

Abstract
Purpose
Gene identification in small families segregating autosomal dominant sensorineural hearing loss presents a significant challenge. To address this challenge, we have developed a machine learning based software tool, AudioGene v2.0, to prioritize candidate genes for mutation screening based on audioprofiling.
Methods
We analyzed audiometric data from a cohort of American families with high frequency autosomal dominant sensorineural hearing loss. Those families predicted to have a DFNA2 audioprofile by AudioGene v2.0 were screened for mutations in the KCNQ4 gene.
Results
Two novel missense mutations and a stop mutation were detected in three American families predicted to have DFNA2-related deafness for a positive predictive value of 6.3%. The false negative rate was 0%. The missense mutations were located in the channel pore region and the stop mutation was in transmembrane domain S5. The latter is the first DFNA2-causing stop mutation reported in KCNQ4.
Conclusions
Our data suggest: (1) that the N-terminal end of the P-loop is crucial in maintaining the integrity of the KCNQ4 channel pore; and, (2) that AudioGene audioprofile analysis can effectively prioritize genes for mutation screening in small families segregating high frequency autosomal dominant sensorineural hearing loss. AudioGene software will be made freely available to clinicians and researchers once it has been fullly validated.
Keywords: KCNQ4, DFNA2, hearing loss, AudioGene v2.0, Audioprofiling
INTRODUCTION
The prevalence of significant hearing loss (≥ 25 decibels) is 15–20% in the adult population and rises to approximately 50% in individuals 80 years of age or older 1. ADSNHL accounts for approximately 15% of inherited hearing loss. To date, 22 genes for autosomal dominant deafness have been identified, and a further 30 autosomal dominant loci mapped to chromosomal regions 2. In most cases the hearing loss is sensorineural (SNHL) and non-syndromic.
Identification of phenotype-genotype correlations is crucial in determining the etiology of ADSNHL (reviewed in 3) and has implications for prognostic and therapeutic outcomes. It is clear that some correlations are very robust, such as the low frequency audioprofile associated with WFS1-related hearing loss (DFNA6/14/38) 4 and the “cookie-bite” audioprofile associated with TECTA-related hearing loss (DFNA8/12) 5, while other correlations are more difficult to define. Autosomal dominant high frequency hearing loss, for example, can be the consequence of mutations in a large number of different genes (ie. KCNQ4 (DFNA2), DFNA5 (DFNA5), COCH (DFNA9), POU4F3 (DFNA15)). It is possible that the cluster of genes that cause high frequency hearing loss can be refined by analyzing additional audiometric data. One such analysis involves multiple regression studies of threshold data with respect to age and/or select frequencies to determine whether these genes fall into identifiable subclusters. If subclustering is possible, audioprofiling would significantly decrease the work required to identify the genetic cause of hearing loss in small families segregating ADSNHL.
To test the feasibility of this concept, we have developed the AudioGene system, which analyzes audiometric data and predicts the likely underlying genetic cause of hearing loss based on known phenotypic parameters. This audioprofiling initiative is important because: 1) in the short term, identifying the etiology of hearing loss is valuable to families segregating ADSNHL 6; and 2) in the long term, establishing causality will have prognostic and therapeutic importance 7, 8. In this study, we report audiometric analysis of the well characterized ADSNHL gene KCNQ4 (DFNA2). Since KCNQ4 mutations at the DFNA2 locus are a common cause of ADSNHL 9–17, genotypic data for KCNQ4 (DFNA2) is provided as validation for this novel approach.
The DFNA2 locus was first identified in 1994 on chromosome 1p34 in Indonesian and American families segregating ADSNHL noticeably more severe in the high frequencies than in the low frequencies 18. Subsequently, two deafness-causing genes were identified at the DFNA2 locus, GJB3 and KCNQ4 14, 19. Mutations in KCNQ4 are the predominant cause of hearing impairment at the DFNA2 locus, and hearing loss at this locus represents a common form of ADSNHL 9–17. KCNQ4 is a member of the voltage-gated potassium channel family and is involved in potassium recycling in the inner ear 14. The 695-amino acid protein contains six transmembrane domains and a hydrophobic P-loop region located between transmembrane domains S5 and S6 (residues 259 to 296). The P-loop domain forms the channel pore that contains a filter selective for potassium ions. Mutations in the pore region affect this selectivity filter and eliminate channel function 14. To date, at least 15 deafness-causing mutations have been identified in KCNQ4. Most of these mutations are missense changes that act via a dominant-negative mechanism to induce progressive, predominantly high frequency hearing impairment 14. However, two families have also been reported with deletions that lead to frameshifts and stop codons, p.Gln71ProfsX64 and p.Gln71SerfsX68 10, 20. In these families, the phenotype reflects a dosage effect and is characterized by better low frequency but more rapid high frequency deterioration as compared to the hearing loss phenotype in families segregating missense mutations 9, 11.
MATERIALS AND METHODS
Subjects
American subjects with the diagnosis of non-syndromic ADSNHL were recruited to this study. DNA was extracted from blood lymphocytes using established procedures. Audiograms and medical histories were obtained to verify high frequency hearing loss. The control group comprised 100 unrelated individuals. This study was approved by the University of Iowa Institutional Review Board and participants gave consent for their involvement.
Audiometric Data
For each subject, basic audiograms that evaluate hearing thresholds at specified frequencies (250, 500, 1000, 2000, 4000, and 8000 Hz) were obtained using universal standards and formatted for audioprofile analysis. Because ADSNHL is typically symmetric 21, data were recorded as binaural averages unless the degree of asymmetry between ears was greater than 30 dB at a given frequency. In those instances, thresholds from the better hearing ear were used. Although additional data from temporal bone imaging and vestibular testing can be extremely valuable in prioritizing genes for mutation screening, these data were not included in this study.
AudioGene v2.0
AudioGene v2.0 was trained using audiograms from subjects known to carry deafness-causing mutations in KCNQ4 (DFNA2), DFNA5 (DFNA5), WFS1 (DFNA6/14/38), TECTA (DFNA8/12), COCH (DFNA9) or COL11A2 (DFNA13). Initially, the oldest and youngest age-related threshold audiograms were chosen, with missing attributes filled in by interpolation using the CPAN module Math::Interpolate. Second and first order polynomials were fitted to the data using CPAN’s PDL::Fit::Polynomial and these coefficients were then included as attributes. Classification was performed in Weka 22 using a Support Vector Machine (SVM) approach and the SMO (sequential minimal optimization) algorithm 23. Accuracy was measured with 10-fold cross validation.
When presented with an “unknown” audiogram, AudioGene v2.0 is designed to select genes for mutation screening based on audioprofiling (Figure 1). For each audiogram or series of audiograms from a subject or multiple subjects segregating ADSNHL in a single family, AudioGene v2.0 initially assigns the audioprofile to a given gene cluster. Within a cluster, AudioGene v2.0 then rank-orders genes based on deafness-causing likelihood. This strategy facilitates the prioritization of candidate genes for mutation screening.
Figure 1.
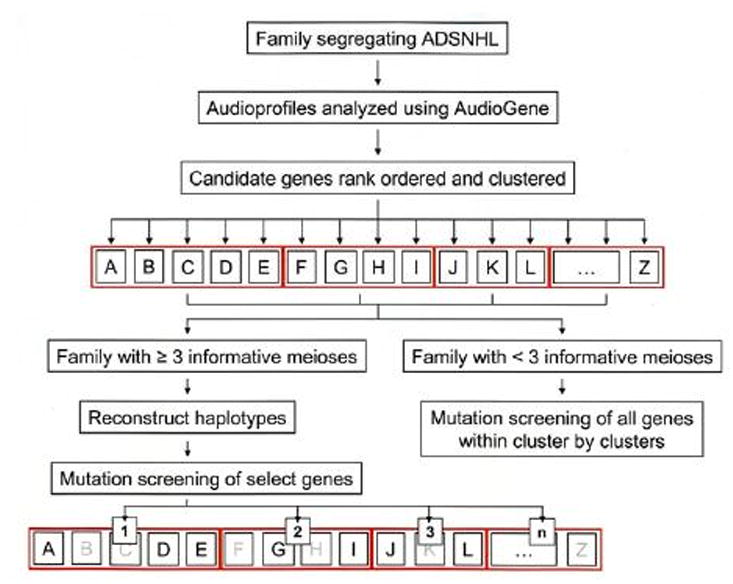
Genetic screening strategy for small families segregating ADSNHL. AudioGene is used to rank order all known genes that cause ADSNHL (letters in black boxes), placing genes that have similar audioprofiles into clusters (red boxes). Haplotyping is used to determine whether any candidate gene within a given cluster can be eliminated; mutation screening is completed on candidate genes that cannot be eliminated. If families are too small to make haplotyping useful, all genes within a cluster will be screened, beginning with the highest ranking cluster.
As dictated by family size and the availability of DNA samples, bidirectional sequencing of candidate genes can be completed or haplotypes can be constructed to quickly rule-in or rule-out a given gene. Once genetic screening identifies a disease-causing mutation, the relevant phenotype-genotype data is incorporated into the AudioGene training dataset to improve its predictive accuracy.
Human Expert Analysis
Blinded otolaryngologists and audiologists based at the University of Iowa Hospitals and Clinics analyzed 50 unlabeled audiograms from ADSNHL patients with previously characterized mutations. There was no time limit imposed on these experts to make their prediction and the same set of audiograms was analyzed by AudioGene v2.0 for comparison. The experts were permitted to use any information at their disposal while making their predictions.
Genotyping and Mutation Detection
The entire coding region of KCNQ4 was PCR amplified and bidirectionally sequenced from the DNA of one proband from each family (or multiple affected subjects if available). All PCR reactions were performed using gene-specific primers (Table 1), 20 ng of genomic DNA, 200 μM of dNTPs and 0.5 μM of primers and the following conditions: denaturation for 3 min at 95°C followed by 35 cycles of 95°C for 30 sec, 54–67°C annealing for 30 sec, 72°C for 30 sec and a terminal extension for 10 min at 72°C. PCR products were analyzed on 2% agarose gels. Amplimers were sequenced bidirectionally on an ABI 3130 automated sequencer using the Big-Dye Terminator Version 3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA).
Table 1.
Oligonucleotides used to amplify human KCNQ4
| Exon | Forward Primer (5′-3′) | Reverse Primer (5′-3′) | TM (°C) a |
|---|---|---|---|
| 1 | ATGCGTCTCTGAGCGCCC | ACGGCCGTGCAGAGCGAA | 66 |
| 2 | ACCTTGGCTGTCCTGTCCCT | CCATGGCTCAGAGAGCAGCTT | 66 |
| 3 | TGGGCCCCAGTTCTGGAGA | GGTGACGCCTTTACTCCCAATC | 66 |
| 4 | GGGTAGGCTGGCTGTGATCTCG | CGGAGAGGGCGAGGTCAGA | 66 |
| 5 | CCTTTATCCCTTTCCCGTGT | CCATCGTGACTCCTGACTCA | 66 |
| 6 | CCAGGAGAGGGAGAATCCAT | GACACCCTTGCAGCCTCTTA | 66 |
| 7 | GACACCCTTGCAGCCTCTTA | GTGTGTCAACCCTGTGTGCT | 66 |
| 8 | TTCTTTCAGGGGAGAGGGCT | TGAGGACAAGTGGCTGAGAC | 66 |
| 9 | CACTGGGTGAGGGGGGTGGT | GGGCTGCCACCTCCTCTTGCT | 66 |
| 10 | TGGCTAACTTGGCTCTCTCC | CGATCGAGGGCCGGCTGAG | 66 |
| 11 | TCTGGGCCTCTGTCTTCCTA | GTGGTGGATGCCTGTAATCC | 66 |
| 12 | TGCTCTAACAGGACACTCCCTCTG | ACCCCAGCCCTGAATGAAGT | 66 |
| 13 | TTGTGGAAGGGAGGGACGGC | TGACATGGGGAGGATGCGTT | 66 |
| 14 | TCCGCCCCGAGACCCAAG | GGAGAGAGACCACACGCAGT | 61 |
Annealing temperature
RESULTS
ADSNHL Families Analyzed by AudioGene v2.0
160 individuals from 77 unrelated American families with apparent ADSNHL of unknown etiology participated in this study. Probands from these families were screened for KCNQ4 (DFNA2) mutations. Audiograms from a further 360 persons with ADSNHL for which a genetic cause had previously been determined were used to train AudioGene v2.0.
Genotype Predictions using AudioGene v2.0
AudioGene v2.0 was trained on nearly 2400 audiograms from 360 persons with ADSNHL caused by mutations in KCNQ4 (DFNA2), DFNA5 (DFNA5), WFS1 (DFNA6/14/38), TECTA (DFNA8/12), COCH (DFNA9) or COL11A2 (DFNA13). These genes were selected for training in part due to the availability of large numbers of audiograms, and in part because they represent two degrees of discernment capability - that is: 1) gross clustering; and 2) gene prioritization within a given cluster. The classification of the 360 individuals with known ADSNHL gene mutations by AudioGene v2.0 is summarized in Figure 2. Analysis of these audiograms showed that AudioGene v2.0 can accurately predict the correct gene based on a classifier trained on two audiograms for each of the members of the training sets for DFNA8 and DFNA9 92.9% of the time (Table 2). Based on clustering, training sets for additional loci were sequentially added and as expected the accuracy of AudioGene was reduced. However, even with training sets for all six loci, the accuracy of the program was still above 77%. It is noteworthy that DFNA8 and DFNA9 are in different clusters (Figure 2). DFNA9 falls into the same cluster as DFNA2 and DFNA5, and within this cluster, AudioGene v2.0 correctly rank-orders genes 86% of the time (Figure 2).
Figure 2.
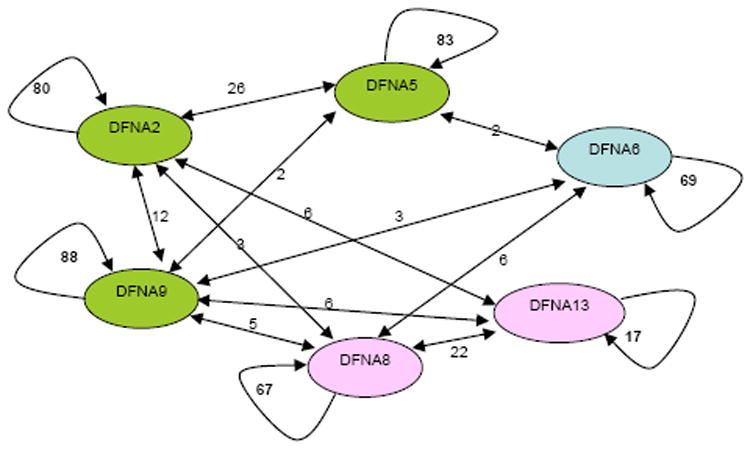
Graphic depiction of the classification (and misclassification) of 2,400 audiograms from 360 patients for six genes. Each node in the graph represents a gene and each arc represents the number of individuals classified as having the audioprofile for that gene by Audiogene v2.0. The number of individuals indicated between the nodes are those misclassified by the program.
Table 2.
Overall accuracy of AudioGene v2.0
| Gene Set a | Accuracy |
|---|---|
| DFNA 8/9 | 92.9% |
| DFNA 8/9/6 | 91.2% |
| DFNA 8/9/6/2 | 84.5% |
| DFNA 8/9/6/2/13 | 77.9% |
| DFNA 8/9/6/2/13/5 | 77.3% |
Each row shows percent accurate gene prediction from the set of genes listed. In each case, the program was trained with audiograms from the list of genes shown and tested with 10-fold cross validation.
Validation of AudioGene v2.0
To determine whether AudioGene-based prioritization of genes within a given cluster is comparable to the accuracy of trained human specialists, we developed a web interface to allow otolaryngologists and audiologists to examine and predict the associated genotype for 50 audiograms (Figure 3). A comparison of the results from 27 human experts versus AudioGene v2.0 demonstrates that human experts had an average accuracy of 55% while the machine classifier accurately distinguished between genotypes 88% of the time. The data also show that machine classification is very consistent, while human expertise is highly variable (Figure 3).
Figure 3.
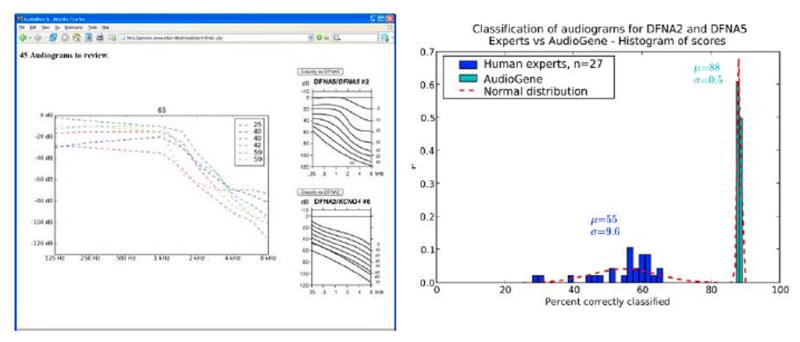
Left: Web interface of a tool to allow human experts to classify audiograms to the likely causal gene. Right: Results of human classification of 50 audiograms for DFNA2 and DFNA5 (in the same cluster) by 27 experts versus machine classification of the same set of audiograms.
To validate AudioGene v2.0 as a clinical and research tool, we then studied a cohort of 77 families segregating presumed ADSNHL represented by audiograms from 160 individuals. When these audiograms were analyzed by AudioGene v2.0, 89 individuals from 48 families were predicted to have a DFNA2 profile (Figure 4). Positive and negative predictive values were 6.3% (3/48) and 100% (29/29) respectively.
Figure 4.
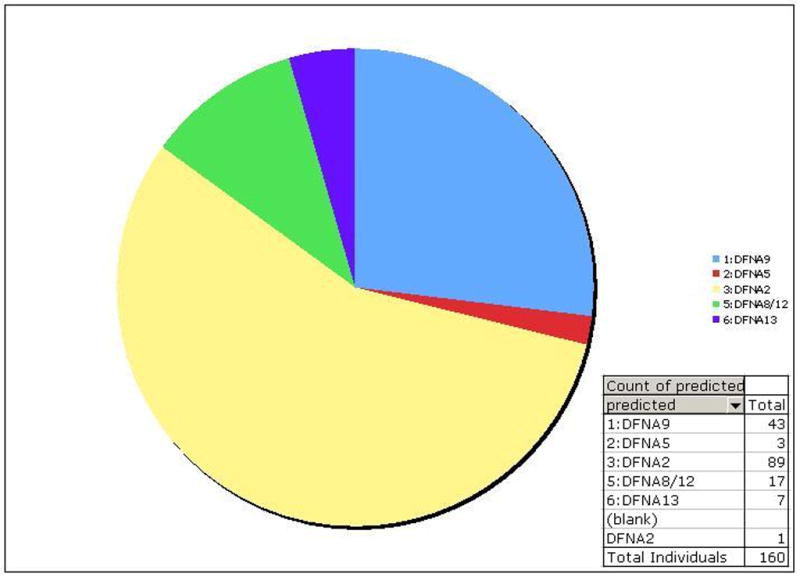
Graphical representation of the gene prediction for each of 160 individuals from 77 ADSNHL families by AudioGene v2.0 audioprofiling
Screening of KCNQ4 in ADSNHL Families Reveals Novel Mutations
Mutation screening of KCNQ4 was completed in at least one subject from each of the 77 families. In three families in the cohort of families predicted by AudioGene v2.0 to have mutations in this gene, novel mutations were found. No mutations were found in KCNQ4 in the cohort of subjects predicted to have deafness of a different gene etiology.
In unrelated American families 3 and 4 we identified two novel missense mutations in exon 5 of KCNQ4 that were located at the N-terminal end of the P-loop, close to transmembrane domain S5. One mutation was an A→G nucleotide change (c.778G>A) resulting in a glutamate-to-lysine substitution (USA 3; p.E260K; Table 1; Figure 5A,B); the other mutation, an A→T nucleotide change (c.785A>T), leads to an aspartate-to-valine subsitution (USA 4; p.D262V; Table 1; Figure 5AC,D). These mutations were numbered based on human KCNQ4 cDNA and protein (NCBI Accession Numbers: NM_172163 and NP_751895) sequences. Both subjects developed high frequency hearing loss during childhood (Figure 6), although the hearing loss was more severe in the subject carrying the p.D262V alteration. This difference may reflect the progressive nature of DFNA2 hearing loss, as there was a seven year difference in ages between subjects at the time of audiological testing. Other members of both families were unavailable for genetic testing and additional pedigree information could not be obtained.
Figure 5.
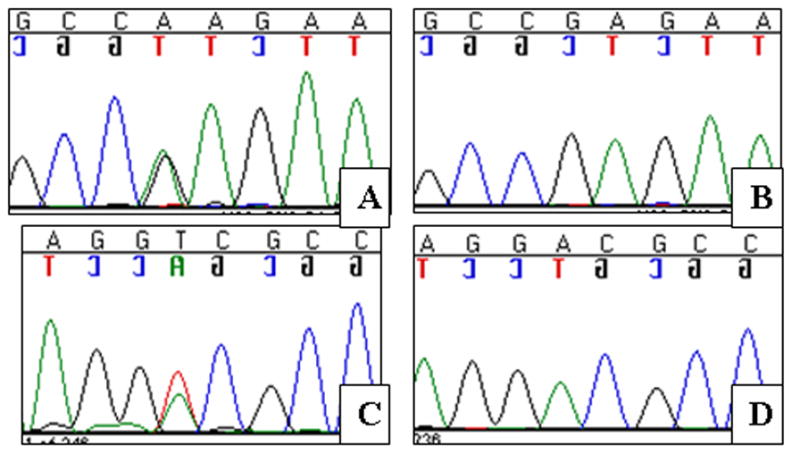
Point mutations in KCNQ4 exon 5. A–B Sequence chromatograms of the patient with the p.E260K mutation and a normal CEPH control. The mutation results in a G/A nucleotide substitution. C–D Sequence chromatograms of the patient with the p.D262V mutation and a normal CEPH control. The mutation results in an A/T nucleotide substitution.
Figure 6.
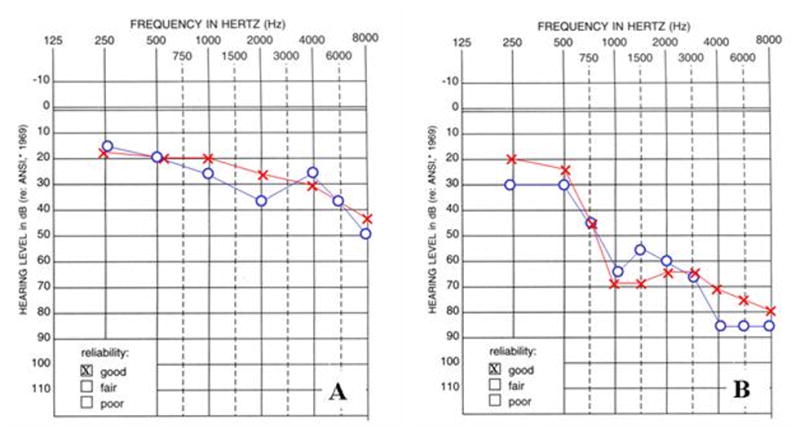
Audiograms of individuals from American families 3 and 4 with hearing loss due to missense mutations in KCNQ4. A Audiogram of the individual carrying the p.E260K mutation measured at five years of age. B Audiogram of the individual carrying the p.D262V mutation measured at twelve years of age.
In a third American family with a DFNA2 profile, a stop mutation was detected in exon 5 of KCNQ4 (Table 1, USA5; Figure 7 and Figure 8). A G→A nucleotide change (c.807G>A) was identified in affected family members III:2 and IV:2 but not in unaffected individual IV:1 (Figure 7 and Figure 9). The heterozygous base change introduces a stop codon (p.W241X) that is predicted to result in a truncated version of the KCNQ4 protein lacking most of transmembrane domain S5, the entire channel pore region, the S6 transmembrane domain and the cytoplasmic C-terminal domain. The DFNA2 phenotype associated with this mutation appears to be more severe than that associated with KCNQ4 missense mutations as affected individual IV:2 had severe-to-profound hearing loss by 3 years of age and has received a cochlear implant.
Figure 7.
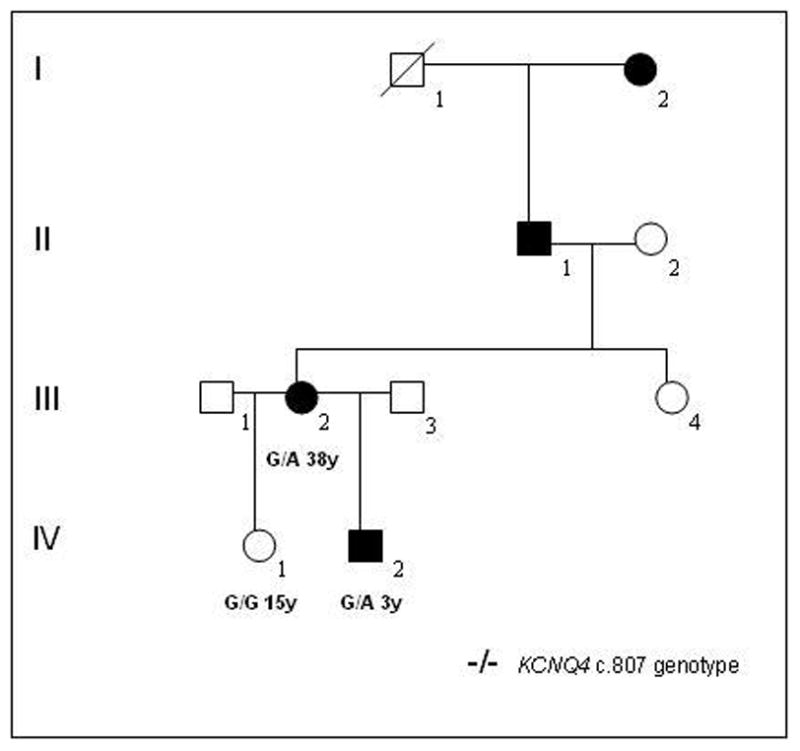
Pedigree of American family 5 with non-syndromic DFNA2 hearing loss due to a stop mutation in KCNQ4. Genotypes for nuceotide c.807 in the KCNQ4 gene and age (years) are shown for those individuals included in the genetic analysis. Open symbols unaffected; filled black symbols affected; diagonal line deceased.
Figure 8.
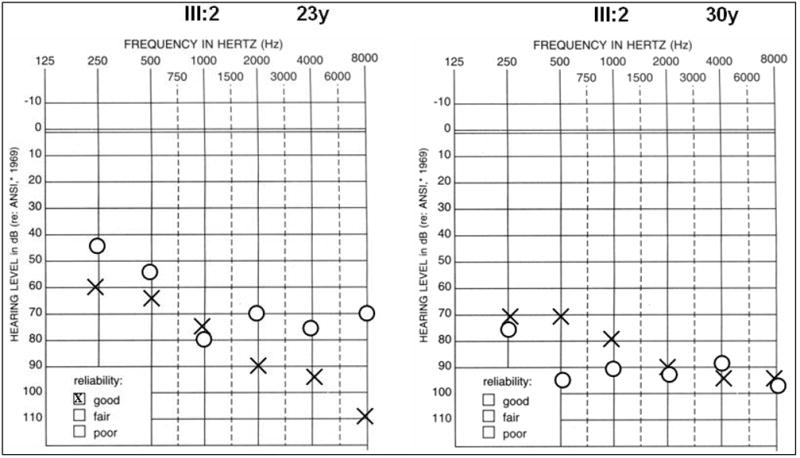
Audiograms of representative affected family member III:2 at 23 and 30 years of age respectively from American family 5. High frequency hearing is more severely affected at both ages, although thresholds are significantly higher at the later age reflecting the progressive nature of the condition.
Figure 9.
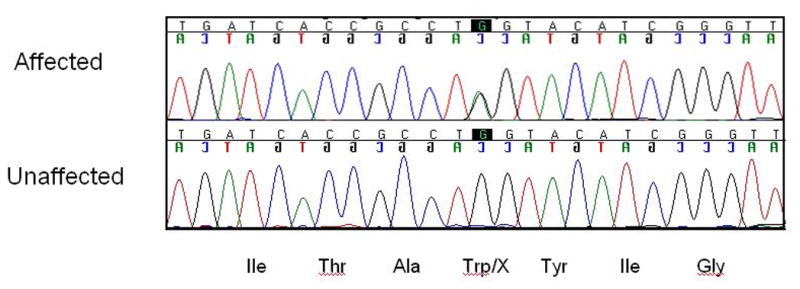
The novel causative stop mutation in the KCNQ4 gene in American family 5. The mutation is a G-to-A nucleotide alteration in exon 5 (c.807G→A) that results in introduction of a stop codon (p.W241X).
A multi-sequence alignment of the KCNQ4 protein sequence was generated using ClustalW24. The p.E260 and p.D262 residues are highly conserved in mammals, consistent with their location in the P-loop of the channel pore region (Figure 10A). Conseq analysis confirmed this conservation with both residues having a score of 9 and predicted that the glutamic acid and aspartic acid residues are exposed and functionally important (Figure 10B). Neither missense mutation was found in a screen of 100 unrelated controls (200 chromosomes).
Figure 10.
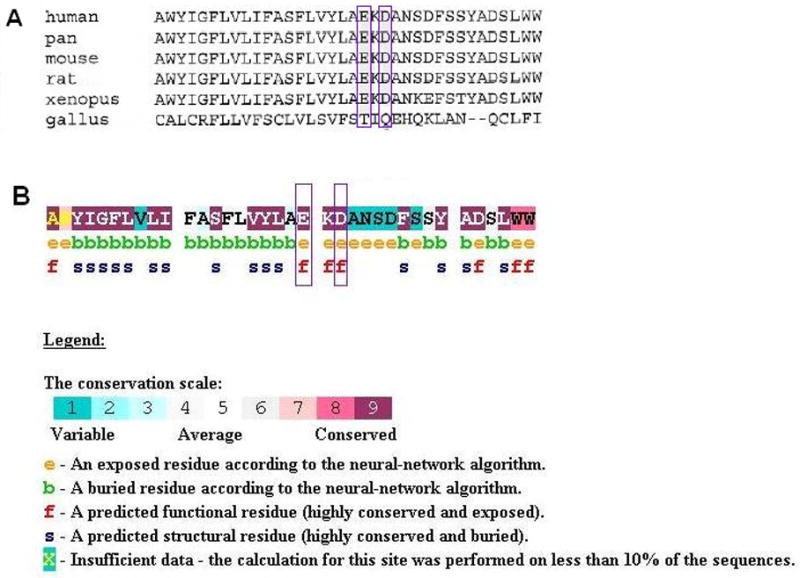
Analysis of the missense mutations in KCNQ4. A Multi-sequence alignment of KCNQ4 sequence that contributes to the 5th transmembrane and P-loop domains. The glutamate and aspartate residues affected by the p.E260K and p.D262V mutations respectively are highly conserved (purple boxes). B Conseq analysis of the residues affected by the mutations showing that they are both predicted to be exposed and functionally important.
DISCUSSION
We believe that there is a fundamental need for a program like AudioGene for several reasons. First, identifying the genetic cause of deafness in small families segregating ADSNHL is very challenging. Clinicians are left with prioritizing genes for mutation screening based on audioprofiling. While it is well recognized that this approach is easily applicable to some audioprofiles (low frequency audioprofile and WFS1-related hearing loss (DFNA6/14/38)25; “cookie-bite” audioprofile and TECTA-related hearing loss (DFNA8/12)5), as the number of dominant loci and associated genes increases, the magnitude of the data becomes vast. Even if this human approach to gene prioritization was feasible, “naked-eye” clustering of audiograms could be used to recognize only a relatively few clusters and then only by a few persons with considerable experience with ADSNHL. We have shown, however, that even with only two genes to consider, naked-eye is very inferior to machine-driven classification. Second, as larger genetic studies become feasible (e.g., 1,000,000 SNP-based associations with 1000s of patients), the need to systematically and consistently phenotype subjects will overwhelm available human expertise. Third, as a translational outcome, the program will be a valuable aid in the genetic diagnosis and management of families segregating ADSNHL. And finally, as machine learning becomes more robust with increasing amounts of data and the employment of multiple different methods of analysis, we hypothesize that the number of discrete clusters that can be recognized will increase.
A major strength of AudioGene v2.0 is that we used nearly 2400 audiograms from 360 persons to create this program. In addition, we have audiometric and genotypic data from approximately 2000 more persons to integrate into the program. We believe this repository of data will represent the most comprehensive set of records for ADSNHL available. It will eventually include most of the originally reported DFNA families together with numerous families that have not been reported. We recognize that even with this vast amount of data, it may not be possible to identify the “correct” gene given a series of audiograms in a family segregating ADSNHL every time, however we believe it will be possible to rank order all known ADSNHL genes for mutation screening and to group genes into clusters of probability. Furthermore, we expect to be able to place the “correct” gene somewhere within the top cluster, and anticipate being able to refine clusters with increasing amounts of data. To obtain more audiograms to generate increasing amounts of phenotype-genotype data, we have developed a secure database system called the Collaborative Phenotypic Database (CPDB) where collaborating groups can deposit and view limited clinical data. This system is also available to collect data for eye, tumor and autism subjects.
We have demonstrated the efficacy of AudioGene by identifying three novel mutations in the known ADSNHL gene KCNQ4 (DFNA2): p.E260K, p.D262V and p.W241X. These mutations are located within the conserved P-loop region and the S5 transmembrane domain. Both glutamic acid and aspartic acid are negatively charged and hydrophilic residues. In contrast, lysine is positively charged and valine is nonpolar and hydrophobic. These amino acid substitutions are predicted to alter the nature of the protein by changing either its polarity or hydrophobicity. These changes could lead to abnormal function of the channel pore and interfere with transport of potassium ions in the inner ear. It is likely that these missense mutations affect the function of KCNQ4 via a dominant-negative mechanism, since proven dominant-negative mutations have been identified in the channel pore region 14, 17. It is thought that dominant-negative mutations lead to hearing impairment by interfering with the function of normal KCNQ4 channel subunits in the inner ear 14, 26. Four subunits must assemble correctly to form a functional channel; if one of these subunits is abnormal then only 1/16 of all channels will function normally 14. The loss of KCNQ4 channel activity leads to progressive outer hair cell degeneration and hearing loss 14, 26.
The p.W241X mutation identified in this study is the first reported DFNA2-causing stop mutation in KCNQ4. Since key domains required for assembly of KCNQ channels are located in the c-terminal cytoplasmic portion of the channel it is possible that the p.W241X truncated version of the protein interferes with normal formation of the KCNQ4 channel. This would represent a dominant-negative effect, although it is unclear whether the reduction in KCNQ4 channel activity would be greater than that induced by missense mutations where only 1/16 channels have normal function. Alternatively, the truncated version may not be incorporated into hetero- or homotetrameric KCNQ channels, leading to a haploinsufficient phenotype. Haploinsufficiency would appear the more likely of these two scenarios given the lack of c-terminal domains required for normal channel assembly. This hypothesis is also consistent with the genotype-phenotype correlation of human KCNQ4 mutations. The p.W41X mutation and the two previously reported deletions that lead to frameshifts and premature stop codons, p.Gln71ProfsX64 and p.Gln71SerfsX68 10, 20, are associated with more severe high frequency hearing loss at an earlier age compared to missense mutations. However, one difference is that these deletions are associated with milder low frequency hearing loss than the p.W241X or missense mutations. This may reflect spatially-distinct requirements for KCNQ4 channel activity in the mammalian cochlea.
Silencing of the p.E260K and p.D262V dominant-negative missense mutations would be predicted to preserve hearing. A recent proof-of-principle study involving another dominant deafness gene has validated this prediction – an siRNA was shown to potently suppress expression of the R75W allele of human GJB2 in a murine model 8. By using a construct containing GJB2R75W, that interferes with function of wild-type gap junction protein 27, Maeda and colleagues were able recapitulate human deafness (DFNA3) in a murine model 8. In subsequent experiments, the same construct and specific anti-GJB2R75W siRNAs were mixed with DOTAP/cholesterol liposomes, soaked in Gelfoam, and applied topically to the murine round window membrane. Although liposome-nucleic acid complexes were detected in non-sensory cells of the cochlea, the siRNA specifically reduced expression of the GJB2R75W allele and reversed the hearing loss phenotype 28. Based on these results, it is likely that the p.E260K and p.D262V alleles of KCNQ4 can be targeted by a similar RNAi approach. Introduction of a wild-type KCNQ4 gene would be expected to protect against the hearing loss induced by the p.W241X allele. Development of mouse models and therapeutic approaches for these alleles would increase our understanding of the role of KCNQ4 in the inner ear and potentially provide better clinical outcomes for DFNA2 patients.
The positive predictive value of 6.3% (3/48) for KCNQ4 mutations in the families predicted to have DFNA2 ADSNHL was relatively low. The specificity of AudioGene DFNA2 predictions was therefore low (39.2%), leading to a high false postive rate (60.8%). These values are consistent with the fact that high frequency ADSNHL is a heterogenous disorder and that at specific ages audioprofiles induced by mutations in different genes can be very difficult to distinguish. Similarity between audioprofiles is also compounded by the variable progression of hearing loss observed within and between most high frequency ADSNHL families. One way to potentially improve the positive predictive value for high frequency ADSNHL families would be to analzye audiometric data from individuals only at specific ages when subtle differences in audioprofile are most discernible. The negative predictive value for the cohort of families we analyzed was 100% (29/29), showing that if a family was not predicted to have a DFNA2 profile by AudioGene then a KCNQ4 mutation was not identified. This high sensitivity (100%) and negligible false postive rate (0%) means that AudioGene is a very effective tool for excluding high frequency ADSNHL families from KCNQ4 screening.
There are several alternatives to the development of a system like AudioGene, but they have important limitations. For example, one option is to use either a mutation detection chip or a resequencing chip to screen all possible ADSNHL genes. We have piloted a mutation detection chip for Usher syndrome (APSER), but its sensitivity is only 0.70 and it does not obviate the need for confirmatory sequencing. For this reason, we have discontinued use of the ASPER chip for Usher syndrome patients in favor of a direct sequencing strategy. Resequencing chips would be a better option however there are also limitations with this approach. First, ADSNHL genes have been identified for less than half of the mapped loci making any chip incomplete 2, and second, many mutations are deletions and insertions. The detection of these mutations is more difficult and requires substantially more control hybridization experiments to determine baseline fluctuations in the hybridization specificities, thereby reducing chip sensitivity29. Another option is to rank order candidate genes for mutation screening in small families segregating ADSNHL using the “naked-eye” to recognize audiometric similarities and dissimilarities. However, subtleties will be missed, data manipulation is limited and tedious, and as more ADSNHL genes are cloned, the problem of rank ordering becomes increasingly difficult. In contrast, with an algorithm like AudioGene, greater amounts of data enhance performance.
In summary, we have developed a novel audioprofiling system, AudioGene v2.0 that can predict genotypic information from audiometric data. We have demonstrated the feasibility of this approach by analyzing ADSNHL families with high frequency hearing loss. Genetic analysis of the families we studied with AudioGene v2.0 confirms the accuracy of the program as three novel mutations in KCNQ4 were identified. We therefore believe that a system like AudioGene will represent an important tool to researchers who are focusing on the identification of novel ADSNHL loci, to clinicians who care for deaf families, and to clinical diagnostic laboratories that offer mutation screening for deafness. To this end AudioGene software will be made freely available to clinicians and researchers once it has been validated for all genes in the training set.
Acknowledgments
RJH Smith is the Sterba Hearing Research Professor, University of Iowa College of Medicine, who supported the project with an NIH-NIDCD grant (RO1 DCO3544) for autosomal dominant non-syndromic hearing loss.
Abbreviations
- ADSNHL
autosomal dominant sensorineural hearing loss
- SNHL
sensorineural hearing loss
Footnotes
No researchers involved in this study report a conflict of interest.
References
- 1.Morton NE. Genetic epidemiology of hearing impairment. Ann N Y Acad Sci. 1991;630:16–31. doi: 10.1111/j.1749-6632.1991.tb19572.x. [DOI] [PubMed] [Google Scholar]
- 2.Van Camp G, Smith RJ. Hereditary Hearing Loss Homepage. 2007. [Google Scholar]
- 3.Huygen P, Pauw R, Cremers C. Genes, Hearing and Deafness. In: Martini M, Stephens D, Read AP, editors. From Molecular Biology to Clinical Practice - Audiometric profiles associated with genetic nonsyndromal hearing impairment: a review and phenotype analysis. London, UK: Informa Healthcare; 2007. [Google Scholar]
- 4.Bespalova IN, Van Camp G, Bom SJ, et al. Mutations in the Wolfram syndrome 1 gene (WFS1) are a common cause of low frequency sensorineural hearing loss. Hum Mol Genet. 2001;10(22):2501–8. doi: 10.1093/hmg/10.22.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verhoeven K, Van Laer L, Kirschhofer K, et al. Mutations in the human alpha-tectorin gene cause autosomal dominant non-syndromic hearing impairment. Nat Genet. 1998;19(1):60–2. doi: 10.1038/ng0598-60. [DOI] [PubMed] [Google Scholar]
- 6.Brunger JW, Murray GS, O’Riordan M, et al. Parental attitudes toward genetic testing for pediatric deafness. Am J Hum Genet. 2000;67(6):1621–5. doi: 10.1086/316901. Epub 2000 Nov 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maeda Y, Fukushima K, Kawasaki A, et al. Cochlear expression of a dominant-negative GJB2R75W construct delivered through the round window membrane in mice. Neurosci Res. 2007;58(3):250–4. doi: 10.1016/j.neures.2007.03.006. Epub 2007 Mar 24. [DOI] [PubMed] [Google Scholar]
- 8.Maeda Y, Fukushima K, Nishizaki K, et al. In vitro and in vivo suppression of GJB2 expression by RNA interference. Hum Mol Genet. 2005;14(12):1641–50. doi: 10.1093/hmg/ddi172. [DOI] [PubMed] [Google Scholar]
- 9.Akita J, Abe S, Shinkawa H, et al. Clinical and genetic features of nonsyndromic autosomal dominant sensorineural hearing loss: KCNQ4 is a gene responsible in Japanese. J Hum Genet. 2001;46(7):355–61. doi: 10.1007/s100380170053. [DOI] [PubMed] [Google Scholar]
- 10.Coucke PJ, Van Hauwe P, Kelley PM, et al. Mutations in the KCNQ4 gene are responsible for autosomal dominant deafness in four DFNA2 families. Hum Mol Genet. 1999;8(7):1321–8. doi: 10.1093/hmg/8.7.1321. [DOI] [PubMed] [Google Scholar]
- 11.De Leenheer EM, Huygen PL, Coucke PJ, et al. Longitudinal and cross-sectional phenotype analysis in a new, large Dutch DFNA2/KCNQ4 family. Ann Otol Rhinol Laryngol. 2002;111(3 Pt 1):267–74. doi: 10.1177/000348940211100312. [DOI] [PubMed] [Google Scholar]
- 12.De Leenheer EM, Ensink RJ, Kunst HP, et al. DFNA2/KCNQ4 and its manifestations. Adv Otorhinolaryngol. 2002;61:41–6. doi: 10.1159/000066802. [DOI] [PubMed] [Google Scholar]
- 13.Ensink RJ, Huygen PL, Van Hauwe P, et al. A Dutch family with progressive sensorineural hearing impairment linked to the DFNA2 region. Eur Arch Otorhinolaryngol. 2000;257(2):62–7. doi: 10.1007/pl00007511. [DOI] [PubMed] [Google Scholar]
- 14.Kubisch C, Schroeder BC, Friedrich T, et al. KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell. 1999;96(3):437–46. doi: 10.1016/s0092-8674(00)80556-5. [DOI] [PubMed] [Google Scholar]
- 15.Talebizadeh Z, Kelley PM, Askew JW, et al. Novel mutation in the KCNQ4 gene in a large kindred with dominant progressive hearing loss. Hum Mutat. 1999;14(6):493–501. doi: 10.1002/(SICI)1098-1004(199912)14:6<493::AID-HUMU8>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 16.Van Camp G, Coucke PJ, Akita J, et al. A mutational hot spot in the KCNQ4 gene responsible for autosomal dominant hearing impairment. Hum Mutat. 2002;20(1):15–9. doi: 10.1002/humu.10096. [DOI] [PubMed] [Google Scholar]
- 17.Van Hauwe P, Coucke PJ, Ensink RJ, et al. Mutations in the KCNQ4 K+ channel gene, responsible for autosomal dominant hearing loss, cluster in the channel pore region. Am J Med Genet. 2000;93(3):184–7. doi: 10.1002/1096-8628(20000731)93:3<184::aid-ajmg4>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 18.Coucke P, Van Camp G, Djoyodiharjo B, et al. Linkage of autosomal dominant hearing loss to the short arm of chromosome 1 in two families. N Engl J Med. 1994;331(7):425–31. doi: 10.1056/NEJM199408183310702. [DOI] [PubMed] [Google Scholar]
- 19.Xia JH, Liu CY, Tang BS, et al. Mutations in the gene encoding gap junction protein beta-3 associated with autosomal dominant hearing impairment. Nat Genet. 1998;20(4):370–3. doi: 10.1038/3845. [DOI] [PubMed] [Google Scholar]
- 20.Kamada F, Kure S, Kudo T, et al. A novel KCNQ4 one-base deletion in a large pedigree with hearing loss: implication for the genotype-phenotype correlation. J Hum Genet. 2006;51(5):455–60. doi: 10.1007/s10038-006-0384-7. Epub 2006 Apr 5. [DOI] [PubMed] [Google Scholar]
- 21.Van Camp G, Willems PJ, Smith RJ. Nonsyndromic hearing impairment: unparalleled heterogeneity. Am J Hum Genet. 1997;60(4):758–64. [PMC free article] [PubMed] [Google Scholar]
- 22.Witten I, Frank E. Practical machine learning tools and techniques. 2. San Francisco: Morgan Kaufmann; 2005. [Google Scholar]
- 23.Platt J. Fast Training of Support Vector Machines using Sequential Minimal Optimization. MIT Press; 1998. [Google Scholar]
- 24.Berezin C, Glaser F, Rosenberg J, et al. ConSeq: the identification of functionally and structurally important residues in protein sequences. Bioinformatics. 2004;20(8):1322–4. doi: 10.1093/bioinformatics/bth070. Epub 2004 Feb 10. [DOI] [PubMed] [Google Scholar]
- 25.Cryns K, Pfister M, Pennings RJ, et al. Mutations in the WFS1 gene that cause low-frequency sensorineural hearing loss are small non-inactivating mutations. Hum Genet. 2002;110(5):389–94. doi: 10.1007/s00439-002-0719-1. Epub 2002 Apr 9. [DOI] [PubMed] [Google Scholar]
- 26.Kharkovets T, Dedek K, Maier H, et al. Mice with altered KCNQ4 K+ channels implicate sensory outer hair cells in human progressive deafness. EMBO J. 2006;25(3):642–52. doi: 10.1038/sj.emboj.7600951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kudo T, Kure S, Ikeda K, et al. Transgenic expression of a dominant-negative connexin26 causes degeneration of the organ of Corti and non-syndromic deafness. Hum Mol Genet. 2003;12(9):995–1004. doi: 10.1093/hmg/ddg116. [DOI] [PubMed] [Google Scholar]
- 28.Maeda Y, Fukushima K, Nishizaki K, et al. In vitro and in vivo suppression of GJB2 expression by RNA interference. Hum Mol Genet. 2005;14(12):1641–50. doi: 10.1093/hmg/ddi172. Epub 2005 Apr 27. [DOI] [PubMed] [Google Scholar]
- 29.Karaman MW, Groshen S, Lee CC, et al. Comparisons of substitution, insertion and deletion probes for resequencing and mutational analysis using oligonucleotide microarrays. Nucleic Acids Res. 2005;33(3):e33. doi: 10.1093/nar/gni034. [DOI] [PMC free article] [PubMed] [Google Scholar]