stained tissues were photographed using a Nikon Eclipse TE2000-U microscope using NIS Elements software.
Western blotting and antibodies
Cells were lysed in a buffer containing 50 mM Tris (pH 7.9), 150 mM NaCl, 1% NP40, 1 mM EDTA, 10% glycerol, 1mM sodium vanadate, and protease inhibitor cocktail (Roche Pharmaceuticals, Nutley, NJ). Proteins were separated by SDS-PAGE with 4–20% gradient gels (Bio-Rad Laboratories, Hercules, CA), transferred to a Hybond-ECL nitrocellulose membrane (GE Healthcare Biosciences, Piscataway, NJ), and blocked in 5% dry milk in PBS. The membrane was then incubated with primary and secondary antibodies, and target proteins were detected with ECL detection reagent (GE Healthcare Biosciences).
The c-Met (C-12), N-Ras, β-actin, and caspase-9 antibodies were obtained from Santa Cruz Biotechnology. Phospho-ERK, phospho-Akt (Thr 308 and Ser 473), ERK, Akt, and caspase-3 antibodies were purchased from Cell Signaling Technology (Beverly, MA).
Immunoprecipitation
Cells were lysed on ice for 30 min, and the lysate incubated with protein-A-agarose (Sigma) pre-conjugated with anti-phospho-tyrosine antibody (Millipore, Billerica, MA). For the conjugation reaction, 10 uL of the resin was incubated with 4 ug antibody in a cold room with gentle shaking in a total volume of 500 uL. The reaction proceeded for 2h, followed by overnight incubation of the conjugated resin with the cell lysates (250 ug total protein). Bound proteins were eluted with SDS PAGE loading buffer by boiling and loaded in a 4–20% gradient gel for western blot analysis.
NRAS siRNA transfection
SMARTpool siRNA directed to wild type NRAS and non-targeting siRNA, as well as custom designed siRNA specific for the Q61R NRAS mutation, were purchased from Dharmacon (Thermo Fisher Scientific, Lafayette, CO). Melanoma cells were plated in six well plates at a density of 1.5 × 105 cells/well and transfection of wild type or mutant NRAS siRNA and control siRNA was performed the next day using Oligofectamine reagent (Invitrogen, Carlsbad, CA). In each case 100 nM siRNA was transfected with 3 uL of oligofectamine. Cell lysates were prepared on day 3 after a 7-min induction with 100 ng/mL HGF, followed by western blotting.
Cell viability assays
Melanoma cells were plated at a density of 1 × 104 cells/well in triplicate in a 24-well plate in RPMI 1640 growth medium supplemented with 100 ng/mL HGF, with or without PHA665752 at a concentration range of 0 to 5 uM for 72 hours.. MTT reagent [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (Sigma-Aldrich, St. Louis, MO), dissolved in PBS, was added to a final concentration of 1 mg/mL. After 3 h, the precipitate formed was dissolved in DMSO, and the color intensity estimated in a MRX Revelation microplate absorbance reader (Dynex Technologies, Chantilly, VA) at 570 nm. IC50 values of PHA665752 were calculated from the data obtained from the MTT assays using Curve Expert 1.3 software (Microsoft Corporation, Redmond, WA).
Cell migration assay
Assays for melanoma cell migration were performed in Boyden chambers using uncoated filters (BD Biocoat control inserts, BD Biocoat, San Jose, CA). 2.5 × 105 cells/well were plated in serum-free medium, with or without a four hour treatment of 0.5 uM PHA665752, and the migration assay performed as described in reference 23. Stained cells were photographed with a Nikon Eclipse TE2000-U microscope at 20X magnification using NIS Elements advanced research software. To quantify migration, the cells in each filter were counted from five independent fields under the microscope at 40X magnification and the mean cell number/field was calculated.
Cell Cycle Analysis
Cells expressing wild type or mutant NRAS were treated with 1 uM PHA665752 and prepared as a suspension of 1 × 106 cells/mL of PBS. After fixation with 90% ethanol for 1 hour, the cells were centrifuged and stained with propidium iodide (PI) (Boehringer Mannheim, Indianapolis, IN) at a final concentration of 5 mg/ml PI and 10 mg/ml RNAse. DNA contents and cell cycle phases were analyzed using a FACScan flow cytometer (Becton Dickinson, San Jose, CA).
Results
Phosphorylation of c-Met in human melanoma varies with genotype
To determine the expression of c-Met and its activation status in vivo, thirty-seven formalin-fixed paraffin-embedded human primary melanomas with known NRAS and BRAF mutational status were examined by IHC for c-Met and phospho-c-Met (Figure 1). IHC staining for phospho-c-Met was scored for percentage of positive cells as described in Methods. The staining was cytoplasmic, and the percentage of involved tumor varied from case to case, as indicated by the IHC scores. There was no particular predilection for positive cells in the superficial vs. deep portions of the tumor. Fifty-four percent of NRAS mutant tumors and 82% of wt/wt tumors were positive for phospho-c-Met, whereas only 29% of BRAF mutant tumors yielded positive results (p = 0.030, Chi square). The distribution of IHC scores, a well as clinical data, are shown in Table 1. The clinical features reflect those of the larger population from which this cohort was taken, including the higher rate of ulceration in BRAF mutants and the preference of NRAS-mutated tumors to develop on the extremities. 21 The only notable difference is the higher median Breslow depth for the BRAF mutated-cases which were selected by chance for the current study (4.0 mm in this study vs. 1.28 mm in the larger population). Clark levels, however, were similar among the three genotypes. These findings confirm the activation of c-Met in melanoma and uniquely suggest that this process may be influenced by the NRAS/BRAF mutational status.
Figure 1.

Detection of c-Met and phospho-c-Met in melanoma tumor tissues. NRAS-mutated, wt/wt, and BRAF-mutated cutaneous melanomas were examined by IHC for c-Met and phospho-c-Met expression. NRAS mutants and wt/wt tumors show high expression of both. 20X, AEC.
Table 1.
IHC scores for percentage of tumor cells staining positively for phospho-C-Met, and clinical data according to genotype
|
NRAS Mutant N (%) |
Wt/Wt N (%) |
BRAF Mutant N (%) |
|
|---|---|---|---|
| N | 13 | 11 | 13 |
| p-C-Met IHC score | |||
| 0 | 6 (46.2) | 2 (18.2) | 9 (69.2) |
| 1 | 1 (7.7) | 1 (9.1) | 2 (15.4) |
| 2 | 3 (23.1) | 5 (45.5) | 0 (0) |
| 3 | 3 (23.1) | 3 (27.3) | 2 (15.4) |
| Median age (yrs) | 52 | 54 | 46 |
| Gender | |||
| Male | 6 (46.1) | 7 (63.3) | 11 (84.6) |
| Female | 7 (53.8) | 4 (36.4) | 2 (15.4) |
| Histology | |||
| SS | 9 (69.2) | 7 (63.6) | 5 (38.5) |
| Nodular | 2 (15.4) | 4 (36.4) | 6 (46.2) |
| Lentigo | 1 (7.7) | 0 (0) | 0 (0) |
| Unclassified | 1 (7.7) | 0 (0) | 2 (15.4) |
| Median Breslow depth (mm) | 1.18 | 1.75 | 4.00 |
| Median Clark Level | 3 | 4 | 4 |
| Site (%) | |||
| Extremity | 12 (92.3) | 6 (54.5) | 3 (23.1) |
| Trunk | 1 (7.7) | 5 (45.5) | 8 (61.5) |
| Head/Neck | 0 (0) | 0 (0) | 2 (15.4) |
| Ulceration | |||
| yes | 1 (7.7) | 1 (9.1) | 6 (46.2) |
| no | 12 (92.3) | 10 (90.9) | 7 (53.8) |
| Solar Elastosis* | |||
| yes | 10 (76.9) | 8 (72.7) | 10 (83.3) |
| no | 3 (23.1) | 3 (27.3) | 2 (16.7) |
| Status | |||
| Alive, NED | 10 (76.9) | 7 (63.6) | 8 (61.5) |
| Dead of disease | 3 (23.1) | 4 (36.4) | 4 (30.8) |
| Dead, cause unknown | 0 (0) | 0 (0) | 1 (7.7) |
| AJCC Stage | |||
| 1 | 7 (53.8) | 5 (45.5) | 0 (0) |
| 2 | 4 (30.8) | 1 (9.0) | 3 (23.1) |
| 3 | 2 (15.4) | 5(45.5) | 10 (76.9) |
SS, superficial spreading; NED, no evidence of disease
One BRAF-mutated tumor was not evaluable for solar elastosis.
To confirm the findings described above, the expression of c-Met and phospho-c-Met protein were examined by western blotting in human melanoma cell lines with known NRAS and BRAF genotypes. Nine of the ten melanoma cell lines expressed detectable levels of c-Met, with highest levels found in the NRAS mutants (Figure 2a). c-Met was absent in one BRAF mutant melanoma line and in cultured melanocytes which carry neither mutation. To optimize detection of phospho-c-Met, immunoprecipitation with anti-phospho-tyrosine antibody preceded western blotting. Constitutive c-Met phosphorylation was not detected in any melanoma cell line grown under serum-free conditions, but was induced in all cases by exogenous HGF, with NRAS mutants and wt/wt cells exhibiting a more robust response than BRAF mutants. Representative experiments are shown in Figure 2b.
Figure 2.
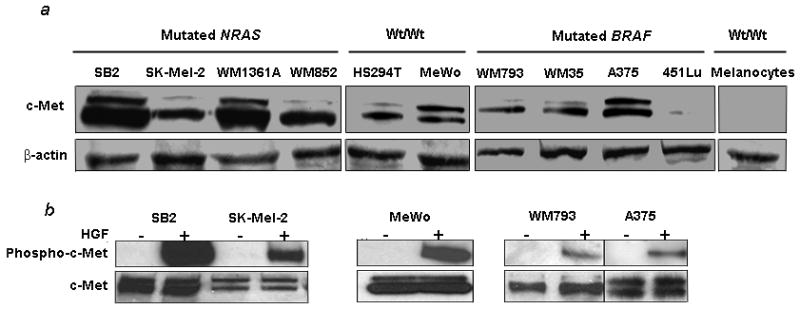
c-Met and phospho-c-Met in melanoma cells. (a) c-Met expression is detected by immunoblotting in all but one melanoma cell line. Melanocytes are negative. (b) HGF induces phosphorylation of c-Met in the same melanoma cells. Cell lines were treated with HGF at a concentration of 100 ng/mL for seven minutes prior to protein isolation and detection of phospho-c-Met.
Effect of mutated NRAS knockdown on c-Met phosphorylation
The above results indicate higher c-Met levels and increased HGF responsiveness in NRAS mutants and possibly wt/wt melanoma cells, in comparison to BRAF mutants. To further examine the influence of NRAS on c-Met activation, we used RNA interference to knock down levels of mutated or wild type N-Ras in cells carrying the one or the other genotype, and compared the ensuing effects on c-Met phosphorylation. All cell lines used in this study carry only mutated or wild type NRAS, i.e., none is heterozygous at that locus, facilitating the interpretation of these experiments. SKMel2, WM852, and WM1361A cells, which carry the Q61R NRAS mutation, were treated with siRNA designed specifically for that mutated sequence. Untransfected and non-targeting siRNA-transfected cells served as controls. Results showed successful reduction of N-Ras protein levels, confirming that the siRNA was functional (Figure 3a). Levels of total c-Met protein remained unchanged with transfection. However, mutant N-Ras knockdown reduced the level of HGF-induced c-Met phosphorylation in all three lines. Transfection of wild type N-Ras-specific siRNA into A375 cells and WM793 cells (BRAF mutants), or MeWo cells (wt/wt) reduced the amount of N-Ras protein, but had only a minor effect on phospho-c-Met levels (Figure 3b). Phospho-c-Met inhibition for each of the six cells lines is depicted in Figure 3c. Levels of phospho-c-Met in cells treated with non-targeting siRNA vs. NRAS-specific siRNA differed significantly between cell lines carrying mutant NRAS and those with wild type NRAS (p < 0.001). These results suggest that c-Met phosphorylation is modulated by N-Ras, and that this effect is greatly enhanced when the N-Ras protein is mutated.
Figure 3.

Effect of NRAS knockdown on c-Met phosphorylation. (a) Knockdown of mutated N-Ras by RNA interference in three cell lines carrying the Q61R NRAS mutation leads to diminished levels of phospho-c-Met. Cells were treated overnight with 100 nM siRNA specific for the NRAS mutation. Total cellular protein was prepared 72 hours after transfection, following a 7 minute exposure to HGF 100 ng/ml, and examined for N-Ras, phospho-c-Met, total c-Met, and actin by western blotting. (b) Knockdown of wild type N-Ras in cells lacking an NRAS mutation has minimal effect on c-Met phosphorylation. Experiments were performed in an identical manner to those described in (a) with the exception that the three cell lines carry wild type NRAS and the siRNA was therefore specific for the wild type message. (c) Densitometry results for phospho-c-Met (normalized to levels of total c-Met) in cells treated with non-targeting siRNA vs. NRAS-specific siRNA are shown. Quantitation of the above-described experimental results was performed with Image J software (NIH) using the ‘gelplot1’ Macro software. Declines in levels of phospho-c-Met in cells treated with non-targeting siRNA vs. NRAS-specific siRNA differed significantly between cell lines carrying mutant NRAS and those with wild type NRAS (p< 0.001). Unt, untreated; NT, non-targeting.
Concentration-dependent effects of PHA665752 on c-Met phosphorylation, pathway induction, and proliferation
We next wished to determine if the presence of an NRAS-mutation translated to enhanced sensitivity of melanoma cells to c-Met inhibitors. Accordingly, we studied the highly specific small molecule c-Met inhibitor, PHA665752, which functions by blocking c-Met phosphorylation. PHA665752, applied to the melanoma cells at concentrations ranging from 1nM to 1uM was found to efficiently block c-Met phosphorylation in response to HGF in a dose dependent manner. Phosphorylation was markedly diminished at concentrations of 25–100 nM in all cell lines and was essentially undetectable at a concentration of 1 uM (Fig. 4a). Under the same conditions for induction of c-Met phosphorylation, HGF induced Akt phosphorylation at Ser 473 (Fig. 4b). This process was similarly inhibited in a concentration dependent manner by PHA665752. In NRAS mutants, inhibition of Akt activation was complete or nearly complete at a concentration of 100 nM. In contrast, both BRAF mutants and one of the two wt/wt cells showed only minor reduction in Akt phosphorylation at this dose level. Phosphorylation of Akt at Thr 308 was not induced by HGF and was unaffected by PHA665752 treatment (data not shown). The impact of c-Met activation on the MAPK pathway, as evidenced by Erk1/2 phosphorylation, varied considerably among cell lines and demonstrated no genotype-specific pattern (data not shown).
Figure 4.

Concentration-dependent effects of c-Met inhibition. (a) Treatment of melanoma cells with PHA665752 results in a concentration-dependent inhibition of c-Met phosphorylation. Cells were treated hours for four hours with PHA665752 at a concentration range of 0 to 1000 nM in the absence or presence of HGF 100 nM. (b) Similar concentrations inhibit phosphorylation of Akt, with the NRAS-mutated lines demonstrating the greatest sensitivity. Cells were treated with PHA665752 for 4 hours at a concentration 0, 50 or 100 nM in the presence or absence of HGF 100 nM. Cell lysates were subsequently examined for effects on Akt phosphorylation by western blotting. (c) Growth inhibitory effects of PHA665752 also appear to be influenced by genotype as demonstrated in the charted IC50 values. Cells were plated in triplicate and treated with PHA665752 at a concentration range of 0 to 5 uM for 72 hours. Cell viability was determined by MTT assay and IC50 values were subsequently calculated. The mean IC50 for NRAS mutants was significantly lower than those of the other two genotypes, with p-values of < 0.001.
Cell growth inhibition required higher concentrations of PHA665752, as evidenced by the IC50 values (Fig. 4c). A mean IC50 of 0.69 uM was determined for NRAS mutants (SB2, SK- Mel-2 and WM82), 0.82 uM for wt/wt cells (MeWo and HS294T), and 1.31 uM for BRAF mutants (A375 and WM35). The IC50 for NRAS mutants was significantly lower than those of the other two genotypes, with p-values of < 0.001 for both (t-test).
Effects of PHA665752 on migration and apoptosis vary with genotype
The role of HGF in cell migration is well documented.1 Accordingly, in vitro cell migration assays using modified Boyden chambers were used to test migration of melanoma cells in response to HGF as a chemo-attractant. The assays were performed in the presence or absence of PHA665752. To assure adequate c-Met inhibition, a PHA665752 concentration of 500 nM was applied to these experiments. The NRAS mutant cell lines SB2 and SKMel-2 migrated briskly in response to HGF, whereas the wt/wt line MeWo showed moderate migration and the BRAF mutant line A375 migrated poorly (Fig. 5a). PHA665752 dramatically reduced the migration of NRAS-mutated cells by an average of 88.2% compared to 19.7% for the other two lines lacking an NRAS mutation (p < 0.001; Figs. 5a and b).
Figure 5.

Effects of c-Met inhibition on migration and apoptosis. (a) NRAS-mutated cell lines (SB2 and SK-Mel-2) migrate efficiently toward HGF; this process is completely inhibited by PHA665752. BRAF mutated cells (A375) migrate poorly and are unaffected by PHA665752. The wt/wt MeWo cells migrate well but are only slightly inhibited by the drug. Cell were untreated or treated with PHA5652 0.5 uM for 4 hours and permitted to migrate overnight in Boyden chambers using uncoated filters. The chemo-attractant in the lower chamber was HGF 100 nM. Photographs are representative of three independent experiments. (b) Charted data represent the mean and standard deviation of cell counts from five independent fields. Inhibition of migration for the two NRAS mutants, SB2 and SK-Mel-2 is significant (p < 0.001). Data shown are representative of three independent experiments.
(c) The percentage of cells in sub-G0/G1 after treatment with PHA665752 is shown. Cells were treated with 1 uM PHA665752 for 72 or 96 hours, stained with PI, and subsequently examined for sub-G0/G1 arrest by flow cytometry. The (*) indicates a significant difference from baseline values, which is limited to SB2 and SK-Mel-2 (p < 0.001). Data from the two NRAS mutant and two NRAS wild type lines were combined for analysis. The figure is representative of two independent experiments. (d) Western blotting for caspase-3 and caspase-9 processing confirms the induction of apoptosis in NRAS mutant cells (SB2 and SK-Mel-2) compared to wt/wt (MeWo) and BRAF mutant (A375) cells after c-Met inhibitor treatment. Cells were untreated or treated with PHA665752 1 uM for 48 or 72 hours prior to western blot analysis for caspase cleavage.
Finally, we examined a differential effect of c-Met inhibition on apoptosis. Consistent with data from other laboratories, preliminary experiments using PHA665752 at nanomolar concentrations failed to yield an effect on any cell line 8; therefore a concentration of 1 uM was applied to these experiments. Cells expressing mutant NRAS or BRAF were treated with PHA665752 for different time intervals and stained with PI for cell cycle analysis (Fig 5c). Whereas the drug showed no effect on mutant BRAF cells, a significantly higher fraction of NRAS mutant cells were found in sub-G0/G1 after 96 hours of treatment (p < 0.001). To confirm these findings, processing of caspase 9 and caspase 3 with PHA665752 treatment was examined by immunoblotting. NRAS-mutated cells showed detectable cleavage of both caspases, whereas mutant BRAF cells failed to show similar processing (Fig. 5d). The wt/wt MeWo cells exhibited a low level of caspase 9 cleavage at baseline that was not enhanced by PHA665752. Taken together, these functional assays demonstrate a unique sensitivity of NRAS-mutated melanoma cells to c-Met inhibition.
DISCUSSION
Personalized cancer therapy based on unique molecular or genetic features of a given individual’s tumor is the future of medical oncology. This approach is both rational and humane, offering the greatest opportunity for response to those patients whose tumors carry the therapeutic target, and sparing toxicity and expense for those with no chance of benefit. In this context, our data support the novel concept that the majority of patients with NRAS-mutated melanomas, and possibly a portion of those with wt/wt genotypes, are most likely to benefit from new drugs targeting c-Met. Conversely, it would appear that patients with BRAF-mutated tumors would be better served by other modalities directed to that oncogene. Consistent with our findings are similar data from lung cancer cells which demonstrate increased sensitivity of KRAS mutants with high phospho-c-Met levels to PHA665752.24 It must be noted that much of our data are pre-clinical and conclusions from patient material are based on a relatively small sample size. However, we believe that if these findings are confirmed in a well-designed study incorporating a larger patient cohort, they will support the application of NRAS and BRAF genotyping to stratification schemes for new clinical trials of c-Met inhibitors as they emerge for the treatment of melanoma. In fact, many approaches directed to c-Met pathways are now under development, including antibodies targeting c-Met or HGF 25, 26; dominant negative forms of c-Met 27; c-Met siRNA 28; and small molecule c-Met inhibitors, most of which target the ATP binding site of the c-Met kinase.23, 29–32 Roughly half of the NRAS-mutated tumors examined in this study stained positively for phospho-c-Met by IHC. It will be important to determine in future studies with larger patient numbers if this finding defines c-Met-directed drug sensitivity in the NRAS-mutated tumors or if the presence of an NRAS mutation is sufficient. Additionally, as some BRAF-mutated tumors also have phosphorylated c-Met, it is imperative to determine if these tumor are sensitive to c-Met inhibition, expanding the therapeutic options for patients whose tumors carry this genotype.
The obvious question raised by our findings involves the mechanism whereby mutated NRAS interacts with c-Met. It is widely accepted that the codon 61 mutation of N-Ras stabilizes the GTP-bound state of this protein, leading to prolonged activation and signaling.33 Our RNA interference experiments, in which knockdown of mutated NRAS, but not wild type NRAS, resulted in diminished levels of phospho-c-Met place c-Met phosphorylation downstream of activated NRAS. Supporting this pathway from a different perspective are data from murine NIH3T3 and C127 cells which over-express c-Met when transformed by the Ras oncogene.34 Other reports, however, suggest the opposite, i.e., that binding of HGF to c-Met leads to activation of N-Ras and the MAPK pathway.35 An attractive model that incorporates all of these findings places HGF expression, and in some cell types, c-Met expression downstream of activated NRAS, providing for an autocrine activation loop. Notably, our findings in cultured melanoma cells argue against this hypothesis as c-Met activation in this system requires exogenous HGF, regardless of NRAS mutational status. It is possible, however, that the NRAS-mutated melanoma cell lines express HGF but do not secrete the protein as an artifact of tissue culture. There is surprisingly little known about HGF expression by human cutaneous melanomas or its association with NRAS and BRAF genotypes; this has become a new focus of research in our laboratory. Interestingly, some wt/wt cells and tumors, while not identical to the NRAS mutants, behaved similarly in several in vitro assays and demonstrated high levels of phospho-c-Met by IHC. One obvious commonality between the wt/wt tumors and NRAS-mutated tumors is the absence of a BRAF mutation, raising the possibility that the BRAF genotypic background also partially determines c-Met expression and activation.
Confounding the in vitro findings are the data in Table 1 showing a higher level of invasiveness for BRAF-mutated tumors compared to those with NRAS mutations. This is contrary to the expectation that BRAF-mutated tumors should be less aggressive as they are less likely to carry activated c-MET. In keeping with the higher Breslow levels in the tumors with BRAF mutations, this patient group also presented with higher AJCC stages. Importantly, these findings differ from our larger, previously published study population in which BRAF tumors were generally intermediate in depth and behavior to those with NRAS mutations and wt/wt genotypes.21 Furthermore, they are contradictory to the general consensus of aggressive behavior of c-MET activated tumors, and are inconsistent with our in vitro data. We therefore suspect that the observed clinical differences between NRAS and BRAF mutated tumors in this small study population are artifacts of sample size and selection. Issues such as these can be better addressed in experiments in which activated c-MET is expressed in melanomas of various genotypes. Clearly this issue must be clarified in future studies with larger patient sample numbers.
Dissecting these proposed interactions between c-Met and mutated NRAS will require precise and diligent molecular analyses. Unfortunately, such efforts are hampered by the paucity of NRAS-mutated melanoma cell lines; wt/wt lines are rarer still. We believe that future research may be better served by the use of primary cell cultures from patients with tumors of known genotype, an approach that is becoming quite feasible as NRAS and BRAF tumor genotyping assays are evolving as part of the standard clinical melanoma evaluation in many research institutions. With both primary cultures and paraffin embedded tissue from the same patient, we would be better enabled to move forward with mechanistic studies exploring the interaction of genotype and c-Met activation with such processes as angiogenesis, invasion, and apoptosis.
In conclusion, we have discovered an association of mutated NRAS with increased HGF-dependent activation of c-Met and with enhanced sensitivity to c-Met inhibition. These data suggest that progression of NRAS-mutated melanoma could be highly dependent on c-Met signaling, and that blocking this pathway may represent an effective therapeutic approach for this patient subset. Such benefits may extend to a portion of patients with wt/wt tumors, as well. To date, the major therapeutic focus in advanced melanoma has been the BRAF mutation. Our findings now uniquely offer the potential for mutation-directed treatment for the remainder of melanoma patients who currently lack options for targeted therapy.
NOVELTY and IMPACT.
This study is the first to demonstrate a preferential utilization of the c-Met pathway by NRAS-mutated melanomas and enhanced sensitivity of these tumors to pharmacologic c-Met inhibition. These findings may translate to c-Met targeted therapy for the subset of melanoma patients whose tumors carry this genotype.
Acknowledgments
Grant support: This work was supported in part by the UT MDACC SPORE in Melanoma P50 093459 (EAG, SE, and JAE; Grimm, PI) and NCI Grant CCSG CA16672 (DNA Analysis Core).
STR DNA fingerprinting was performed by the Cancer Center Support Grant funded Characterized Cell Line core, NCI # CA16672. We are grateful for the support of the Mirium and Jim Mulva Melanoma Research Foundation, Houston, Texas. We also thank Ms. Sandra A. Kinney for IHC staining, Dr. Wuguo (Mark) Deng for calculating IC50 values, Dr. Kevin Kim for providing mutant NRAS specific siRNA, and Pfizer for providing PHA665752.
Abbreviations
- c-Met
cellular mesenchymal to epithelial transition factor
- HGF
hepatocyte growth factor
- PI3K
phosphatidylinositol-3 kinase
- MAPK
mitogen-activated protein kinase
- IHC
immunohistochemistry
Footnotes
Michael A. Davies declares a potential COI due to research support from Glaxo Smithkline, Astra Zeneca, Merck and Roche.
Reference List
- 1.To CT, Tsao MS. The roles of hepatocyte growth factor/scatter factor and met receptor in human cancers (Review) Oncol Rep. 1998;5:1013–24. doi: 10.3892/or.5.5.1013. [DOI] [PubMed] [Google Scholar]
- 2.Gherardi E, Sharpe M, Lane K, Sirulnik A, Stoker M. Hepatocyte growth factor/scatter factor (HGF/SF), the c-met receptor and the behaviour of epithelial cells. Symp Soc Exp Biol. 1993;47:163–81. [PubMed] [Google Scholar]
- 3.Park M, Dean M, Cooper CS, Schmidt M, O’Brien SJ, Blair DG, Vande Woude GF. Mechanism of met oncogene activation. Cell. 1986;45:895–904. doi: 10.1016/0092-8674(86)90564-7. [DOI] [PubMed] [Google Scholar]
- 4.Ma PC, Jagadeeswaran R, Jagadeesh S, Tretiakova MS, Nallasura V, Fox EA, Hansen M, Schaefer E, Naoki K, Lader A, Richards W, Sugarbaker D, et al. Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Res. 2005;65:1479–88. doi: 10.1158/0008-5472.CAN-04-2650. [DOI] [PubMed] [Google Scholar]
- 5.Peruzzi B, Bottaro DP. Targeting the c-Met signaling pathway in cancer. Clin Cancer Res. 2006;12:3657–60. doi: 10.1158/1078-0432.CCR-06-0818. [DOI] [PubMed] [Google Scholar]
- 6.Furge KA, Zhang YW, Vande Woude GF. Met receptor tyrosine kinase: enhanced signaling through adapter proteins. Oncogene. 2000;19:5582–89. doi: 10.1038/sj.onc.1203859. [DOI] [PubMed] [Google Scholar]
- 7.Ma PC, Tretiakova MS, Nallasura V, Jagadeeswaran R, Husain AN, Salgia R. Downstream signalling and specific inhibition of c-MET/HGF pathway in small cell lung cancer: implications for tumour invasion. Br J Cancer. 2007;97:368–377. doi: 10.1038/sj.bjc.6603884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Puri N, Ahmed S, Janamanchi V, Tretiakova M, Zumba O, Krausz T, Jagadeeswaran R, Salgia R. c-Met is a potentially new therapeutic target for treatment of human melanoma. Clin Cancer Res. 2007;13:2246–53. doi: 10.1158/1078-0432.CCR-06-0776. [DOI] [PubMed] [Google Scholar]
- 9.Halaban R, Rubin JS, Funasaka Y, Cobb M, Boulton T, Faletto D, Rosen E, Chan A, Yoko K, White W, et al. Met and hepatocyte growth factor/scatter factor signal transduction in normal melanocytes and melanoma cells. Oncogene. 1992;7:2195–2206. [PubMed] [Google Scholar]
- 10.Cruz J, Reis-Filho JS, Silva P, Lopes JM. Expression of c-met tyrosine kinase receptor is biologically and prognostically relevant for primary cutaneous malignant melanomas. Oncology. 2003;65:72–82. doi: 10.1159/000071207. [DOI] [PubMed] [Google Scholar]
- 11.Saitoh K, Takahashi H, Sawada N, Parsons PG. Detection of the c-met proto-oncogene product in normal skin and tumours of melanocytic origin. J Pathol. 1994;174:191–9. doi: 10.1002/path.1711740308. [DOI] [PubMed] [Google Scholar]
- 12.Riker AI, Enkemann SA, Fodstad O, Liu S, Ren S, Morris C, Xi Y, Howell P, Metge B, Samant RS, Shevde LA, Li W, et al. The gene expression profiles of primary and metastatic melanoma yields a transition point of tumor progression and metastasis. BMC Med Genomics. 2008;1:13. doi: 10.1186/1755-8794-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Otsuka T, Takayama H, Sharp R, Celli G, LaRochelle WJ, Bottaro DP, Ellmore N, Vieira W, Owens JW, Anver M, Merlino G. c-Met autocrine activation induces development of malignant melanoma and acquisition of the metastatic phenotype. Cancer Res. 1998;58:5157–67. [PubMed] [Google Scholar]
- 14.Goydos JS, Mann B, Kim HJ, Gabriel EM, Alsina J, Germino FJ, Shih W, Gorski DH. Detection of B-RAF and N-RAS mutations in human melanoma. J Am Coll Surg. 2005;200:362–70. doi: 10.1016/j.jamcollsurg.2004.10.032. [DOI] [PubMed] [Google Scholar]
- 15.Weber J. Immunotherapy for melanoma. Curr Opin Oncol. 2011;23:163–9. doi: 10.1097/CCO.0b013e3283436e79. [DOI] [PubMed] [Google Scholar]
- 16.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Drummer R, Garbe C, Testori A, Maio M, Hogg D, Lorogan P, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davies MA, Samuels Y. Analysis of the genome to personalize therapy for melanoma. Oncogene. 2010;29:5545–55. doi: 10.1038/onc.2010.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ball NJ, Yohn JJ, Morelli JG, Norris DA, Golitz LE, Hoeffler JP. RAS mutations in human melanoma: a marker of malignant progression. J Invest Dermatol. 1994;102:285–90. doi: 10.1111/1523-1747.ep12371783. [DOI] [PubMed] [Google Scholar]
- 19.Uffort DG, Grimm EA, Ellerhorst JA. NF-kappaB mediates mitogen-activated protein kinase pathway-dependent iNOS expression in human melanoma. J Invest Dermatol. 2009;129:148–54. doi: 10.1038/jid.2008.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Romano P, Manniello A, Aresu O, Armento M, Cesaro M, Parodi B. Cell line data base: structure and recent improvements towards molecular authentication of human cell lines. Nucleic Acids Res. 2009;37:D925–32. doi: 10.1093/nar/gkn730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ellerhorst JA, Greene VR, Ekmekcioglu S, Warneke CL, Johnson MM, Cooke CP, Wang LE, Prieto VG, Gershenwald JE, Wei Q, Grimm EA. Clinical correlates of NRAS and BRAF mutations in primary human melanoma. Clin Cancer Res. 2011;17:229–35. doi: 10.1158/1078-0432.CCR-10-2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ellerhorst JA, Prieto VG, Ekmekcioglu S, Broemeling L, Yekell S, Chada S, Grimm EA. Loss of MDA-7 expression with progression of melanoma. J Clin Oncol. 2002;20:1069–74. doi: 10.1200/JCO.2002.20.4.1069. [DOI] [PubMed] [Google Scholar]
- 23.Chattopadhyay C, el-Naggar AK, Williams MD, Clayman GL. Small molecule c-MET inhibitor PHA665752: effect on cell growth and motility in papillary thyroid carcinoma. Head Neck. 2008;30:991–1000. doi: 10.1002/hed.20816. [DOI] [PubMed] [Google Scholar]
- 24.Matsubara D, Ishikawa S, Oguni S, Aburatani H, Fukayama M, Niki T. Molecular predictors of sensitivity to the MET inhibitor PHA665752 in lung carcinoma cells. J Thorac Oncol. 2010;5:1317–24. doi: 10.1097/JTO.0b013e3181e2a409. [DOI] [PubMed] [Google Scholar]
- 25.Burgess T, Coxon A, Meyer S, Sun J, Rex K, Tsuruda T, Chen Q, Ho SY, Li L, Kaufman S, McDorman K, Cattley RC, et al. Fully human monoclonal antibodies to hepatocyte growth factor with therapeutic potential against hepatocyte growth factor/c-Met-dependent human tumors. Cancer Res. 2006;66:1721–29. doi: 10.1158/0008-5472.CAN-05-3329. [DOI] [PubMed] [Google Scholar]
- 26.Jin H, Yang R, Zheng Z, Romero M, Ross J, Bou-Reslan H, Carano RA, Kasman I, Mai E, Young J, Zha J, Zhang Z, et al. MetMAb, the one-armed 5D5 anti-c-Met antibody, inhibits orthotopic pancreatic tumor growth and improves survival. Cancer Res. 2008;68:4360–8. doi: 10.1158/0008-5472.CAN-07-5960. [DOI] [PubMed] [Google Scholar]
- 27.Firon M, Shaharabany M, Altstock RT, Horev J, Abramovici A, Resau JH, Vande Woude GF, Tsarfaty I. Dominant negative Met reduces tumorigenicity-metastasis and increases tubule formation in mammary cells. Oncogene. 2000;19:2386–97. doi: 10.1038/sj.onc.1203557. [DOI] [PubMed] [Google Scholar]
- 28.Shinomiya N, Gao CF, Xie Q, Gustafson M, Waters DJ, Zhang YW, Vande Woude GF. RNA interference reveals that ligand-independent met activity is required for tumor cell signaling and survival. Cancer Res. 2004;64:7962–70. doi: 10.1158/0008-5472.CAN-04-1043. [DOI] [PubMed] [Google Scholar]
- 29.Christensen JG, Zou HY, Arango ME, Li Q, Lee JH, McDonnell SR, Yamazaki S, Alton GR, Mroczkowski B, Los G. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol Cancer Ther. 2007;6:3314–22. doi: 10.1158/1535-7163.MCT-07-0365. [DOI] [PubMed] [Google Scholar]
- 30.Zou HY, Li Q, Lee JH, Arango ME, McDonnell SR, Yamazaki S, Koudriakova TB, Alton G, Cui JJ, Kung PP, Nambu MD, Los G, et al. An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007;67:4408–17. doi: 10.1158/0008-5472.CAN-06-4443. [DOI] [PubMed] [Google Scholar]
- 31.Buchanan SG, Hendle J, Lee PS, Smith CR, Bounaud PY, Jessen KA, Tang CM, Huser NH, Felce JD, Froning KJ, Peterman MC, Aubol BE, et al. SGX523 is an exquisitely selective, ATP-competitive inhibitor of the MET receptor tyrosine kinase with antitumor activity in vivo. Mol Cancer Ther. 2009;8:3181–90. doi: 10.1158/1535-7163.MCT-09-0477. [DOI] [PubMed] [Google Scholar]
- 32.Munshi N, Jeay S, Li Y, Chen CR, France DS, Ashwell MA, Hill J, Moussa MM, Leggett DS, Li CJ, et al. ARQ197, a highly selective small molecule inhibitor of c-Met, with selective antitumor properties in a broad spectrum of human cancer cells. 2007. AACR Annual Meeting; 2007. Ref type: abstract. [Google Scholar]
- 33.Polakis P, McCormick F. Structural requirements for the interaction of p21ras with GAP, exchange factors, and its biological effector target. J Biol Chem. 1993;268:9157–60. [PubMed] [Google Scholar]
- 34.Webb CP, Taylor GA, Jeffers M, Fiscella M, Oskarsson M, Resau JH, Vande Woude GF. Evidence for a role of Met-HGF/SF during Ras-mediated tumorigenesis/metastasis. Oncogene. 1998;17:2019–25. doi: 10.1038/sj.onc.1202135. [DOI] [PubMed] [Google Scholar]
- 35.Ueoka Y, Kato K, Wake N. Hepatocyte growth factor modulates motility and invasiveness of ovarian carcinomas via ras mediated pathway. Mol Cell Endocrinol. 2003;202:81–8. doi: 10.1016/s0303-7207(03)00067-4. [DOI] [PubMed] [Google Scholar]