

Abstract
Atopic dermatitis (AD) has long been associated with Staphylococcus aureus skin colonization or infection and is typically managed with regimens that include antimicrobial therapies. However, the role of microbial communities in the pathogenesis of AD is incompletely characterized. To assess the relationship between skin microbiota and disease progression, 16S ribosomal RNA bacterial gene sequencing was performed on DNA obtained directly from serial skin sampling of children with AD. The composition of bacterial communities was analyzed during AD disease states to identify characteristics associated with AD flares and improvement post-treatment. We found that microbial community structures at sites of disease predilection were dramatically different in AD patients compared with controls. Microbial diversity during AD flares was dependent on the presence or absence of recent AD treatments, with even intermittent treatment linked to greater bacterial diversity than no recent treatment. Treatment-associated changes in skin bacterial diversity suggest that AD treatments diversify skin bacteria preceding improvements in disease activity. In AD, the proportion of Staphylococcus sequences, particularly S. aureus, was greater during disease flares than at baseline or post-treatment, and correlated with worsened disease severity. Representation of the skin commensal S. epidermidis also significantly increased during flares. Increases in Streptococcus, Propionibacterium, and Corynebacterium species were observed following therapy. These findings reveal linkages between microbial communities and inflammatory diseases such as AD, and demonstrate that as compared with culture-based studies, higher resolution examination of microbiota associated with human disease provides novel insights into global shifts of bacteria relevant to disease progression and treatment.
Atopic dermatitis (AD, “eczema,” OMIM 603165) is a chronic, relapsing, intensely pruritic inflammatory skin disorder that can be successfully treated with varying combinations of topical or systemic antibiotics, corticosteroids, and dilute bleach baths (Huang et al. 2009). The prevalence of AD has more than doubled in industrialized countries with no clear cause (Asher et al. 2006; Shaw et al. 2011) and at a high cost (Bickers et al. 2006). More than half of children with moderate to severe AD develop allergic rhinitis and/or asthma, atopic disorders associated with significant morbidity and rare mortality. Genetic as well as environmental factors affect the expression of allergic diseases, including AD (von Mutius 2000; Cramer et al. 2010). Mutations in FLG, the gene encoding the skin barrier protein filaggrin, are associated with AD, particularly in patients who subsequently develop asthma and/or allergic rhinitis, suggesting that epicutaneous sensitization may contribute to atopic disease (Palmer et al. 2006; Sandilands et al. 2007).
AD patients experience frequent cutaneous infections, and Staphylococcus aureus is commonly cultured from lesional and nonlesional AD skin (Leyden et al. 1974). Reduced antimicrobial peptide expression in the skin of AD patients may contribute to this susceptibility (Ong et al. 2002). While individual microbes causative of common AD skin infections have been studied, it is increasingly clear that individual microbes function within larger bacterial communities (Cogen et al. 2010b).
High-throughput DNA sequencing of the bacterial 16S rRNA gene has revealed a vastly greater bacterial diversity on healthy human skin than demonstrated by culture-based methods (Costello et al. 2009; Grice et al. 2009). The skin's topography and microenvironments are powerful determinants of microbial community structure at particular skin sites (Gao et al. 2007; Costello et al. 2009; Grice et al. 2009). AD preferentially involves the antecubital and popliteal regions, sites that harbor similar groups of organisms and share distinct compositions of microbial communities (Grice et al. 2009). These findings suggest that microbial communities might underlie or contribute to observed predilections of some dermatologic disorders for stereotypical sites.
To examine the role of the skin microbiome in AD, we performed a skin microbiome study of AD disease states (baseline disease state, disease flare, and post-treatment for disease flare) in 12 pediatric patients with moderate-to-severe AD and 11 healthy controls, aged 2–15 yr (Table 1; Supplemental Table S1). In this study, we demonstrate how different disease states, severity, and treatments relate to microbial dynamics in this common skin disorder. We report dramatic reductions in the skin microbial diversity during AD flares, which is restored with common AD therapies.
Table 1.
Clinical characteristics of the participantsa

Results
Characteristics of bacterial communities associated with AD
To investigate the role of bacterial communities in AD, we obtained skin samples from 12 children with moderate-to-severe AD and from 11 controls recruited to the NIH Clinical Center between August 2008 and July 2010. Disease severity was assessed at predetermined sampling timepoints: baseline, stable disease state (B); acute disease flare (F); and 10–14-d post-flare treatment (PF). Disease severity was assessed quantitatively with SCORAD (SCORing AD), a well-validated clinical assessment tool (Williams et al. 1994b; Kunz et al. 1997; Oranje et al. 2007). We obtained skin samples from the left and right antecubital (Ac) and popliteal creases (Pc)—sites of predilection for AD in this age group (Fig. 1A). We also sampled volar forearms (Vf) as control skin sites that are adjacent to, but not a classical site of disease predilection itself, and the nares (N) as a reservoir for S. aureus colonization (Archer and Climo 2001; von Eiff et al. 2001). We sequenced skin samples from 36 AD patient visits and 24 control visits for a total of 151,924 near-full-length 16S ribosomal RNA (rRNA) genes (Supplemental Table S2). Study design is detailed in the Methods section.
Figure 1.

AD disease severity. (A) Representative clinical images of the antecubital (Ac, left) and popliteal creases (Pc, right) in two patients with overall disease severity scores (objective SCORAD). (B) Longitudinal objective SCORAD trend for each patient (n = 12) at baseline, flare, and postflare.
Mean objective SCORAD scores designating AD disease severity were significantly elevated during flares (42 ± 2.2) as compared with baseline (22 ± 3.1, P < 7.3 × 10−4) and postflare (18 ± 3.0, P < 0.0038) (Fig. 1B). Categorization of baseline, flare, and postflare was based on patient clinical status and specified frequency of AD treatment administration (see Methods section for detailed classification criteria). Flare was defined by worsening disease irrespective of use of therapy >24 h prior to sampling. We examined the recent treatment history prior to flares because we predicted that the timing of treatments in relation to sampling might influence the skin microbiome. We found that while no significant differences were detected in objective SCORAD scores (P < 0.22), flare timepoints could be grouped into patients that used (1) “no-treatment” (n = 7), defined as patients who reported no use of topical medications (corticosteroids, calcineurin inhibitors, or antibiotics) for ≥7 d, and no ingestion of oral antibiotics ≥4 wk prior to flare skin sampling, or (2) “intermittent-treatment” (n = 5), defined as patients who reported use of topical medications in the previous 7 d and/or ingestion of oral antibiotics in the previous 4 wk before skin sampling (Table 2).
Table 2.
Atopic dermatitis treatments prior to flare sampling

To investigate a possible relationship, we analyzed the association between AD disease severity and Shannon diversity, an ecological measure of microbial communities that considers: (1) richness, or the total number of bacterial types, and (2) evenness, or the relative proportion of these bacterial types. The antecubital and popliteal creases are considered sites of disease predilection and showed similar results. We averaged these sites per subject (“AcPc”) for subsequent analyses, unless otherwise indicated, to avoid treating related samples from the same individual as biological replicates. The partial correlation between objective SCORAD and Shannon diversity of AcPc, adjusting for disease state, was significantly inversely correlated (r = −0.57, P < 3.6 × 10−4) (Fig. 2A), indicating that severe AD was associated with lower skin bacterial diversity at sites of disease predilection.
Figure 2.

Microbial community-level statistics in the AD microbiome. (A) Relationship between objective SCORAD and Shannon diversity in the Ac of AD patients. Partial correlation (adjusting for disease state). (B) Longitudinal Shannon diversity trend in AD grouped by no-treatment (trt) and intermittent-trt flares (n = 12, Ac). (C) Mean Shannon diversity ± SEM in controls and all AD disease states (Ac, Pc, volar forearm [Vf], nares [N]). (D) Mean theta (θ) similarity coefficients ± SEM. Pairwise comparisons of community structure between individuals within a control or AD disease group. Bars represent average of all pairwise comparisons of community structure for control individuals to other controls, baselines to baselines, flares(no-trt)-flares(no-trt), flare(intermittent-trt)-flare(intermittent-trt), and postflares-postflares.
AD patients using no treatments during flares exhibited markedly reduced Shannon diversity in the antecubital creases as compared with intermittent-treatment use (P < 0.025) (Fig. 2B,C; Supplemental Table S3). Shannon diversity during intermittent-treatment flares was not significantly different from baseline, postflares, or controls. Similar trends were observed for popliteal creases, another site of disease predilection (Fig. 2C; Supplemental Table S3). These results suggest that cutaneous microbial diversity is associated with disease status in the context of recent treatment history, and that AD treatments can prevent or reverse the change in diversity associated with flares even before clinical improvement is seen. In contrast to the antecubital and popliteal creases, diversity in the nares as well as the volar forearm was relatively constant, demonstrating that changes in bacterial diversity were specific to sites of disease predilection (Fig. 2C; Supplemental Table S3).
To assess how distinct the skin microbiomes for each AD disease state and controls were from each other, we compared the level of similarity of bacterial community structures with the Yue-Clayton theta similarity coefficient (θ) (Yue and Clayton 2005). The θ index takes into consideration the number of bacterial species present and their relative abundances in the two communities being compared (Yue and Clayton 2005; Schloss and Handelsman 2006) with θ = 1 indicating identical community structure and θ = 0 indicating absolute dissimilarity. AD patients within the no-treatment flare group (θ = 0.91 ± 0.029) were statistically significantly more similar to each other than baseline, intermittent-treatment flares, and postflare (Fig. 2D; Supplemental Fig. S1; Supplemental Table S4), far exceeding the reference point of interpersonal variation observed in the within-group θ for controls (θ = 0.26 ± 0.025, P < 4.3 × 10−10). Our results underscored that no-treatment flares were distinguishable from intermittent-treatment flares and likely reflected the natural disease course unaltered by recent medications.
To investigate whether these bacterial community shifts across timepoints were unique to the AD cohort, we calculated both Shannon and Theta indices across visits for the healthy controls, who were sampled at similar time intervals as AD patients (Supplemental Table S5). Mean changes in Shannon diversity between visits were significantly higher for no-treatment AD flare patients (baseline/flare, and flare/postflare) as compared with healthy controls (P < 0.002 and P < 0.02, respectively, for Ac and Pc). Similarly, mean theta values between visits were significantly lower in the no-treatment AD flare group than healthy controls in the Ac (P < 0.03). We conclude from this analysis that the skin bacterial community shifts across timepoints are significantly different in AD patients as compared with similarly sampled healthy controls.
Selective bacterial shifts and AD disease states
To identify bacteria contributing to the reduction in AD microbial diversity, we classified the phylum, order, and genus of the sequences (Supplemental Fig. S2; Supplemental Table S6) at each site. To determine the similarity of the bacterial communities between sites at a taxonomic level, we first performed cross-site correlations of the abundances of each taxa at the phylum-order level (Supplemental Fig. S3). In AD patients, the antecubital and popliteal creases were highly similar (partial correlation adjusting for disease state r = 0.80), exceeding the similarity of these sites in controls (r = 0.68, Fisher r-to-z transformation, P < 0.0002). The similarity between the antecubital and popliteal creases relates to the observed localization of AD in these sites and further justifies combining these sites for analysis. Antecubital creases were similar to the adjacent volar forearm in both AD and control individuals (r = 0.79 and 0.81, respectively), suggesting that the typically unaffected skin of the volar forearm significantly correlated with sites of disease predilection. In general, the nares taxa were poorly correlated with the skin sites for all individuals.
Examining the relative abundance of specific taxa at each timepoint, we observed that the antecubital and popliteal creases were strikingly dominated by the genus Staphylococcus. The no-treatment flares had the highest abundance of bacterial 16S genes classified as Staphylococcus at 90 ± 1.6% as compared with other AD patients and controls (35 ± 2.2%, P < 0.013 [baseline]; 31 ± 5.0%, P < 0.050 [intermittent-treatment flares]; 20 ± 1.3%, P < 2.0 × 10−4 [postflare]; 16 ± 1.8%, P < 6.2 × 10−4 [controls]) (Fig. 3A; Supplemental Table S7).
Figure 3.

Bacterial taxonomic classifications in the AD skin microbiome. (A) Mean relative abundance of the 14 major phyla-order in the antecubital (Ac) and popliteal creases (Pc) for controls and AD disease states: baseline, flare (no-treatment [trt] and intermittent-trt), and postflare (Supplemental Table S13 for order of subjects). (B) Mean relative abundances for Ac and Pc of species-level classifications of staphylococcal species. Order of subjects follows A.
The genus Staphylococcus includes several bacterial species relevant to clinical disease, including S. epidermidis as a skin commensal (Wisplinghoff et al. 2004; Hidron et al. 2008) and S. aureus as a pathogen with a known association with AD. To distinguish among Staphylococcal species, we generated a custom 16S rRNA sequence database to uniquely classify the Staphylococcal sequences (Fig. 3B). The proportion of S. aureus in the no-treatment flares was 65 ± 3.5%, statistically significantly higher than baseline, postflare, and controls (Fig. 4A; Supplemental Table S7). The proportion of S. aureus in the intermittent-treatment flares (15 ± 2.5%) was significantly higher than controls (P < 0.020), but not significantly higher than baseline, when some AD patients carry large amounts of S. aureus.
Figure 4.

Relationship between staphylococcal species and AD. (A) Longitudinal trend of mean proportion of S. aureus in AD in antecubital and popliteal creases (AcPc, n = 12) grouped by no-treatment (trt) and intermittent-trt flares. (B) Proportion of S. aureus and Shannon diversity index in AcPc. Partial correlation (adjusting for disease state, AcPc). (C) Longitudinal trend of mean proportion of S. epidermidis in AcPc. (D) Correlation of proportion of S. aureus versus objective SCORAD for each site (Ac, Pc, Volar forearm/Vf, Nares/N). Partial correlation (adjusting for disease state and site).
With significant shifts in S. aureus abundance observed in AD, we then examined the relationship between the relative abundances of S. aureus and Shannon diversity. Adjusting for disease status, the partial correlation between S. aureus abundance and Shannon diversity was r = −0.68, P < 6.3 × 10−6, indicating that increases in S. aureus accounted for reductions in overall diversity observed in this AD cohort (Fig. 4B; Supplemental Table S8).
We then analyzed the relationship between the proportion of S. aureus and disease severity. Given the heterogeneity of the microbiome over the human skin surface (Grice et al. 2009), we evaluated the contribution of skin site. Partial correlation between S. aureus abundance and disease severity, adjusting for disease state and site, was significant: antecubital (r = 0.59, P < 2.1 × 10−4) and popliteal creases (r = 0.57, P < 4.3 × 10−4) (Fig. 4D; Supplemental Table S9). Volar forearm sites also showed a positive partial correlation (r = 0.70, P < 1.4 × 10−5), possibly related to a field effect, or extension of changes in the microbiome to adjacent skin. This positive partial correlation observed for the volar forearms appeared to be influenced by the increase in body surface area of affected skin extending to the volar forearms during AD disease flares. The nasal site did not demonstrate a positive correlation. These findings suggest that overabundance of cutaneous S. aureus and the associated loss of microbiome diversity are intimately tied to the pathogenesis of AD. Moreover, these data emphasize the regional variation of the skin microbiome at distinct sites and the importance of rational selection of sampling sites in microbiome studies.
Given the dominance of S. aureus during AD flares, we sought to determine whether this reduction in diversity was attributed solely to a relative overexpansion of S. aureus in the local microbiome. Interestingly, trends in decreased microbial diversity and the similarities of the microbiomes of flaring patients persisted even when the abundance of S. aureus sequences was excluded from the analysis and sequence numbers were normalized post-removal (Supplemental Fig. S4; Supplemental Table S10). After subtracting S. aureus sequences, we recalculated Shannon and θ diversity metrics. The decrease in Shannon diversity during the no-treatment flares remained significantly different in Ac and Pc (P < 0.05 flare vs. baseline, postflare, and controls), and was significantly correlated with increasing disease severity (Ac: r = −0.48, P = 0.004; Pc: r = −0.44, P = 0.009) as compared with the other timepoints and with controls. After removal of S. aureus sequences, the no-treatment flares in Ac and Pc remained similar to each other based on Theta index as well. These results suggested that similarities in microbiomes extended beyond S. aureus during no-treatment AD flares.
Therefore, in addition to shifts in S. aureus, we sought to determine whether the presence of novel bacteria or absence of protective bacteria could play a role in AD flares. While the abundance of S. epidermidis in AD is conflicting in the literature (Gloor et al. 1982; Williams et al. 1990; Higaki et al. 1999), the proportion of S. epidermidis during flares was significantly higher than during postflares and in controls (P < 0.034 and P < 0.006, respectively) (Fig 4C). S. epidermidis was also the dominant staphylococcal species in the AcPc of controls (Spearman correlation, r = 0.98, P < 8.9 × 10−8; Supplemental Table S7). These findings demonstrate distinct selective shifts among at least two staphylococcal species relative to disease status.
We also identified additional genera over-represented at baseline and postflare. We used the Wilcoxon rank-sum test for comparisons of no-treatment flares with baseline and postflare. We observed a significant relative increase in Streptococcus, Corynebacterium, and Propionibacterium species post-treatment (Supplemental Table S11). With the sequencing depth in this study (Supplemental Fig. S5; Supplemental Table S12) we also identified several lower-abundance bacteria such as the Rothia species, which were also statistically significantly increased between disease timepoints (Supplemental Table S11). To compare the taxa present during baseline and postflare, we used matched-pairs analyses to examine whether the same taxa were abundant, or whether new taxa appeared during postflare. In total, 58% of taxa (272 of 469) observed at either baseline or postflare were present at both timepoints. Of the taxa present in >9/12 patients (Supplemental Table S11), all taxa were present at both baseline and postflare, none at statistically significantly different quantities. We also determined that the abundance of genera between baseline and postflare was significantly correlated (partial correlation adjusting for patient, r = 0.61, P < 2.2 × 10−16), indicating that there is a relative expansion of taxa already present in the microbiome with a relatively low number of newly introduced bacteria detected at our level of sampling.
Genomic analyses and culture data comparison
Since cultivation studies have demonstrated a higher likelihood of S. aureus colonization of AD skin in positive nasal carriers (Williams et al. 1998; Breuer et al. 2002), we examined the results from S. aureus cultures concurrently obtained with genomic sampling. Assessment of S. aureus nares bioburden (measured as colonies per sample: 0, 1–10, >10) was significantly associated with relative abundance of S. aureus nares genomic sequences P < 0.0072 (0 vs. 1–10 colonies/sample); P < 2.1 × 10−6 (0 vs. >10 colonies/sample) (Supplemental Fig. S6; Supplemental Table S13), suggesting that genomic identification tracked with traditional cultivation methods for this easily cultured microbe. All AD patients had ≥1 nares culture positive for S. aureus as compared with 55% of controls (Table 1). Genomic analysis of nares samples did not detect an association between mean objective SCORAD and the relative abundance of S. aureus genomic sequences (Fig. 4D; Supplemental Table S9).
In an effort to determine whether a particular S. aureus subtype predominated during flares, we examined the microbial population dynamics of unique S. aureus 16S rRNA sequences. The heterogeneity of the 16S rRNA sequences supports earlier studies demonstrating absence of predominant S. aureus clones or biotypes (Hoeger et al. 1992; Lomholt et al. 2005; Kim et al. 2009; Yeung et al. 2011).
Associations with treatment
Prior studies have shown conflicting results regarding the effect of topical steroids and antimicrobials on the abundance of S. aureus (Lever et al. 1988; Stalder et al. 1994; Williams et al. 1998; Ravenscroft et al. 2003; Gong et al. 2006; Hung et al. 2007; Bath-Hextall et al. 2010). One study demonstrated that use of dilute sodium hypochlorite baths did not completely eliminate S. aureus, yet resulted in improved clinical outcomes for AD patients (Huang et al. 2009). Since bacterial diversity in AD skin differed between intermittent-treatment and no-treatment flares, we sought to confirm whether treatment within a defined period of time prior to flares influenced Shannon diversity. We examined samples obtained from four additional AD patients during disease flares. Other sampling timepoints for these four patients did not fulfill strict predetermined criteria for classification as baseline and flare, and were not included in original analyses. For the patients who used no treatment prior to flare, relative Shannon diversity was low, similar to findings from the no-treatment flare group (Table 2; Supplemental Table S14). In contrast, AD patients who used any treatment prior to flare sampling exhibited the higher diversity characteristic of the intermittent-treatment flare group. One particular AD patient was sampled during two different flares, but with two different treatment regimens prior to flare sampling. During the first flare, the patient used no topical treatments in the week prior to sampling and demonstrated reduced Shannon diversity (0.626). Before the second flare sampling, the patient used intermittent dilute bleach baths and had a higher Shannon diversity (1.650). Using the Wilcoxon rank-sum test, the mean Shannon diversity for all flares was statistically significantly different between all no-treatment and intermittent-treatment flares (Fig. 5) (P < 0.0025, n = 16). These findings suggest that antimicrobial or anti-inflammatory medications decreased S. aureus predominance (Supplemental Table S14), affecting bacterial diversity during flares.
Figure 5.

Shannon diversity index during flares. Circles indicate AD patients with a full longitudinal cycle (baseline, flare, and postflare). Triangles indicate flares from four additional (add'l) patients. Means between no-treatment (trt) and intermittent-trt tested with Wilcoxon rank-sum test (P < 0.0025, n = 16).
Discussion
AD represents a classic inflammatory skin disorder associated with a specific bacterial species. It is often managed with antimicrobial approaches and as such, AD serves as an example of a disorder in which the contribution of microorganisms to disease course can be examined via metagenomic and microbial community analyses. Using direct 16S rRNA gene sequencing, we defined and characterized the fluctuation of skin bacterial communities during the representative basal (baseline), exacerbation (flare), and resolution (postflare) phases of AD. Our microbiome analyses demonstrated a topographic and temporal shift in the predominant bacterial species and the bacterial diversity in the skin of pediatric AD patients.
A hallmark of AD is the chronic, recurrent flaring of intensely itchy skin. By serially analyzing AD patients, we demonstrated a strong association between worsening disease severity and lower skin bacterial diversity. We also determined that this microbiome shift is primarily localized to sites of disease predilection. Because many dermatologic disorders present with a stereotypical body distribution, the site-specificity of skin bacterial communities suggests that not only do particular ecological niches of the skin favor the growth of certain bacteria, but that communities of microbes are also important in the initiation and perpetuation of certain skin diseases.
Direct 16S rRNA gene sequencing of samples obtained during different disease states of AD provided a less-biased microbiological survey of the selective shifts of Staphylococcus, particularly of S. aureus. While S. aureus skin colonization and infection is a common feature of AD (Leyden et al. 1974), we have expanded these observations by demonstrating concurrent fluctuations of other skin bacteria. For example, S. epidermidis is the predominant Staphylococcus species in the skin of controls and has been considered a commensal in healthy skin (Iwase et al. 2010; Lai et al. 2010), with the ability to inhibit S. aureus (Iwase et al. 2010). Although results from previous culture-based studies are conflicting (Gloor et al. 1982; Williams et al. 1990; Higaki et al. 1999), our genomic analysis showed that the proportion of S. epidermidis consistently increased during no-treatment flares. The simultaneous prevalence of both S. aureus and S. epidermidis that we observed during disease flares provides new insights into the relationship between staphylococci. These staphylococcal species may share a mutualistic or commensal relationship to enhance common resistance to antimicrobial peptides (Peschel et al. 2001; Sieprawska-Lupa et al. 2004; Lai et al. 2007; Li et al. 2007) or enhance binding to exposed extracellular matrix proteins in inflamed AD skin (Nilsson et al. 1998; McCrea et al. 2000; Cho et al. 2001; Williams et al. 2002). Alternatively, the observed concordance may represent a compensatory or antagonistic mechanism of S. epidermidis, increasing in an attempt to control S. aureus.
In addition to the selective shifts in Staphylococcus species, the relative abundance of Streptococcus, Corynebacterium, and Propionibacterium species varied across the AD disease states, highlighting the complex relationships among bacteria and the importance of comprehensive investigation of microbial communities. S. aureus and S. epidermidis production of antibacterial compounds, including bacteriocins and antimicrobial peptides (Cogen et al. 2010a,b; Iwase et al. 2010; Lai et al. 2010; Joo et al. 2011), may contribute to the relative decrease in Streptococcus, Corynebacterium, and Propionibacterium species observed during AD flares. In addition to identifying changes in the skin commensal population, we determined that low-abundance genera including Rothia species were over-represented during different disease states in all patients. Furthermore, our data suggested that following treatment, increasing diversity of the microbiome, similar to the diversity observed in healthy skin, arises from a relative expansion of taxa already present in the skin microbial community with colonization of few additional new genera not present at baseline. Greater sampling depth in future studies may demonstrate that the majority of taxa present at baseline persist in low numbers during flares, then re-expand following treatment. Overall, the ability to globally examine bacterial community dynamics highlights a major advantage of microbial genomics, defining parameters needed for the development of new methods to maintain or enhance beneficial microbes and microbial diversity to promote health.
While bacterial culture methods are readily implemented and are relatively inexpensive in comparison to 16S rRNA gene sequencing, genomic methods are highly complementary, are rapidly dropping in cost and turnaround time, and can identify bacteria that are not easily cultivatable, yet may be important contributors to disease pathophysiology or progression. For easily cultured bacteria such as S. aureus, we have shown that the relative abundance of genomic sequences from nasal samples corresponds to the bacterial bioburden assessed using traditional cultivation techniques. However, these results strengthen assertions that genomic analyses of more difficult-to-cultivate bacteria represent the true relative abundance of those bacteria in a sample. No association was identified between the relative abundance of S. aureus genomic sequences in nasal samples and disease severity, suggesting that although the nares may serve as a reservoir for S. aureus, there may not be a dose-response between the abundance of nasal S. aureus and AD severity. Culture-based methods remain important and provide vital information on bioburden. However, the higher resolution and reduced bias of sequence-based and culture-independent microbiome studies in human diseases is of great benefit.
We determined that AD flares are characterized by low bacterial diversity in the absence of recent treatment. In contrast, intermittent or active treatment is associated with higher bacterial diversity. Although the impact of common AD therapies on S. aureus levels has been debated (Stalder et al. 1994; Williams et al. 1998; Ravenscroft et al. 2003), our results and prior studies (Huang et al. 2009) indicate that the clinical effectiveness of AD treatments does not rely on elimination of S. aureus. However, AD therapeutic modalities may act to recalibrate the diversity of the skin microbiome. For intermittent-treatment flare patients who used sporadic AD treatments in the week preceding flare sampling, the early shift toward a diverse microbiome in the presence of active clinical disease suggests that known effective AD treatments diversify the AD skin bacterial community. Intermittent-treatment flares likely represent a point in the continuum of the disease during transition from flare to postflare. However, the presence of active clinical disease during intermittent-treatment flares suggests that lesional skin requires continued intensive treatment to sufficiently reduce the inflammatory response toward that observed in the postflare state. Increases in diversity associated with AD treatments may be due to therapies that preferentially kill S. aureus, allowing relative expansion of other bacteria, promotion of microbes that control S. aureus predominance, or total reduction of skin microbes, followed by rapid repopulation with a diverse community.
In summary, specific AD disease states are characterized by concurrent and anticorrelated shifts in microbial diversity and proportion of Staphylococcus. Since untreated flares have reduced diversity and high Staphylococcus proportions as compared with baseline, postflares, and, notably, intermittent-treatment flares, we propose that increases in the proportion of Staphylococcus and reductions in microbial diversity precede worsening of AD disease severity as observed in no-treatment flares (Fig. 6). Relatively high Staphylococcus proportions observed in a few of the baseline timepoints could represent the pre-flare stage, in which staphylococcal levels increase, yet clinical disease has not begun to dramatically worsen. In contrast, reduction in S. aureus proportions and restoration of microbial diversity was observed in the intermittent-treatment flares, which are characterized as having clinically worsened disease. Based on this, we propose that use of AD treatments modify microbial diversity and proportions of Staphylococcus, but that consistent and continued treatment over a period of time is required to induce the “resolving flare” stage, which transitions into a restoration of full microbial diversity and low population levels of Staphyloccocus, typical of a true postflare.
Figure 6.

AD microbiome progression hypothesis. (*) Proposed relationship among shifts in skin microbial diversity, the proportion of Staphylococcus, and disease severity.
With public health concerns regarding the development of antibiotic resistance, understanding how current treatments affect bacteria and subsequent disease activity will allow us to identify and develop directed therapies for AD that modify the skin microbiome, potentially reducing use of systemic antibiotics. Our findings highlight the importance of performing longitudinal microbiome studies to examine the influence of microbial communities on health and disease. Advances in technologies including whole-genome shotgun sequencing will continue to enhance our ability to probe microbial communities via metagenomics and functional metabolomics. The ability to interrogate microbial communities en toto and in situ has begun to provide insights regarding the important and diverse roles of microbes in human disorders and how shifts in microbial communities may improve human health.
Methods
Study design
A prospective natural history study was approved by the institutional review board of NHGRI, NIH. The study was designed to evaluate pediatric AD patients at three timepoints (baseline, flare, and postflare) to capture clinically distinct timepoints that characterize this chronic relapsing, remitting skin disease. Age-matched children were sampled at similar time intervals to serve as healthy pediatric controls.
Inclusion criteria for AD patients included ages 2–18 yr, moderate-to-severe disease, presence of more than or equal to one affected antecubital or popliteal crease at enrollment, and ability to tolerate >7 d without topical AD treatments and >4 wk off both systemic antibiotics and corticosteroids. The diagnosis of AD was based on the UK Working Party definition (Williams et al. 1994a). Objective SCORAD as assessed by one individual (HHK) was used to determine study eligibility and disease status at each clinic visit (Williams et al. 1994b; Kunz et al. 1997; Oranje et al. 2007). Moderate-to-severe disease was defined by objective SCORAD ≥15 (range 0–83) (Oranje et al. 2007).
Exclusion criteria for all subjects included receiving investigational new treatments, ultraviolet light therapy, monoclonal antibodies, systemic immunosuppressants within 7 d or five half-lives (taking the longer time period) of skin sampling; a history of bone-marrow transplant or gene therapy; and clinically apparent underlying immunodeficiency. Exclusion criteria specific for AD patients included systemic antibiotics during the preceding 2 wk (except for post-flare timepoint). Exclusion criteria for healthy controls also included current or prior chronic skin disorders such as AD or psoriasis; asthma and allergic rhinitis, via International Study of Asthma and Allergies in Childhood questionnaire (Asher et al. 1995); other chronic medical conditions; and use of systemic antibiotics in the preceding 6 mo.
For AD patients, baseline was defined as usual and stable disease state and ability to tolerate >7 d without topical AD treatments to intended sample sites and >4 wk off both oral antibiotics and corticosteroids. Flare was defined as acute exacerbation of disease on any skin region prior to initiation of intensified AD treatment and without restriction of usual treatments >24 h prior to sampling. Subjects were instructed to promptly contact the research team when skin disease worsening was apparent. Flares were evaluated and sampled within 24 h of notification to avoid delay in intensified treatment. Postflare was defined as 10–14 d after the initiation of intensified AD treatment. Intensified AD treatment recommendations could include the following based on their typical regimen: dilute bleach baths (0.25 cup of 6% bleach into bath half filled with water for a final concentration of 0.0005%) three to four times per week, higher potency topical steroids twice daily, bland emollients twice daily, and, in certain cases of suspected clinical infection, systemic antibiotics prescribed by primary care physicians (Supplemental Table S1).
Subjects
AD patients aged 2–15 yr of age, and similarly aged healthy controls were recruited from the larger Washington, DC metropolitan region. Written informed consent was obtained from parents or guardians of all participating children. Complete medical and medication history and skin examination was performed at all clinic visits.
Sample collection
Skin preparation instructions included avoiding bathing and application of topical medications and emollients for 24 h at sampling sites prior to all sampling timepoints (baseline, flare, and postflare). Samples were obtained as described (Grice et al. 2009). Nasal cultures were obtained at each timepoint. Blood was drawn during the first visit.
Cultures
Nasal cultures were performed by the Department of Laboratory Medicine (DLM), Clinical Center, NIH. Cultures were obtained with swabs (BBLtm CultureSwabTM, made by Copan for Becton, Dickinson and Company) and plated on three different growth mediums (Remel Products). After incubation, the colonies were counted and identified on the basis of gross and microscopic assessment after treatment with Gram's stain. Culture results were grouped into 0, 1–10, and >10 colonies of Staphylococcus aureus per sample.
DNA extraction and sequencing
Skin swabs were incubated in enzymatic lysis buffer and lysozyme (20 mg/mL) for 30 min at 37°C. Two 5-mm stainless steel beads (Qiagen) were added to the solution, placed in a Tissuelyser (Qiagen), and processed for 2 min at 30Hz.The standard protocol for the PureLink Genomic DNA kit (Invitrogen) was followed for all subsequent steps. rRNA genes (16S) were amplified from purified genomic DNA using primers 8F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1391R (5′-GACGGGCGGTGWGTRCA-3′). PCR amplification, cloning, and sequencing were performed as described (Grice et al. 2009). Sequence assembly, alignment, and chimera elimination were performed as described, with specified modifications (Grice et al. 2009; Schloss et al. 2009; Huse et al. 2010).
Analysis pipeline
Community analysis
Operational taxonomic units (OTUs) were identified and a distance matrix calculated using mothur software v14.0 (Schloss et al. 2009). No lane masking was applied. The distance matrix was then analyzed to calculate OTUs using the average neighbor algorithm. Community evenness and richness (Shannon diversity index “shannon”), shared community membership, and shared community structure (theta index “thetayc”) at a 98% similarity cutoff were calculated using summary.shared() and summary.single() in mothur. Shannon diversity was calculated as follows: 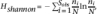 , where Sobs
= number of observed OTUs, ni = number of individuals in OTU i, and N = total number of individuals in the community. The theta index, or Yue and Clayton measure of similarity between two community structures (Yue and Clayton 2005), was calculated as follows:
, where Sobs
= number of observed OTUs, ni = number of individuals in OTU i, and N = total number of individuals in the community. The theta index, or Yue and Clayton measure of similarity between two community structures (Yue and Clayton 2005), was calculated as follows:
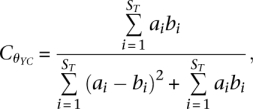 |
where ST = total number of OTUs in communities A and B, ai = relative abundance of OTU i in community A, and bi = relative abundance of OTU i in community B. To calculate alpha and beta diversity metrics minus S. aureus, we removed sequences classified as S. aureus from the analysis. Resulting samples were then subsampled 100× to the lowest number of sequences per group (Ac = 129, Pc = 31, N = 68, Vf = 133) and subsampled results were averaged. Results without subsampling yielded similar results (data not shown).
Because left and right symmetric sites generally exhibit low variability (Grice et al. 2009), the sites were grouped prior to analysis. For timepoint categories baseline, no-treatment flare, intermittent-treatment flare, and control calculations of Shannon diversity and theta indices, values were averaged for the antecubital and popliteal crease unless otherwise indicated. To estimate depth of sequence sampling, rarefaction curve data for each individual at each site was calculated using rarefaction.single in mothur, and individuals for each group were averaged to obtain group-wide rarefaction curves.
Genus-level analysis
Following sequence trimming, alignment, and other pre-processing steps as previously described, 16S rRNA sequences were classified to the genus level using the Ribosomal Database Project (RDP) classifications v1.0, training set 4 (Cole et al. 2009). For 14 major order-genera that represented >1% of total 16S rRNA sequences, relative abundances were computed by dividing sequence counts by the total sequences obtained for that site, with proportions for antecubital and popliteal crease averaged. Finally, custom scripts were generated for taxonomic classifications to the species level for staphylococcal species. Staphylococcal sequences were speciated by alignment to a curated collection of staphylococcal reference sequences from complete genome sequences and type strains. Each sequence was assigned a label based on the consensus call of sequence alignments with the lowest edit distance between a query and reference.
Statistics
All data are represented as mean ± SEM unless otherwise indicated. For controls with >1 timepoint, the relevant data was averaged for each control. As disease severity differed minimally from left to right symmetric sites, left and right values were averaged prior to statistical comparisons. AcPc indicates the mean of values obtained from the antecubital and popliteal crease for each individual (post-averaging of left and right symmetric sites, and for controls, also post-averaging of timepoints).
Analyses involved calculating partial correlations by using the relevant variables for each subject over all timepoints. Calculations were performed on Fisher-transformed r values. For statistical testing of comparisons of differences between groups, the nonparametric Wilcoxon rank sum test was used for all comparisons (wilcox.test in the statistical software program R). Where indicated, within-subjects analysis was performed by using the option “paired = T” in wilcox.test. All P-values were adjusted using p.adjust in R using Bonferroni (# comparisons ≤ 10) or false discovery rate (# comparisons > 10) corrections. For testing of significantly over-represented genera between the baseline, flare, and post-flare groups, we reduced the number of multiple comparisons by prefiltering the search to the seven genera that were present in 100% of baseline or no-treatment flares (for baseline-flare comparisons), or the 12 genera that were present in 100% of no-treatment flares or postflare (for flare-postflare comparisons) prior to performing a paired Wilcoxon rank sum test. For comparisons between baseline and postflare, we limited the search to genera that were present in >75% of individuals and performed a paired Wilcoxon rank sum test.
Data access
The sequence data from this study have been submitted to GenBank (http://www.ncbi.nlm.nih.gov/genbank) and can be accessed through BioProject ID 46333. Patient and sample metadata have been deposited in the controlled access database dbGaP under study accession phs000266.v1.p1.
NISC Comparative Sequencing Program investigators
Jim Mullikin, Jim Thomas, Robert Blakesley, Alice Young, Grace Chu, Colleen Ramsahoye, Sean Lovett, Joel Han, Richelle Legaspi, Christina Sison, Casandra Montemayor, Michael Gregory, April Hargrove, Taccara Johnson, Nancy Riebow, Brian Schmidt, Betsy Novotny, Jyoti Gupta, Betty Benjamin, Shelise Brooks, Holly Coleman, Shi-ling Ho, Karen Schandler, Mal Stantripop, Quino Maduro, Gerry Bouffard, Mila Dekhtyar, Xiaobin Guan, Cathy Masiello, Baishali Maskeri, Jenny McDowell, Morgan Park, and Meg Vemulapalli.
Acknowledgments
We thank Mark C. Udey, Eric Green, and Evan Snitkin for helpful discussions; Pamela Thomas, Deborah Schoenfeld, Joie Davis, Roselyn Epps, Celeste Nelson, and Donna Gaskins for underlying contributions; and especially the patients and volunteers. This work was supported by NIH CC, NCI, and NHGRI Intramural Research Programs, and in part by 1K99AR059222 (H.H.K.). Sequencing and clinical research support was funded by grants from the National Institutes of Health Common Fund Human Microbiome Project (1UH2AR057504-01 and 4UH3AR057504-02).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.131029.111.
References
- Archer GL, Climo MW 2001. Staphylococcus aureus bacteremia–consider the source. N Engl J Med 344: 55–56 [DOI] [PubMed] [Google Scholar]
- Asher MI, Keil U, Anderson HR, Beasley R, Crane J, Martinez F, Mitchell EA, Pearce N, Sibbald B, Stewart AW, et al. 1995. International Study of Asthma and Allergies in Childhood (ISAAC): rationale and methods. Eur Respir J 8: 483–491 [DOI] [PubMed] [Google Scholar]
- Asher MI, Montefort S, Bjorksten B, Lai CK, Strachan DP, Weiland SK, Williams H 2006. Worldwide time trends in the prevalence of symptoms of asthma, allergic rhinoconjunctivitis, and eczema in childhood: ISAAC phases one and three repeat multicountry cross-sectional surveys. Lancet 368: 733–743 [DOI] [PubMed] [Google Scholar]
- Bath-Hextall FJ, Birnie AJ, Ravenscroft JC, Williams HC 2010. Interventions to reduce Staphylococcus aureus in the management of atopic eczema: an updated Cochrane review. Br J Dermatol 163: 12–26 [DOI] [PubMed] [Google Scholar]
- Bickers DR, Lim HW, Margolis D, Weinstock MA, Goodman C, Faulkner E, Gould C, Gemmen E, Dall T 2006. The burden of skin diseases: 2004: A joint project of the American Academy of Dermatology Association and the Society for Investigative Dermatology. J Am Acad Dermatol 55: 490–500 [DOI] [PubMed] [Google Scholar]
- Breuer K, HÄussler S, Kapp A, Werfel T 2002. Staphylococcus aureus: colonizing features and influence of an antibacterial treatment in adults with atopic dermatitis. Br J Dermatol 147: 55–61 [DOI] [PubMed] [Google Scholar]
- Cho SH, Strickland I, Boguniewicz M, Leung DY 2001. Fibronectin and fibrinogen contribute to the enhanced binding of Staphylococcus aureus to atopic skin. J Allergy Clin Immunol 108: 269–274 [DOI] [PubMed] [Google Scholar]
- Cogen AL, Yamasaki K, Muto J, Sanchez KM, Crotty Alexander L, Tanios J, Lai Y, Kim JE, Nizet V, Gallo RL 2010a. Staphylococcus epidermidis antimicrobial δ-toxin (phenol-soluble modulin-γ) cooperates with host antimicrobial peptides to kill group A Streptococcus. PLoS ONE 5: e8557 doi: 10.1371/journal.pone.0008557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogen AL, Yamasaki K, Sanchez KM, Dorschner RA, Lai Y, MacLeod DT, Torpey JW, Otto M, Nizet V, Kim JE, et al. 2010b. Selective antimicrobial action is provided by phenol-soluble modulins derived from Staphylococcus epidermidis, a normal resident of the skin. J Invest Dermatol 130: 192–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, et al. 2009. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37: D141–D145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R 2009. Bacterial community variation in human body habitats across space and time. Science 326: 1694–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer C, Link E, Horster M, Koletzko S, Bauer CP, Berdel D, von Berg A, Lehmann I, Herbarth O, Borte M, et al. 2010. Elder siblings enhance the effect of filaggrin mutations on childhood eczema: results from the 2 birth cohort studies LISAplus and GINIplus. J Allergy Clin Immunol 125: 1254–1260 [DOI] [PubMed] [Google Scholar]
- Gao Z, Tseng CH, Pei Z, Blaser MJ 2007. Molecular analysis of human forearm superficial skin bacterial biota. Proc Natl Acad Sci 104: 2927–2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloor M, Peters G, Stoika D 1982. On the resident aerobic bacterial skin flora in unaffected skin of patients with atopic dermatitis and in healthy controls. Dermatologica 164: 258–265 [DOI] [PubMed] [Google Scholar]
- Gong JQ, Lin L, Lin T, Hao F, Zeng FQ, Bi ZG, Yi D, Zhao B 2006. Skin colonization by Staphylococcus aureus in patients with eczema and atopic dermatitis and relevant combined topical therapy: a double-blind multicentre randomized controlled trial. Br J Dermatol 155: 680–687 [DOI] [PubMed] [Google Scholar]
- Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, Bouffard GG, Blakesley RW, Murray PR, Green ED, et al. 2009. Topographical and temporal diversity of the human skin microbiome. Science 324: 1190–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidron AI, Edwards JR, Patel J, Horan TC, Sievert DM, Pollock DA, Fridkin SK 2008. NHSN annual update: antimicrobial-resistant pathogens associated with healthcare-associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006-2007. Infect Control Hosp Epidemiol 29: 996–1011 [DOI] [PubMed] [Google Scholar]
- Higaki S, Morohashi M, Yamagishi T, Hasegawa Y 1999. Comparative study of staphylococci from the skin of atopic dermatitis patients and from healthy subjects. Int J Dermatol 38: 265–269 [DOI] [PubMed] [Google Scholar]
- Hoeger PH, Lenz W, Boutonnier A, Fournier JM 1992. Staphylococcal skin colonization in children with atopic dermatitis: prevalence, persistence, and transmission of toxigenic and nontoxigenic strains. J Infect Dis 165: 1064–1068 [DOI] [PubMed] [Google Scholar]
- Huang JT, Abrams M, Tlougan B, Rademaker A, Paller AS 2009. Treatment of Staphylococcus aureus colonization in atopic dermatitis decreases disease severity. Pediatrics 123: e808–e814 [DOI] [PubMed] [Google Scholar]
- Hung SH, Lin YT, Chu CY, Lee CC, Liang TC, Yang YH, Wang LC, Chiang BL 2007. Staphylococcus colonization in atopic dermatitis treated with fluticasone or tacrolimus with or without antibiotics. Ann Allergy Asthma Immunol 98: 51–56 [DOI] [PubMed] [Google Scholar]
- Huse SM, Welch DM, Morrison HG, Sogin ML 2010. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ Microbiol 12: 1889–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwase T, Uehara Y, Shinji H, Tajima A, Seo H, Takada K, Agata T, Mizunoe Y 2010. Staphylococcus epidermidis Esp inhibits Staphylococcus aureus biofilm formation and nasal colonization. Nature 465: 346–349 [DOI] [PubMed] [Google Scholar]
- Joo HS, Cheung GY, Otto M 2011. Antimicrobial activity of community-associated methicillin-resistant Staphylococcus aureus is caused by phenol-soluble modulin derivatives. J Biol Chem 286: 8933–8940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DW, Park JY, Park KD, Kim TH, Lee WJ, Lee SJ, Kim J 2009. Are there predominant strains and toxins of Staphylococcus aureus in atopic dermatitis patients? Genotypic characterization and toxin determination of S. aureus isolated in adolescent and adult patients with atopic dermatitis. J Dermatol 36: 75–81 [DOI] [PubMed] [Google Scholar]
- Kunz B, Oranje AP, Labreze L, Stalder JF, Ring J, Taieb A 1997. Clinical validation and guidelines for the SCORAD index: consensus report of the European Task Force on Atopic Dermatitis. Dermatology 195: 10–19 [DOI] [PubMed] [Google Scholar]
- Lai Y, Villaruz AE, Li M, Cha DJ, Sturdevant DE, Otto M 2007. The human anionic antimicrobial peptide dermcidin induces proteolytic defence mechanisms in staphylococci. Mol Microbiol 63: 497–506 [DOI] [PubMed] [Google Scholar]
- Lai Y, Cogen AL, Radek KA, Park HJ, Macleod DT, Leichtle A, Ryan AF, Di Nardo A, Gallo RL 2010. Activation of TLR2 by a small molecule produced by Staphylococcus epidermidis increases antimicrobial defense against bacterial skin infections. J Invest Dermatol 130: 2211–2221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lever R, Hadley K, Downey D, Mackie R 1988. Staphylococcal colonization in atopic dermatitis and the effect of topical mupirocin therapy. Br J Dermatol 119: 189–198 [DOI] [PubMed] [Google Scholar]
- Leyden JJ, Marples RR, Kligman AM 1974. Staphylococcus aureus in the lesions of atopic dermatitis. Br J Dermatol 90: 525–530 [DOI] [PubMed] [Google Scholar]
- Li M, Lai Y, Villaruz AE, Cha DJ, Sturdevant DE, Otto M 2007. Gram-positive three-component antimicrobial peptide-sensing system. Proc Natl Acad Sci 104: 9469–9474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomholt H, Andersen KE, Kilian M 2005. Staphylococcus aureus clonal dynamics and virulence factors in children with atopic dermatitis. J Invest Dermatol 125: 977–982 [DOI] [PubMed] [Google Scholar]
- McCrea KW, Hartford O, Davis S, Eidhin DN, Lina G, Speziale P, Foster TJ, Hook M 2000. The serine-aspartate repeat (Sdr) protein family in Staphylococcus epidermidis. Microbiology 146: 1535–1546 [DOI] [PubMed] [Google Scholar]
- Nilsson M, Frykberg L, Flock JI, Pei L, Lindberg M, Guss B 1998. A fibrinogen-binding protein of Staphylococcus epidermidis. Infect Immun 66: 2666–2673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, Gallo RL, Leung DY 2002. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med 347: 1151–1160 [DOI] [PubMed] [Google Scholar]
- Oranje AP, Glazenburg EJ, Wolkerstorfer A, de Waard-van der Spek FB 2007. Practical issues on interpretation of scoring atopic dermatitis: the SCORAD index, objective SCORAD and the three-item severity score. Br J Dermatol 157: 645–648 [DOI] [PubMed] [Google Scholar]
- Palmer CN, Irvine AD, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee SP, Goudie DR, Sandilands A, Campbell LE, Smith FJ, et al. 2006. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet 38: 441–446 [DOI] [PubMed] [Google Scholar]
- Peschel A, Jack RW, Otto M, Collins LV, Staubitz P, Nicholson G, Kalbacher H, Nieuwenhuizen WF, Jung G, Tarkowski A, et al. 2001. Staphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor MprF is based on modification of membrane lipids with l-lysine. J Exp Med 193: 1067–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravenscroft JC, Layton AM, Eady EA, Murtagh MS, Coates P, Walker M, Cove JH 2003. Short-term effects of topical fusidic acid or mupirocin on the prevalence of fusidic acid resistant (FusR) Staphylococcus aureus in atopic eczema. Br J Dermatol 148: 1010–1017 [DOI] [PubMed] [Google Scholar]
- Sandilands A, Terron-Kwiatkowski A, Hull PR, O'Regan GM, Clayton TH, Watson RM, Carrick T, Evans AT, Liao H, Zhao Y, et al. 2007. Comprehensive analysis of the gene encoding filaggrin uncovers prevalent and rare mutations in ichthyosis vulgaris and atopic eczema. Nat Genet 39: 650–654 [DOI] [PubMed] [Google Scholar]
- Schloss PD, Handelsman J 2006. Introducing SONS, a tool for operational taxonomic unit-based comparisons of microbial community memberships and structures. Appl Environ Microbiol 72: 6773–6779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537–7541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw TE, Currie GP, Koudelka CW, Simpson EL 2011. Eczema prevalence in the United States: data from the 2003 National Survey of Children's Health. J Invest Dermatol 131: 67–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieprawska-Lupa M, Mydel P, Krawczyk K, Wojcik K, Puklo M, Lupa B, Suder P, Silberring J, Reed M, Pohl J, et al. 2004. Degradation of human antimicrobial peptide LL-37 by Staphylococcus aureus-derived proteinases. Antimicrob Agents Chemother 48: 4673–4679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalder JF, Fleury M, Sourisse M, Rostin M, Pheline F, Litoux P 1994. Local steroid therapy and bacterial skin flora in atopic dermatitis. Br J Dermatol 131: 536–540 [DOI] [PubMed] [Google Scholar]
- von Eiff C, Becker K, Machka K, Stammer H, Peters G 2001. Nasal carriage as a source of Staphylococcus aureus bacteremia. Study Group. N Engl J Med 344: 11–16 [DOI] [PubMed] [Google Scholar]
- von Mutius E 2000. The environmental predictors of allergic disease. J Allergy Clin Immunol 105: 9–19 [DOI] [PubMed] [Google Scholar]
- Williams RE, Gibson AG, Aitchison TC, Lever R, Mackie RM 1990. Assessment of a contact-plate sampling technique and subsequent quantitative bacterial studies in atopic dermatitis. Br J Dermatol 123: 493–501 [DOI] [PubMed] [Google Scholar]
- Williams HC, Burney PG, Pembroke AC, Hay RJ 1994a. The U.K. working party's diagnostic criteria for atopic dermatitis. III. Independent hospital validation. Br J Dermatol 131: 406–416 [DOI] [PubMed] [Google Scholar]
- Williams HC, Burney PG, Strachan D, Hay RJ 1994b. The U.K. working party's diagnostic criteria for atopic dermatitis. II. Observer variation of clinical diagnosis and signs of atopic dermatitis. Br J Dermatol 131: 397–405 [DOI] [PubMed] [Google Scholar]
- Williams JV, Vowels BR, Honig PJ, Leyden JJ 1998. S. aureus isolation from the lesions, the hands, and the anterior nares of patients with atopic dermatitis. Pediatr Dermatol 15: 194–198 [DOI] [PubMed] [Google Scholar]
- Williams RJ, Henderson B, Sharp LJ, Nair SP 2002. Identification of a fibronectin-binding protein from Staphylococcus epidermidis. Infect Immun 70: 6805–6810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB 2004. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis 39: 309–317 [DOI] [PubMed] [Google Scholar]
- Yeung M, Balma-Mena A, Shear N, Simor A, Pope E, Walsh S, McGavin MJ 2011. Identification of major clonal complexes and toxin producing strains among Staphylococcus aureus associated with atopic dermatitis. Microbes Infect 13: 189–197 [DOI] [PubMed] [Google Scholar]
- Yue JC, Clayton MK 2005. A similarity measure based on species proportions. Comm Statist Theory Methods 34: 2123–2131 [Google Scholar]