

Abstract
Background and aims
Acute liver failure (ALF) is frequently complicated by cerebral edema, systemic inflammation and multi-organ dysfunction. Vascular endothelial growth factor (VEGF) may stimulate liver regeneration but can also be pro-inflammatory, activating endothelial cells and increasing permeability, actions mediated through Src kinase signalling. We therefore examined whether a Src inhibitor could have therapeutic potential in ALF.
Methods
Murine ALF was induced with azoxymethane. Liver pathology was graded by a blinded examiner and apoptosis quantified by immunohistochemistry. Cerebral VEGF expression was imaged using VEGF-GFP transgenic mice. Circulating and macrophage-secreted VEGF levels were measured. Experimental animals received a Src inhibitor or vehicle controls.
Results
VEGF was undetectable in normal plasma but reached a mean of 835pg/ml at grade III encephalopathy (p<0.001). Ammonia, lipopolysaccharide and interferon-gamma acted synergistically to enhance VEGF secretion by macrophages. Production of VEGF by cerebral cortical astrocytes increased with disease progression. Late treatment with inhibitors of Src or VEGF did not improve liver histology, encephalopathy or survival. However, early use of a Src kinase inhibitor significantly reduced hepatic injury, delayed encephalopathy and allowed 25% of mice to survive an otherwise lethal insult.
Conclusion
Systemic and cerebral VEGF levels are significantly elevated during experimental ALF and may be exacerbated by hyperammonemia and macrophage activation. Early use of a Src inhibitor reduced hepatocellular injury and enabled survival, indicating such agents may have some promise in the treatment of ALF.
Keywords: Acute liver failure, ammonia, azoxymethane, bosutinib, encephalopathy, VEGF, Src
Introduction
Acute liver failure (ALF) has a reported mortality rate of 50-80% without liver transplantation [1,2]. Cerebral edema and multi-organ failure remain the most common causes of death [3,4]. These complications may be worsened by the systemic inflammatory response syndrome (SIRS) in patients with ALF [5,6].
Potential therapeutic strategies include reducing the systemic complications of ALF or stimulating liver regeneration [7]. Recently, vascular endothelial growth factor (VEGF) was shown to have hepatotropic effects, releasing hepatocyte growth factor (HGF) from sinusoidal endothelial cells [8]. However, such beneficial actions of VEGF might be accompanied by adverse effects elsewhere.
Importantly, VEGF is a pro-inflammatory cytokine and may contribute to the development of SIRS through endothelial activation. Macrophages secrete VEGF in response to interferon gamma (IFN-γ) or lipopolysaccharide (LPS) [9] and the injection of LPS into human subjects elevates plasma VEGF levels, the apparent result of neutrophil degranulation [10]. In endothelial cells, VEGF increases expression of E-selectin, intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) [11], encouraging leukocyte adhesion and chemokine release [12]. Furthermore, VEGF increases the permeability of endothelial cell-cell junctions in several disease states [13].
The endothelial activation and vascular leak responses induced by VEGF depend largely on VEGFR-2/Flk-1 [14] and the Src family kinases, Src and Yes (but not Fyn) [15]. Accordingly, inhibitors of VEGF or Src kinases can ameliorate ischemic injury in the brain [16] and heart [17].
We therefore wished to study the therapeutic effects of a Src inhibitor in ALF. Unfortunately, small animal models of acetaminophen (APAP) toxicity, the commonest cause of ALF in the United States [18], may be complicated by variable reproducibility and by direct injury to other organs [19]. However, the hepatotoxin azoxymethane (AOM) represents a significant advance over earlier models. A single intraperitoneal injection leads to centrilobular hepatic necrosis, apparently due to mitochondrial injury, without histological evidence of damage to other organs apart from the brain, where cerebral edema occurs in mice with Grade IV encephalopathy [20]. This model has been validated by an investigation of clotting factor levels in ALF [21] and replicates many features of the human syndrome. Mice with AOM induced ALF reproducibly develop hepatic encephalopathy, developing elevated blood and cerebral levels of ammonia as well as abnormal amino acid profiles with an excess of cortical glutamine [22].
We therefore used this model to examine the roles of VEGF and Src in experimental ALF and test the hypothesis that Src inhibition could be beneficial.
Materials and methods
Materials
Azoxymethane was purchased from Sigma Aldrich, Missouri. Lipopolysaccharide from E.coli 011:B4 was obtained from Sigma Aldrich. The NIH thioglycollate broth 225710 and the Thermo Electron ALT/GPT assay reagents were purchased from VWR International, West Chester, Pennsylvania. The VEGF inhibitor Cyclo-VEGI was purchased from Calbiochem, San Diego, California. The Src kinase inhibitor SKI-606 was developed by Wyeth-Ayerst (Pearl River, New York) [23] and administered in a vehicle consisting of 50% PEG-400 (Sigma Aldrich), 40% water and 10% ethanol.
Animals
Adult mice aged 8-12 weeks were used for all studies with appropriate age and sex-matched littermate controls. C57BL/6 and Balb/c mice were obtained from Harlan, Indianapolis, Indiana. Dr Brian Seed provided the VEGF-GFP transgenic mice [24]. Animals were housed in the vivaria of The Scripps Research Institute (TSRI) or UCSD Cancer Center, La Jolla, California. Experimental protocols were carried out with Institutional Animal Care and Use Committee approval.
Experimental ALF
The azoxymethane model was used as previously described [20]. Mice were injected intraperitoneally (i.p.) with AOM at doses of 50 or 100μg/g in a volume of 100μl sterile phosphate buffered saline (PBS). Mouse body temperatures were maintained in isothermic conditions at 37°C. Intraperitoneal injections of pre-warmed sterile dextrose were given against hypoglycemia and dehydration. Fluid supplementation was triggered by an observed fall in intake, reduced mobility, decreased skin turgor or by a decline in body weight and was delivered in aliquots of no more than 0.2ml per hour based on a total 24 hour fluid requirement of 0.1ml per gram body weight.
Hepatic encephalopathy (HE) was determined using a quantitative reflex scoring system [25] and allocated to four grades, equating to I = lethargy and loss of scatter reflex, II = ataxia, III = loss of righting reflex and IV = coma [20].
Biological samples
After sacrifice, livers and brains were rapidly removed for histology, immunohistochemistry or water content analysis. Blood was obtained by cardiac puncture. Histology specimens were fixed in 10% formalin solution in PBS overnight and paraffin-embedded 6μm sections were cut, mounted on glass slides and stained with hematoxylin and eosin (H&E). Plasma VEGF was measured with a murine VEGF ELISA kit from Biosource, Camarillo, California used according to manufacturer’s instructions. Transaminase levels were measured using a Thermo Electron ALT/GPT kit.
Liver histopathology
Liver injury was assessed by a blinded pathologist using a semi-quantitative index [26]. For each section, a minimum of 5 lobules were examined and graded as:
0 Normal
½ Individual necrotic cells adjacent to the central veins with hyaline degeneration
1 Necrotic cells extending 2-3 cell layers from the central veins
2 Necrotic cells 4-6 layers from the central veins but limited in peripheral distribution
3 As above but with necrosis extending from one central vein to another
4 More severe than above with extensive centrilobular necrosis throughout the section
Histochemical detection of apoptosis
Snap frozen liver samples were sectioned using a cryostat and fixed in 1% paraformaldehyde in PBS at pH 7.4 for 10 minutes at room temperature. Hepatic apoptosis was determined using an ApopTag Plus Detection Kit S7111 (Chemicon, Temecula, California) as per the manufacturer’s instructions. This is a Terminal Deoxynucleotide Transferase dUTP Nick End Label (TUNEL) method, detecting free 3’-OH termini present in the DNA of apoptotic cells by enzymatic labelling.
Slides were mounted using GelMount (Fisher Scientific, Pittsburgh) containing 0.5μg/ml propidium iodide and stored at 4°C in the dark before viewing under a confocal microscope.
Measurement of brain water content
Whole brains were carefully excised and surface blood removed by blotting with tissue paper. Brains were immediately weighed then frozen at -80°C, transfered to a vacuum freeze drier system (VirTis, Gardiner, New York) overnight and re-weighed. Water content was calculated as a percentage based on the wet and dry weights.
Primary macrophages and cultured macrophage cell lines
Peritoneal macrophages were harvested from 8-12 week old animals using a published technique [9]. Briefly, mice were injected i.p. with 1ml of 4% NIH thioglycollate broth. Four days later, mice were sacrificed and macrophages collected by exposing the peritoneum under aseptic conditions, injecting 10ml of sterile PBS into the peritoneal cavity and aspirating. After washing twice in serum-free MEM medium, cells were resuspended in MEM plus 10% fetal calf serum (FCS) and transferred to 6cm wells in 4ml aliquots at a density of 0.25×106 cells per cm2 and left to attach overnight in a 5% CO2 humidified incubator at 37°C. The following day, non-adherent cells were removed by washing with serum-free MEM and the medium was changed to MEM supplemented with 1% FCS along with NH4Cl, IFN-γ and/or LPS depending on the particular experiment. LPS was used at a final concentration of 100ng/ml and IFN-γ at 10ng/ml. After incubation for 24 hours, 1ml aliquots of media were aspirated, centrifuged and supernatants frozen at -20°C before VEGF levels were determined by ELISA.
The murine macrophage cell line RAW 264.7 was obtained from the American Type Culture Collection, Manassas, Virginia and maintained according to ATCC instructions. Cells were passaged by scraping and were used under the same conditions as the primary macrophages.
Statistical analysis
Unpaired data were analysed using Student’s t test. A p value of <0.05 was considered to be statistically significant.
Results
Characterization of AOM-induced acute liver failure
Preliminary experiments were conducted to characterize the development of ALF, encephalopathy and cerebral edema after the administration of AOM. Harvested livers at serial timepoints confirmed that mice injected with 100μg/g AOM developed centrilobular microvesicular steatohepatitis after approximately 8-10 hours that progressed to widespread haemorhhagic necrosis by 28-32 hours. Mice predictably developed HE and became comatose at 30-34 hours post injection (Fig. 1A). Water content analysis detected cerebral edema in mice with Grade IV encephalopathy (Fig. 1B) but not at earlier grades of HE.
Figure 1. HE and cerebral edema in mice with ALF.

(A) Progression through the 4 stages of HE in a group of 6 mice injected with 100μg/g AOM.
Control mice, without acute liver failure, maintained a baseline HE score of 0 (data not shown).
(B) Cerebral edema as detected by total brain water content analysis. Mice that developed grade IV HE were sacrificed at 32 hours post-azoxymethane and had significantly higher cerebral water levels than age and sex-matched controls injected with PBS. Values represent mean +/- SEM. ** = p<0.01
Circulating VEGF is significantly increased during AOM-induced ALF
VEGF was undetectable in the plasma of control mice. However, the administration of AOM was followed by a significant rise to a mean of approximately 20pg/ml after just 8 hours, prior to the development of hepatic encephalopathy and corresponding to the early liver injury characterized by microvesicular steatohepatitis (Fig. 2A). After 32 hours, non-comatose mice with severe liver damage and advanced (grade III) encephalopathy had extremely high plasma VEGF levels, reaching a mean of 835pg/ml.
Figure 2. Significantly enhanced circulating and cerebral cortical levels of VEGF in mice with ALF.
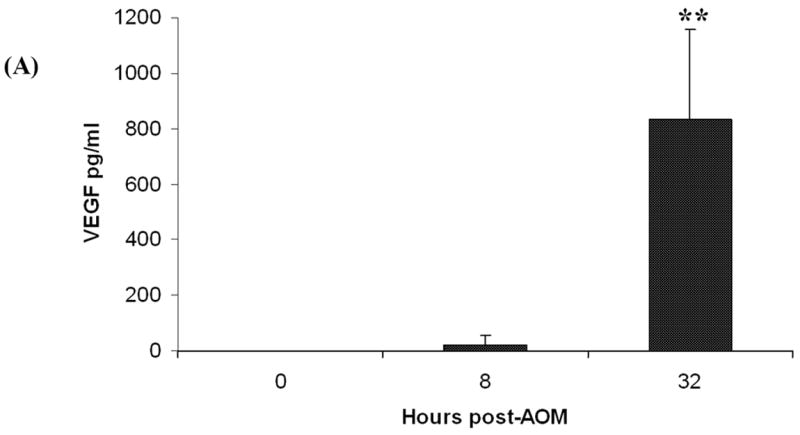

(A) Plasma VEGF levels measured by ELISA. Levels were undetectable in control mice (n = 5). However, in groups of mice administered AOM, levels increased to a mean of 20pg/ml after 8 hours (n = 5) and reached a mean of 835pg/ml at 32 hours (n = 8). Results shown are the mean +/- SEM; ** = highly significant (p < 0.001).
(B-D) Confocal microscopic images of coronal sections through the frontoparietal cortical regions of VEGF-GFP transgenic mouse brains. To visualize the endothelium, mice underwent whole body perfusion with rhodamine lectin (red) immediately after sacrifice. Normal mice (B) showed virtually no visible VEGF-GFP (green) in the cerebral cortex. However, there was evidence of VEGF production within 8 hours of severe liver injury induced by 100μg/g AOM (C). As ALF ensued and encephalopathy progressed, more fluorescence (i.e. VEGF production) was demonstrated in astrocytes close to intracortical blood vessels (D).
Mice with severe encephalopathy have elevated cerebral cortical levels of VEGF
We found locally increased VEGF levels in the brains of mice that developed HE and cerebral edema following AOM. We demonstrated this using VEGF-GFP transgenic mice, which express green fluorescent protein (GFP) under the VEGF promoter [24]. These mice exhibited the same morbidity and mortality as C57BL/6 and BALB/C mice following AOM. Confocal microscopy revealed significant VEGF-GFP fluorescence was not detected in the forebrains of healthy control VEGF-GFP mice (Fig. 2B). However, after inducing ALF, astrocytes in the frontal and parietal cortices of VEGF-GFP mice demonstrated increased fluorescence (Fig. 2C). During the early stages of liver injury, corresponding to the microvesicular steatosis seen around 8-10 hours post-AOM, increased fluorescence was mild but became more pronounced as HE progressed (Fig. 2D).
Ammonia acts synergistically with LPS and IFN-γ to enhance macrophage secretion of VEGF
Hyperammonemia is common in patients with ALF [1]. We therefore examined whether clinically relevant ammonia levels increased VEGF secretion from macrophages activated by pro-inflammatory stimuli. We found that exposure of murine macrophages to E. coli LPS and IFN-γ in vitro led to increased VEGF secretion as previously described [9]. However, supplementing the medium with 50-100μM NH4Cl significantly enhanced VEGF secretion in response to LPS and IFN-γ (Fig. 3). This effect was abolished at higher, cytotoxic concentrations of 0.5-1.0 mM, reflected in reduced cell viability. Conversely, ammonia alone did not raise VEGF levels, suggesting synergy with the pro-inflammatory effects of LPS and IFN-γ that appeared maximal in the presence of 100μM NH4Cl. Similar in vitro results were obtained using primary peritoneal macrophages (data not shown).
Figure 3. Synergistic effect of ammonia, LPS and IFN-γ on macrophage VEGF secretion.
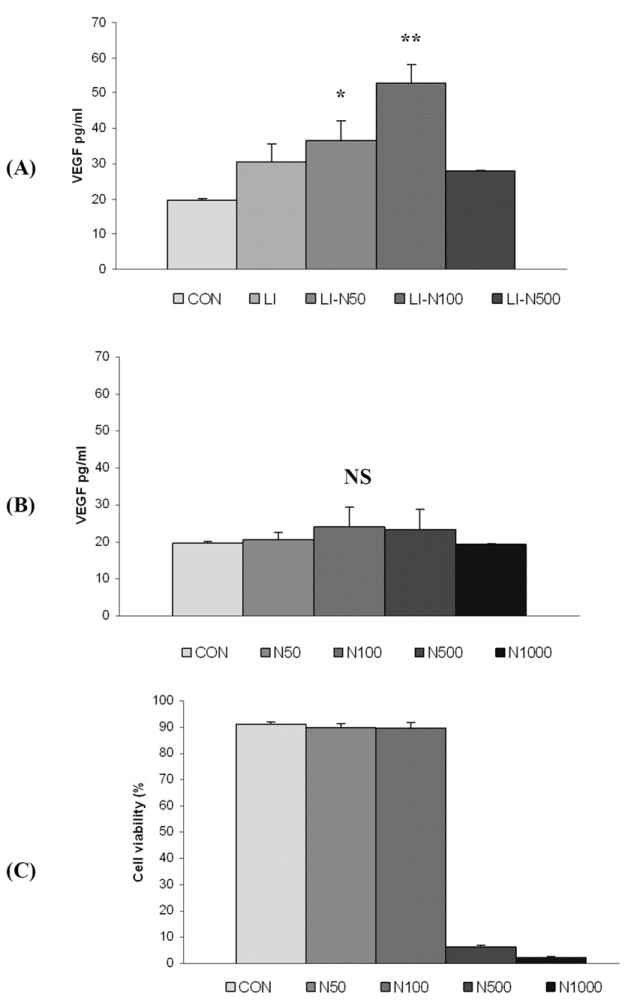
Culture medium VEGF levels from murine RAW 264.7 macrophages maintained at a density of 0.125×106 cells/cm2 in the presence of 100ng/ml E. coli LPS and 10ng/ml IFN-γ (“LI”). (A) The addition of LI resulted in a significant increase in VEGF levels (p<0.05) that was further enhanced by the addition of NH4Cl at concentrations of 50 and 100 μM (“LI-N50” and “LI-N100”). (B) In the absence of LI, adding 50-100μM NH4Cl did not enhance VEGF secretion. Higher ammonia concentrations of 500-1000μM (“N500” and “N1000”) failed to increase VEGF secretion, regardless of the presence or absence of LI. (C) RAW 264.7 macrophage cell viability, as determined by trypan blue exclusion, demonstrating toxic effects of higher quantities of ammonia. * = Statistically significant (p<0.05 vs control); ** = highly significant (p<0.01 vs control); NS = not significant.
Early administration of a Src kinase inhibitor prolongs survival in experimental ALF
We examined the effect of a Src family kinase inhibitor on the severity of ALF. SKI-606 (bosutinib) is a potent and selective inhibitor of Src kinase activity in the nanomolar range [23]. Mice received an i.p. 100μL injection of 10mg/kg SKI-606, at a range of time points following AOM and continuing at 12 hourly intervals. Other mice received 100μl i.p. injections of the drug delivery vehicle alone or the VEGF antagonist Cyclo-VEGI at a dose of 2mg/kg given at 18 hour intervals.
Following AOM-induced liver injury, Cyclo-VEGI had no effect on survival when administered after the onset of HE. Similarly, the SKI-606 had no effect on HE grade or survival from ALF if given late, following the onset of HE. However, early administration of SKI-606 delayed progression of encephalopathy, prolonged survival and enabled 25% of mice to recover from otherwise universally lethal ALF (Fig. 4).
Figure 4. Effect of SKI-606 on survival.

Survival data at 48 hours (A) from groups of mice with ALF induced by AOM. Mice received either vehicle alone (“CON”), the VEGF antagonist Cyclo-VEGI (“c-VEGI”) or SKI-606 at 2 hours or 16 hours post-AOM (“Early SKI-606” and “Late SKI-606” respectively). Only early administration of SKI-606 given 2 hours post-AOM enabled survival from AOM-induced ALF. Even in those that did not survive, SKI-606 substantially delayed the progression of HE to coma and death (B).
Reduced severity of ALF following administration of a Src inhibitor
Early administration of the SKI-606 was associated with a highly significant reduction in the severity of liver injury (Fig. 5). Following AOM administration, ALT levels in controls reached a mean of 3199 IU/L (+/- 298) at 18 hours. In contrast, animals given SKI-606 had significantly smaller ALT rises, with a mean of only 200.5 IU/L (+/- 72) at 18 hours (Fig. 5A).
Figure 5. Severity of liver injury in mice treated with VEGF and Src inhibitors.


(A) Transaminase levels, measured using an ALT/GPT kit, of samples from control mice or at 18 hours following AOM with or without early SKI-606 given 2 hours after AOM. (B) Grading of H&E-stained liver sections from controls or at 18 hours post-AOM, reviewed by a blinded histopathologist, with a semi-quantitative index [26] and scoring at least five lobules per section. *** = p<0.001. (C) H&E-stained light microscopy views of representative 18-hour liver sections from mice administered AOM alone or treated with cVEGI or SKI-606. Early use of the Src inhibitor ameliorated liver injury leaving just centrilobular hepatitis but cVEGI had no obvious effect.
Histological grading revealed amelioration of hepatocellular injury in the early SKI-606-treated mice, the main effect being reduced pan-lobular necro-inflammation, leaving just focal necrosis and inflammation around the central veins (Fig. 5B, 5C). In contrast, mice administered AOM alone had widespread hepatocellular injury with loss of the normal lobular architecture. Late administration of SKI-606 or cyclo-VEGI, subsequent to developing HE, did not reduce liver damage. In contrast to SKI-606, Cyclo-VEGI had no significant effect on the grade of liver injury at either early or late timepoints.
Reduced TUNEL staining after treatment with the Src kinase inhibitor SKI-606
Src family kinases are involved in initiating hepatocyte apoptosis in response to ligation of the CD95 (Apo-1/Fas) death receptor [27]. We therefore wished to measure the extent of apoptosis in the livers of mice with ALF. Using confocal microscopy to examine sections stained with the TUNEL-based ApopTag assay, we found AOM resulted in widespread TUNEL positive staining after 18 hours (Fig. 6A, 6B). However, sections taken from mice after the early administration of SKI-606 demonstrated marked reductions in staining (Fig. 6C). In contrast, neither late administration of SKI-606 nor use of the delivery vehicle alone affected the grade of TUNEL staining.
Figure 6. TUNEL staining of liver sections following azoxymethane-induced liver injury.

Confocal microscopic images of representative liver sections from either control mice or at 18 hours post-AOM. The ApopTag assay stains normal cells green and apoptotic cells red. Control sections (A) showed little apoptosis. However, induction of ALF by AOM lead to widespread numbers of TUNEL positive cells (B) but this was markedly attenuated by early administration of SKI-606 given 2 hours after AOM (C).
Discussion
In a study of 31 patients with acute hepatic necrosis and coma conducted four decades ago, Ritt and colleagues concluded there was “little evidence that any current form of therapy other than supportive measures significantly improves the chances of survival” [28]. Since then, there have been many improvements in supportive care, but there are still few specific therapies for patients with ALF [29]. Due to its beneficial effects on liver regeneration, VEGF has been identified as a putative hepatoprotective agent, based mainly on work in the partial hepatectomy model [30]. However, this model does not fully recreate the toxic milieu of ALF seen in patients with chemical, viral or immunological injury [19]. Furthermore, the consequences of excessive VEGF in sepsis and ischemia suggest that its effects may not be universally beneficial [13].
In this study, we demonstrated that VEGF levels are progressively elevated following AOM liver injury, both in the circulating plasma and within the cerebral cortex. We have also shown that ammonia, in the range of concentrations seen in the blood of patients ALF, potentiates VEGF secretion from macrophages in response to LPS and IFN-γ. This novel finding adds further weight to links between hyperammonemia, sepsis and endothelial dysfunction in ALF.
Importantly, we found the therapeutic response to a Src kinase inhibitor to be highly significant. SKI-606, when given in the early stages of ALF, markedly reduced the severity of liver damage. Progression of HE was significantly delayed and approximately 25% of mice were able to survive an otherwise lethal episode of ALF (Fig. 4).
The basis for this appeared to be a reduced severity of liver injury. Previously, Src family kinases have been shown to mediate hepatocyte apoptosis and levels of Yes, c-Src and Fyn increase in response to CD95 (Apo-1/Fas) ligand, with Yes appearing to play the major role [31].
However, the distinction between hepatocellular apoptosis and necrosis is blurred by the recognition that they share similar initiators and signalling pathways and perhaps they should be considered different points on the same spectrum of cell death [32]. For example, mitochondrial injury may lead either to necrosis due to depletion of ATP or to caspase-dependent apoptosis following cytochrome c release [33]. In APAP toxicity, there is a “second hit” from the innate immune system with neutrophils [34] and NK/NKT cells [35] further exacerbating the initial injury. A positive amplification loop may develop with reactive oxygen species activating c-Jun (NH2) terminal kinase (JNK) and inducing TNF-α expression [36]. In keeping with this role in the second phase of hepatotoxicity, inhibition of JNK reduced injury in a murine APAP model and appeared most effective when administered during the later time points [37].
Like APAP, azoxymethane is metabolized by Cytochrome p450 Cyp2e1 to toxic derivatives that form covalent adducts and cause mitochondrial oxidative stress [38]. The molecular pathogenesis of AOM injury is less well documented, but mitochondrial injury appears evident based on the presence of microvesicular steatosis and profound cristae damage on electron microscopy [20].
Our finding of a significant survival benefit seen with early Src kinase inhibition points to a possible role for Src in the earlier phases of AOM liver injury. Interestingly, while Src activity appears to be anti-apoptotic in malignant cells this is not the case in normal hepatocytes where pro-apoptotic actions of Src can be demonstrated [39]. In our ALF experiments, mice treated with SKI-606 had a definite reduction in TUNEL staining of liver sections, suggestive of reduced hepatocellular apoptosis. However, given that DNA damage in severe necroinflammation may lead to false positive staining with ApopTag TUNEL kits, we cannot exclude the possibility of a reduction in overall hepatotoxicity rather than a specific effect on the apoptotic pathway.
In contrast to the protective effects of the Src inhibitor, we did not detect any survival benefit with VEGF antagonism, despite finding increased circulating and cerebral VEGF levels. This is perhaps to be expected, given the balance between beneficial and deleterious roles of VEGF in organ injury and repair.
For example, in the cecal ligation and punction (CLP) model of sepsis, antagonising VEGF with an adenovirus-delivered sFlt-1 construct ameliorated cardiovascular dysfunction, diminished endothelial permeability and significantly reduced mortality [40]. A recombinant sFlt-1-Fc fusion protein had similar efficacy, suggesting VEGF antagonism as a potential therapy for sepsis [41].
Conversely, mice with APAP-induced ALF who received the VEGF antagonist SU5416 (a Flk-1/KDR receptor tyrosine kinase inhibitor), while showing no initial differences, fared worse than controls during the later stages of toxicity and demonstrated impaired hepatocellular regeneration [42].
These contrasting outcomes in the sepsis and ALF models raise the possibility that VEGFR1/Flt-1 might dominate the pathological actions of VEGF on systemic vascular endothelium in SIRS. VEGFR2/Flk-1 may be more important for liver regeneration in addition to the finding that activation of VEGFR1/Flt-1 stimulates hepatocyte proliferation [8].
Within the liver itself, one cannot assume uniform responses to VEGF by different cell populations. For instance, new light has been thrown on the opposing actions of VEGF on endothelial cells and their supporting vascular smooth muscle cells, pericytes. In contrast to its effect on endothelial cells, VEGF suppressed pericyte function via a VEGF-R2/PDGF-Rβ complex in a mechanism that could be overcome using a VEGF-R2 kinase inhibitor [43]. Stellate cells are the equivalents of pericytes in the liver [44] and their activation plays a key role in sinusoidal injury in both the LPS/beta-galactosamine and carbon tetrachloride models of ALF [45]. Whether VEGF antagonism increases stellate cell activation in AOM-induced ALF remains to be seen. Nonetheless, these disparate roles of VEGF may explain the lack of protection seen with the non-selective VEGF antagonist cyclo-VEGI in our mice with ALF.
In summary, we have demonstrated that early administration of a Src kinase inhibitor decreased the severity of liver injury, delayed progression of encephalopathy and enabled 25% of mice to survive an otherwise universally lethal insult. Src kinase inhibitors may have some therapeutic potential in ALF and deserve further evaluation in other experimental models.
Acknowledgments
RJA received fellowships from the American Liver Foundation and the Scripps Health Foundation. Work in DAC’s laboratory is funded by the National Institutes of Health.
List of Abbreviations
- ALF
acute liver failure
- SIRS
systemic inflammatory response syndrome
- VEGF
vascular endothelial growth factor
- HGF
hepatocyte growth factor
- IFN-γ
interferon gamma
- LPS
lipopolysaccharide
- APAP
acetaminophen
- AOM
azoxymethane
- i.p.
intraperitoneal
- ALT
alanine aminotransferase
- ELISA
enzyme-linked immunosorbent assay
- GFP
green fluorescent protein
- H&E
hematoxylin and eosin
- HE
hepatic encephalopathy
- TNF-α
tumor necrosis factor alpha
- TUNEL
terminal deoxynucleotide transferase dUTP nick end labelling
References
- 1.Lee WM. Acute liver failure. N Eng J Med. 1993;329:1862–1872. doi: 10.1056/NEJM199312163292508. [DOI] [PubMed] [Google Scholar]
- 2.Ostapowicz G, Fontana RJ, Schiodt FV, Larson A, Davern TJ, Han SH, et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med. 2002;137:947–954. doi: 10.7326/0003-4819-137-12-200212170-00007. [DOI] [PubMed] [Google Scholar]
- 3.Ellis A, Wendon J. Circulatory, respiratory, cerebral, and renal derangements in acute liver failure:pathophysiology and management. Semin Liver Dis. 1996;16:379–388. doi: 10.1055/s-2007-1007251. [DOI] [PubMed] [Google Scholar]
- 4.Boeckx NK, Haydon G, Rusli F, Murphy N. Multiorgan failure is the commonest cause of death in fulminant hepatic failure:a single centre experience. Liver Int. 2004;24:702–703. doi: 10.1111/j.1478-3231.2004.0968.x. [DOI] [PubMed] [Google Scholar]
- 5.Rolando N, Wade J, Davalos M, Wendon J, Philpott-Howard J, Williams R. The systemic inflammatory response syndrome in acute liver failure. Hepatology. 2000;32:734–739. doi: 10.1053/jhep.2000.17687. [DOI] [PubMed] [Google Scholar]
- 6.Jalan R, Williams R. The inflammatory basis of intracranial hypertension in acute liver failure. J Hepatol. 2001;34:940–942. doi: 10.1016/s0168-8278(01)00038-1. [DOI] [PubMed] [Google Scholar]
- 7.Palmes D, Skawran S, Spiegel HU. Acute liver failure:from bench to bedside. Transplant Proc. 2005;37:1628–1631. doi: 10.1016/j.transproceed.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 8.LeCouter J, Moritz DR, Li B, et al. Angiogenesis-independent endothelial protection of liver:role of VEGFR-1. Science. 2003;299:890–893. doi: 10.1126/science.1079562. [DOI] [PubMed] [Google Scholar]
- 9.Xiong M, Elson G, Legarda D, Leibovich SJ. Production of vascular endothelial growth factor by murine macrophages:regulation by hypoxia, lactate, and the inducible nitric oxide synthase pathway. Am J Pathol. 1998;153:587–598. doi: 10.1016/S0002-9440(10)65601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mittermayer F, Pleiner J, Schaller G, et al. Marked increase in vascular endothelial growth factor concentrations during Escherichia coli endotoxin-induced acute inflammation in humans. Eur J Clin Invest. 2003;33:758–61. doi: 10.1046/j.1365-2362.2003.01192.x. [DOI] [PubMed] [Google Scholar]
- 11.Kim I, Moon SO, Kim SH, Kim HJ, Koh YS, Koh GY. Vascular endothelial growth factor expression of intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin through nuclear factor-kappa B activation in endothelial cells. J Biol Chem. 2001;276:7614–7620. doi: 10.1074/jbc.M009705200. [DOI] [PubMed] [Google Scholar]
- 12.Boulday G, Haskova Z, Reinders ME, Pal S, Briscoe DM. Vascular endothelial growth factor-induced signaling pathways in endothelial cells that mediate overexpression of the chemokine IFN-gamma-inducible protein of 10 kDa in vitro and in vivo. J Immunol. 2006;176:3098–3107. doi: 10.4049/jimmunol.176.5.3098. [DOI] [PubMed] [Google Scholar]
- 13.Weis SM, Cheresh D. Pathophysiological consequences of VEGF-induced vascular permeability. Nature. 2005;437:497–504. doi: 10.1038/nature03987. [DOI] [PubMed] [Google Scholar]
- 14.Mason JC, Steinberg R, Lidington EA, Kinderlerer AR, Ohba M, Haskard DO. Decay-accelerating factor induction on vascular endothelium by vascular endothelial growth factor (VEGF) is mediated via a VEGF receptor-2 (VEGF-R2) and protein kinase C-alpha/epsilon (PKCalpha/epsilon)-dependent cytoprotective signaling pathway and is inhibited by cyclosporin A. J Biol Chem. 2004;279:41611–41618. doi: 10.1074/jbc.M407981200. [DOI] [PubMed] [Google Scholar]
- 15.Eliceiri BP, Paul R, Schwartzberg PL, Hood JD, Leng J, Cheresh DA. Selective requirement for Src kinases during VEGF-induced angiogenesis and vascular permeability. Mol Cell. 1999;4:915–294. doi: 10.1016/s1097-2765(00)80221-x. [DOI] [PubMed] [Google Scholar]
- 16.Paul R, Zhang ZG, Eliceiri BP, et al. Src deficiency or blockade of Src activity in mice provides cerebral protection following stroke. Nat Med. 2001;7:222–227. doi: 10.1038/84675. [DOI] [PubMed] [Google Scholar]
- 17.Weis S, Shintani S, Weber A, et al. Src blockade stabilizes a Flk/cadherin complex, reducing edema and tissue injury following myocardial infarction. J Clin Invest. 2004;113:885–894. doi: 10.1172/JCI20702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Larson AM, Polson J, Fontana RJ, et al. Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure:results of a United States multicenter, prospective study. Hepatology. 2005;42:1364–1372. doi: 10.1002/hep.20948. [DOI] [PubMed] [Google Scholar]
- 19.Newsome PN, Plevris JN, Nelson LJ, Hayes PC. Animal models of fulminant hepatic failure:a critical evaluation. Liver Transpl. 2000;6:21–31. doi: 10.1002/lt.500060110. [DOI] [PubMed] [Google Scholar]
- 20.Matkowskyj KA, Marrero JA, Carroll RE, Danilkovich AV, Green RM, Benya RV. Azoxymethane-induced fulminant hepatic failure in C57BL/6J mice:characterization of a new animal model. Am J Physiol. 1999;277:G455–462. doi: 10.1152/ajpgi.1999.277.2.G455. [DOI] [PubMed] [Google Scholar]
- 21.Doering CB, Josephson CD, Craddock HN, Lollar P. Factor VIII expression in azoxymethane-induced murine fulminant hepatic failure. Blood. 2002;100:143–147. doi: 10.1182/blood.v100.1.143. [DOI] [PubMed] [Google Scholar]
- 22.Belanger M, Cote J, Butterworth RF. Neurobiological characterization of an azoxymethane mouse model of acute liver failure. Neurochem Int. 2006;48:434–440. doi: 10.1016/j.neuint.2005.11.022. [DOI] [PubMed] [Google Scholar]
- 23.Boschelli DH, Ye F, Wang YD, et al. Optimization of 4-phenylamino-3-quinolinecarbonitriles as potent inhibitors of Src kinase activity. J Med Chem. 2001;44:3965–3977. doi: 10.1021/jm0102250. [DOI] [PubMed] [Google Scholar]
- 24.Fukumura D, Xavier R, Sugiura T, et al. Tumor induction of VEGF promoter activity in stromal cells. Cell. 1998;94:715–725. doi: 10.1016/s0092-8674(00)81731-6. [DOI] [PubMed] [Google Scholar]
- 25.Zimmermann C, Ferenci P, Pifl C, et al. Hepatic encephalopathy in thioacetamide-induced acute liver failure in rats:characterization of an improved model and study of amino acid-ergic neurotransmission. Hepatology. 1989;9:594–601. doi: 10.1002/hep.1840090414. [DOI] [PubMed] [Google Scholar]
- 26.Wood M, Berman ML, Harbison RD, Hoyle P, Phythyon JM, Wood AJ. Halothane-induced hepatic necrosis in triiodothyronine-pretreated rats. Anesthesiology. 1980;52:470–476. doi: 10.1097/00000542-198006000-00003. [DOI] [PubMed] [Google Scholar]
- 27.Reinehr R, Becker S, Eberle A, Grether-Beck S, Haussinger D. Involvement of NADPH oxidase isoforms and Src family kinases in CD95-dependent hepatocyte apoptosis. J Biol Chem. 2005;280:27179–27194. doi: 10.1074/jbc.M414361200. [DOI] [PubMed] [Google Scholar]
- 28.Ritt DJ, Whelan G, Werner DJ, Eigenbrodt EH, Schenker S, Combes B. Acute hepatic necrosis with stupor or coma. An analysis of thirty-one patients. Medicine. 1969;48:151–72. doi: 10.1097/00005792-196903000-00003. [DOI] [PubMed] [Google Scholar]
- 29.Stravitz RT, Kramer AH, Davern T, et al. Intensive care of patients with acute liver failure: Recommendations of the U.S. Acute Liver Failure Study Group. Crit Care Med. 2007;10:1–11. doi: 10.1097/01.CCM.0000287592.94554.5F. [DOI] [PubMed] [Google Scholar]
- 30.Assy N, Spira G, Paizi M, et al. Effect of vascular endothelial growth factor on hepatic regenerative activity following partial hepatectomy in rats. J Hepatol. 1999;30:911–915. doi: 10.1016/s0168-8278(99)80147-0. [DOI] [PubMed] [Google Scholar]
- 31.Reinehr R, Becker S, Wettstein M, Häussinger D. Involvement of the Src family kinase yes in bile salt-induced apoptosis. Gastroenterology. 2004;127:1540–1557. doi: 10.1053/j.gastro.2004.08.056. [DOI] [PubMed] [Google Scholar]
- 32.Malhi H, Gores GJ, Lemasters JJ. Apoptosis and necrosis in the liver: a tale of two deaths? Hepatology. 2006;43(2 Suppl 1):S31–44. doi: 10.1002/hep.21062. [DOI] [PubMed] [Google Scholar]
- 33.Kim JS, Qian T, Lemasters JJ. Mitochondrial permeability transition in the switch from necrotic to apoptotic cell death in ischemic rat hepatocytes. Gastroenterology. 2003;124:494–503. doi: 10.1053/gast.2003.50059. [DOI] [PubMed] [Google Scholar]
- 34.Liu ZX, Govindarajan S, Kaplowitz N. Innate immune system plays a critical role in determining the progression and severity of acetaminophen hepatotoxicity. Gastroenterology. 2004;127:1760–1774. doi: 10.1053/j.gastro.2004.08.053. [DOI] [PubMed] [Google Scholar]
- 35.Liu ZX, Han D, Gunawan B, Kaplowitz N. Neutrophil depletion protects against murine acetaminophen hepatotoxicity. Hepatology. 2006;43:1220–1230. doi: 10.1002/hep.21175. [DOI] [PubMed] [Google Scholar]
- 36.Schwabe RF, Brenner DA. Mechanisms of Liver Injury. I. TNF-alpha-induced liver injury:role of IKK, JNK, and ROS pathways. Am J Physiol Gastrointest Liver Physiol. 2006;290:G583–589. doi: 10.1152/ajpgi.00422.2005. [DOI] [PubMed] [Google Scholar]
- 37.Henderson NC, Pollock KJ, Frew J, et al. Critical role of c-jun (NH2) terminal kinase in paracetamol-induced acute liver failure. Gut. 2007;56:982–990. doi: 10.1136/gut.2006.104372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sohn OS, Fiala ES, Requeijo SP, Weisburger JH, Gonzalez FJ. Differential effects of CYP2E1 status on the metabolic activation of the colon carcinogens azoxymethane and methylazoxymethanol. Cancer Res. 2001;61:8435–8440. [PubMed] [Google Scholar]
- 39.De Toni EN, Kuntzen C, Gerbes AL, et al. p60-c-src suppresses apoptosis through inhibition of caspase 8 activation in hepatoma cells, but not in primary hepatocytes. J Hepatol. 2007;46:682–691. doi: 10.1016/j.jhep.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 40.Yano K, Liaw PC, Mullington JM, et al. Vascular endothelial growth factor is an important determinant of sepsis morbidity and mortality. J Exp Med. 2006;203:1447–1458. doi: 10.1084/jem.20060375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsao PN, Chan FT, Wei SC, et al. Soluble vascular endothelial growth factor receptor-1 protects mice in sepsis. Crit Care Med. 2007;35:1955–60. doi: 10.1097/01.CCM.0000275273.56547.B8. [DOI] [PubMed] [Google Scholar]
- 42.Donahower B, McCullough SS, Kurten R, et al. Vascular endothelial growth factor and hepatocyte regeneration in acetaminophen toxicity. Am J Physiol Gastrointest Liver Physiol. 2006;291:G102–109. doi: 10.1152/ajpgi.00575.2005. [DOI] [PubMed] [Google Scholar]
- 43.Greenberg JI, Shields DJ, Barillas SG, et al. A role for VEGF as a negative regulator of pericyte function and vessel maturation. Nature. 2008;456:809–813. doi: 10.1038/nature07424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sato M, Suzuki S, Senoo H. Hepatic stellate cells: Unique characteristics in cell biology and phenotype. Cell Struc Func. 2003;28:105–112. doi: 10.1247/csf.28.105. [DOI] [PubMed] [Google Scholar]
- 45.Yan C, Zhou L, Han YP. Contribution of hepatic stellate cells and matrix metalloproteinase 9 in acute liver failure. Liver Int. 2008;28:959–971. doi: 10.1111/j.1478-3231.2008.01775.x. [DOI] [PubMed] [Google Scholar]