

Abstract
The ingestion of alcohol during pregnancy can result in a group of neurobehavioral abnormalities collectively known as fetal alcohol spectrum disorders (FASD). During the past decade, studies using animal models indicated that early alcohol exposure can dramatically affect neuronal plasticity, an essential property of the central nervous system responsible for the normal wiring of the brain and involved in processes such as learning and memory. The abnormalities in neuronal plasticity caused by alcohol can explain many of the neurobehavioral deficits observed in FASD. Conversely, improving neuronal plasticity may have important therapeutic benefits. In this review, the author discuss the mechanisms that lead to these abnormalities and comment on recent pharmacological approaches that have been showing promising results in improving neuronal plasticity in FASD.
Keywords: fetal alcohol syndrome, AMPA, NMDA, CREB, phosphodiesterase, neuronal plasticity, development
Fetal alcohol spectrum disorders (FASD) is an umbrella term describing a spectrum of effects that can occur in an individual whose mother abused alcohol during pregnancy. It is the leading known cause of mental retardation in the Western world, with approximately 40,000 cases of FASD reported each year just in the United States—far higher than the combined figures of Down syndrome, spina bifida, and muscular dystrophy (Abel 1995; Klug and Burde 2003; May and Gossage 2001). Along with deficits in higher order functions such as learning, memory, attention, and problem solving, children with FASD show altered somatosensory, auditory, and visual processing and often autistic behavior (Guerri 1998; Mattson and others 1996; Mattson and Riley 1998; O’Connor and Paley 2009) (see Box 1).
Box 1.
The harmful effects of alcohol ingestion during gestation have been noted since ancient times. There are citations in the Bible (“Behold, thou shalt conceive and bear a son: And now, drink no wine or strong drink”) and in Aristotle’s work Problematta (“Foolish, drunken, or harebrain women most often bring forth children like unto themselves”), suggesting that alcohol can be a teratogen. However, it was only in the 20th century that the detrimental effects of alcohol during pregnancy were recognized in clinical practice and by the scientific community. Recognition of fetal alcohol syndrome happened with the studies done by Philip Lemoine in France (Lemoine and others 1968) and Kenneth Jones and David Smith (1973) in the United States, who described a series of abnormalities in the offspring of alcoholic mothers. These abnormalities include growth deficiency, CNS-related abnormalities, and a specific pattern of facial features (Fig. 1). The neurological problems observed in FAS can be variable, ranging from severe mental retardation to subtle attention deficits. The reason for this variability is probably related to multiple factors such as differences in timing of the alcohol exposure, amount of alcohol ingested, and genetic factors. For instance, the facial features (Fig. 1), one of the most important elements for FAS diagnosis, are due to alcohol ingestion during the first trimester of development (Sulik and others 1981). Although the prevalence of FAS in the United States (0.1–0.7 per 1000 births; Abel 1995; May and others 2009) is high compared to many other conditions, it is estimated that the number of children who are not diagnosed as having FAS (i.e., because of a lack of facial features) but may still exhibit neurobehavioral problems caused by prenatal exposure to alcohol is 10 to 20 times higher (Centers for Disease Control and Prevention 1994). Because of the diversity of outcomes caused by prenatal alcohol exposure, the National Task Force on FAS/FAE adopted the umbrella term fetal alcohol spectrum disorders (FASD; Floyd and others 2005).
Figure 1.
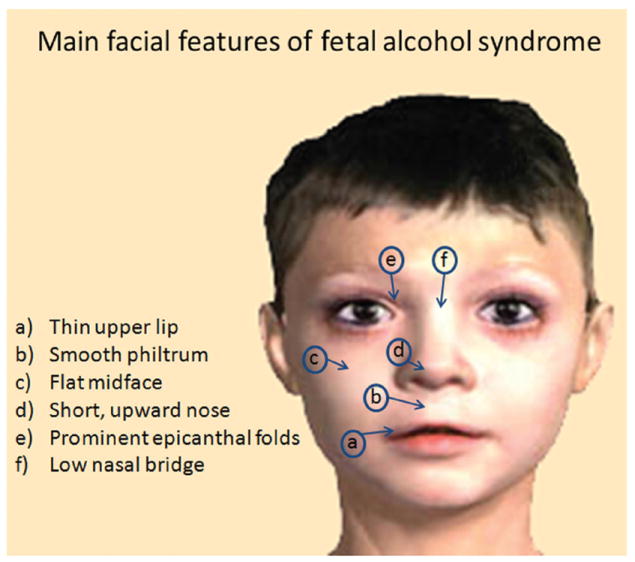
Typical facial features observed in fetal alcohol syndrome.
The behavioral abnormalities observed due to prenatal alcohol exposure can be seen as early as the toddler years and can be displayed as jitteriness, irritability, and disruptions of sleep patterns (Mattson and Riley 1998; O’Connor and Paley 2009). During the school years, children with FASD frequently present with symptoms of attention deficit hyperactivity disorder, depression, learning problems, visuospatial deficits, and impairments in fine and gross motor skills (Mattson and Riley 1998; O’Connor and Paley 2009). During adolescence, aggressive behavior and addiction may be also be added to the group of behavioral problems (Mattson and Riley 1998; O’Connor and Paley 2009).
The effects of alcohol exposure can be different throughout the gestation. During the first-trimester equivalent of human gestation, alcohol exposure can alter normal development of the neural tube and crest, leading to microcephaly (Miller 1996), hydrocephaly (Webster and others 1980), ocular malformations (Cook and others 1987; Sulik and Johnston 1983), and the facial dysmorphology that characterizes fetal alcohol syndrome (Sulik and others 1981), a particular type of FASD. During the second trimester, alcohol exposure strongly affects the proliferation of glia and neuronal precursors probably by altering expression of neurotrophic factors such as transforming growth factor (TGF)–β (Luo and Miller 1998; Miller and Luo 2002; Siegenthaler and Miller 2005), leading to abnormal migration of cortical neurons (Miller and Robertson 1993; Siegenthaler and Miller 2004).
During the third-trimester equivalent of human gestation, the brain goes through a period of fast growth often called “brain growth spurt,” and neurons are more susceptible to the apoptotic effects of alcohol exposure (Ikonomidou and others 2000) (see Box 2). Alcohol exposure during the brain growth spurt affects synaptogenesis and may lead to persistent deficits on neuronal plasticity.
Box 2.
Alcohol-triggered apoptosis is one of the most studied consequences of ethanol exposure (Fig. 2). In particular, ethanol exposure during the third-trimester equivalent of development has been shown to result in a dramatic reduction in total cell number (Ikonomidou and others 2000). Because of this remarkable effect, it was quickly assumed that cell death was the main cause of most neuronal abnormalities observed in models of FASD, and many methods to prevent alcohol-induced neuroapoptosis have been proposed (Marino and others 2004; Ieraci and Herrera 2006; Saito and others 2007; Dong and others 2008).
Figure 2.
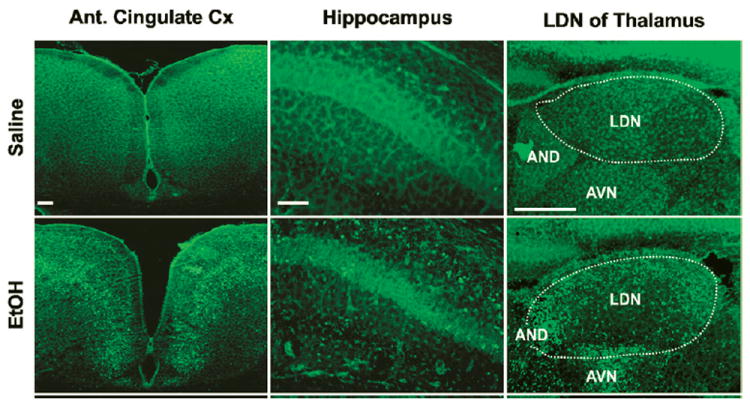
Alcohol-triggered apoptosis revealed by Fluoro-Jade B staining 24 hours after ethanol exposure (5 g/kg subcutaneously). Modified from Ieraci and Herrera (2006). PLoS One, open access.
How does alcohol cause neuroapoptosis? There are two major cascades related to programmed cell death: the extrinsic and the intrinsic pathways. The extrinsic pathway is related to binding of cytokines to death receptors, activation of caspase-8, and cleavage and activation of effector caspase-3, -6, or -7. In the intrinsic pathway, the translocation of the Bax protein from the cytosol to the outer mitochondrial membrane results in an increase of membrane permeability and release of cytochrome C. The involvement of each of the pathways on alcohol-triggered apoptosis has been recently studied. Ethanol exposure in neonates causes widespread apoptosis and reduction in brain mass with blood alcohol levels ranging from 50 to 500 mg/dL (Ikonomidou and others 2000; Young and others 2003). In P7 mice treated with ethanol, no activated caspase-8 is detected, although cleaved caspase-3 is seen (Young and others 2003). However, when caspase-3 knockout mouse pups are treated with ethanol during development, there is still widespread apoptosis, although it occurs much later than in wild-type mice and without characteristic DNA fragmentation (Young and others 2005). These results suggest that ethanol must be acting through the intrinsic (mitochondrial) pathway via a Bax-mediated mechanism. In fact, the Bax null mouse does not show signs of apoptosis, including lack of cytochrome C release, even when treated with robust amounts of ethanol during the neonatal period (Young and others 2003). Furthermore, alcohol exposure does not lead to changes in cell number/density in this mutant. Surprisingly, although an increase in neuroapoptosis is one of the most evident teratogenic effects of early alcohol exposure, the empirical demonstration of a causal link between alcohol-triggered apoptosis and the neurobehavioral pathology associated with FASD remains largely unexplored.
Neuronal plasticity is the brain’s capacity to be shaped by experience by making and breaking connections. This ability is essential during development, when circuits are refined by selective pruning, as well as throughout our lives, in common processes such as learning and memory (Katz and Shatz 1996; White and Fitzpatrick 2007). There is growing evidence that neuronal plasticity is persistently impaired in animal models of FASD (see Table 1). These impairments have been demonstrated by several labs and in different plasticity paradigms such as long-term potentiation and depression (Izumi and others 2005; Richardson and others 2002; Servais and others 2007; Sutherland and others 1997), learning and memory tests (Clements and others 2005; Girard and others 2000; Hamilton and others 2003; Marino and others 2004), barrel cortex plasticity (Rema and Ebner 1999), ocular dominance plasticity (Medina and others 2003; Medina and Ramoa 2005), and eye-blink conditioning (Johnson and others 2008; Stanton and Goodlett 1998). The fact that plasticity is impaired in these models suggests that other types of plasticity such as the refinement of neuronal circuits during development could be disrupted as well. This could explain why some cortical maps are altered in FASD models (Chappell and others 2007; Margret and others 2006; Medina and others 2005; Powrozec and Zhou 2005).
Table 1.
Several Examples of Neuronal Plasticity Paradigms That Are Altered in Models of Fetal Alcohol Spectrum Disorders
| Type of Neuronal Plasticity | Timing of Exposurea | Speciesb | Region Affected | References | ||
|---|---|---|---|---|---|---|
| Long-term potentiation | I-II, III, I-III | R, GP | Hippocampus | 1,2,3 | ||
| Long-term depression | I-II, III | M | Hippocampus, cerebellum | 4,5 | ||
| Ocular dominance plasticity | III | F, M | Visual cortex | 6,7 | ||
| Barrel cortex plasticity | I-II | R | Somatosensory cortex | 8 | ||
| Eyeblink conditioning | III | R | Hippocampus, cerebellum, inferior olivary nucleus | 9 | ||
| Water maze performance | III | R, GP | Hippocampus | 3,10,11 | ||
| Fear conditioning | I-III, III | R, M | Hippocampus, amygdala | 12,13,14 | ||
| Lesion-induced plasticity | I-II | R | Olfactory tubercle, hippocampus | 15,16 | ||
| Olfactory response to alcohol | I-II | R | Olfactory bulb, trigeminal system | 17 |
Equivalents to trimesters of human gestation.
R = rats; M = mice; GP = guinea pig; F = ferret.
Downward arrows = Decrease of function. Upward arrows = Increase of function. Gray star = Servais and others (2007) showed that LTD at the parallel fiber-Purkinje cell synapses was converted to LTP.
References: 1 = Puglia and Valenzuela (2010); 2 = Sutherland and others (1997); 3 = Richardson and others (2002); 4 = Servais and others (2007); 5 = Izumi and others (2005); 6 = Medina and others (2003); 7 = Medina and Ramoa (2005); 8 = Rema and Ebner (1999); 9 = Stanton and Goodlett (1998); 10 = Goodlett and Johnson (1997); 11 = Girard and others (2000); 12 = Ieraci and Herrera (2006); 13 = Murawski and Stanton (2010); 14 = Savage and others (2010); 15 = Gottesfeld and others (1989); 16 = West and others (1984); 17 = Youngentob and Glendinning (2009).
During the past decade, several groups have been developing pharmacological approaches to improve neuronal plasticity (Ghavami and others 2006; Lynch 2002; Nicholson 1990; Rose and others 2005; Staubli and others 1994; Tully and others 2003). Although the ultimate goal of these studies is to develop drugs that can improve cognition in normal subjects, these nootropics (aka smart drugs) have been considered good candidates for treating several conditions that are related to poor neuronal plasticity (Alt and others 2006; Asanuma and others 1996; Black 2005; Blokland and others 2006; Ghavami and others 2006; Johnston 2003, 2004; Kelley and others 2007; Lynch 2002; Nicholson 1990; Rose and others 2005; Staubli and others 1994). Here we discuss the mechanisms that underlie impaired plasticity in FASD and suggest pharmacological approaches that could help in ameliorating these deficits.
Molecular Mechanisms of Neuronal Plasticity
Over the years, it has been demonstrated that neuronal plasticity requires NMDAr and AMPAr function (Berardi and others 2003; Malenka and Bear 2004; Malinow and Malenka 2002; Platenik and others 2000), an appropriate level of GABAergic inhibition (Fagiolini and others 2004; Mohler 2007), and the activation of transcription factors (Atkins and others 1998; Etkin and others 2006; Frank and Greenberg 1994; Josselyn and Nguyen 2005; Lamprecht 2005; Silva and others 1998). Binding of transcription factors on the cAMP-responsive element (CRE) and serum response element (SRE) leads to the expression of genes that execute the functional and morphological changes necessary for neuronal plasticity to occur (Atkins and others 1998; Etkin and others 2006; Frank and Greenberg 1994; Josselyn and Nguyen 2005; Lamprecht 2005; Silva and others 1998). Examples of these changes are modification of receptor trafficking (Keifer and others 2007), alteration of cytoskeleton protein complexes (Lavaur and others 2008), and remodeling of dendritic spines (Suzuki and others 2007).
The prevailing theory to explain activity-dependent plasticity suggests that mechanisms exist to strengthen synapses whose activity coincides with target depolarization beyond some threshold level (Hebb 1949) and to eliminate synapses whose activity is not correlated with postsynaptic activation (Stent 1973). The biophysical properties of the NMDA receptor suggest that it may function as a correlation detector, which would signal synchronous pre- and postsynaptic depolarization (Bourne and Nicoll 1993). The ionic channel linked to the NMDA receptor is blocked by Mg++ at the resting membrane potential (Mayer and others 1984; Nowak and others 1984). Synchronous activity of several presynaptic terminals may activate AMPA receptors depolarizing the postsynaptic membrane. As a result, the Mg++ blockade is released, and the NMDA receptor becomes functional (Mayer and others 1984; Nowak and others 1984). Therefore, NMDA activation can be facilitated or reduced respectively by insertion/removal of AMPA receptors in/from the post-synaptic membrane (Malenka and Bear 2004; Malinow and Malenka 2002). Activation of NMDA receptors leads to a rise in intracellular Ca++ levels, which ultimately leads to changes in gene expression through the following cascades.
cAMP
Cyclic nucleotide levels are regulated by cyclases and phosphodiesterases. Although activation of the former increases the levels of the cyclic form of the nucleotide, the latter decreases it. Therefore, following NMDA activation, calcium influx increases adenyl cyclase activity, which in turn increases cAMP (Fig. 3, red) levels activating protein kinase A (PKA; Waltereit and Weller 2003). Active forms of PKA may translocate to the nucleus, where it phosphorylates CREB at Ser133. Thus, CREB binds to CREs, triggering the expression of plasticity-related genes (Curtis and Finkbeiner 1999; Platenik and others 2000).
Figure 3.
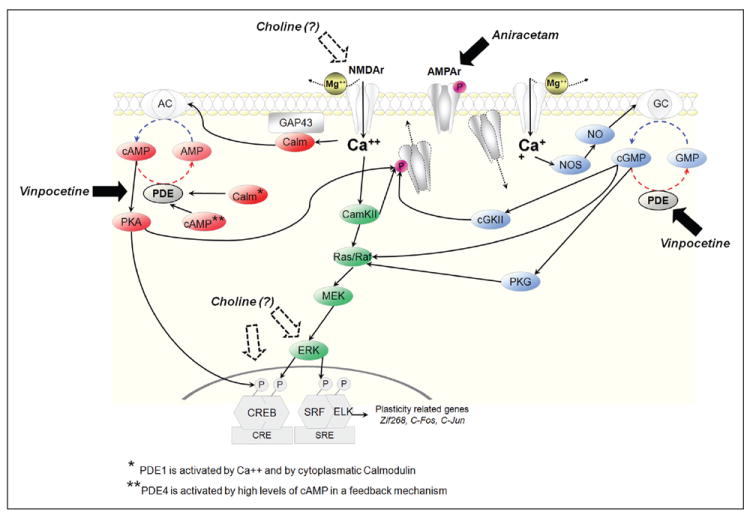
Activation of the CREB/serum response factor (SRF), a crucial cascade for neuronal plasticity. It has been demonstrated that vinpocetine, aniracetam, and choline have successfully improved neuronal plasticity in models of fetal alcohol spectrum disorder (FASD) by different mechanisms. Vinpocetine, as a phosphodiesterase (PDE) type 1 inhibitor, increases levels of cAMP and cGMP. Aniracetam, as an AMPA receptor modulator, facilitates glutamatergic transmission. Although its specific mechanism is poorly understood, it is known that there is a correlation between choline supplementation and facilitation of NMDA function and an increase in phosphorylation of ERK and CREB.
CamKII
Activation of CamKII (Fig. 3, green) by calcium can lead to phosphorylation of AMPA receptors, leading to its incorporation on the postsynaptic membrane and further facilitation of NMDA receptor activation (Barria and others 1997). In addition, Ca++ influx can lead to CREB phosphorylation through ERK, via the CamKII–MEK cascade (Bito and others 1996; Kornhauser and others 2002). Activation of ERK can phosphorylate SRF and ELK, promoting the functional binding of these transcription factors to the SRE (Chai and Tarnawski 2002).
cGMP
Ca++ influx can increase activity of nitric oxide synthase (NOS). The consequent raise in nitric oxide levels enhances guanylyl cyclase activity and the production of cGMP (Fig. 3, blue; Contestabile 2008). Thus, cGMP can activate the Ras/Raf pathway (Fig. 3, green) either directly or through protein kinase G (PKG; Pilz and Broderick 2005).
Different Ways to Enhance Plasticity Through the NMDA-CREB Cascade
Over the years, studies from several groups have indicated that alcohol exposure can affect many of the steps of the aforementioned cascades. For instance, NMDA receptors are down-regulated for several weeks and into adulthood following early alcohol exposure (Rema and Ebner 1999; Savage and others 1992). In addition, alcohol can lead to profound alterations on AMPA receptor function (Bellinger and others 2002; Costa and others 2000; Mameli and others 2005; Valenzuela and others 2008), which could further alter glutamatergic transmission. Downstream from glutamate receptor, CREB expression and phosphorylation can be severely affected by alcohol (Pandey and others 2001; Yang and others 1988, 1998), which would disrupt the expression of genes that are important for neuronal plasticity. In fact, rodents exposed to alcohol during the last week of gestation show less expression of the CREB/SRF-related genes cFOS and JUNb when compared to controls (Nagahara and Handa 1995). Consistent with these findings, Clements and colleagues (2005) showed that animals exposed to alcohol during the third-trimester equivalent of human gestation fail to display an increase in cFOS expression in the hippocampus after Morris water maze testing. Similar findings were recently observed in frontal cortex. Hamilton and colleagues (2010a, 2010b) observed that although the expression of cFOS and ARC after social experience (playing behavior) was increased in the frontal cortex of animals treated with saccharin, the expression of these immediate early genes remained unaltered in animals that were exposed to alcohol throughout gestation.
In summary, alcohol can produce a long-lasting alteration in cascades that are essential to neuronal plasticity, which could lead to a constellation of problems in brain function. Accordingly, targeting elements of these cascades may be important for restoration of neuronal plasticity and improvement of brain function. Here we focus on drugs that can improve plasticity by acting through two different mechanisms: (1) modulating glutamate receptors and (2) increasing cAMP/gGMP levels by phosphodiesterase inhibition.
Modulation of Glutamate Receptors
It is well established that glutamate receptors are crucial for the refinements of neuronal connections that take place during brain development and for learning and memory as well (Malenka and Bear 2004; Malinow and Malenka 2002; Platenik and others 2000; White and Fitzpatrick 2007). Early alcohol exposure can lead to a long-lasting alteration in NMDAr (Hughes and others 1998; Rema and Ebner 1999; Savage and others 1992; Toso and others 2005) and AMPAr (Bellinger and others 2002; Mameli and others 2005), which could explain some of the plasticity deficits seen in FASD models. The modulation of the AMPA receptor has been extensively used to facilitate glutamatergic transmission and improve plasticity (Malinow and Malenka 2002; Staubli and others 1994) and recently is being considered as a treatment for depression and other mood disorders (Alt and others 2006; Mattew and others 2008). One the advantages of targeting this receptor is that modulation of AMPAr can facilitate glutamatergic transmission without inducing seizures and excitotoxic damage, which are commonly seen when NMDA agonists are used (Lynch 2006). There are two strategies that are commonly used to facilitate glutamatergic transmission by acting on the AMPAr: (1) increasing the numbers of AMPArs on the postsynaptic membrane and (2) potentiating AMPAr function.
Incorporation of New AMPAr into the Postsynaptic Membrane
AMPArs are not restricted to the synapse and can be found also on both surfaces and intracellular regions of dendrites. These localizations are not static, and AMPArs can actively move in and out the synaptic membrane (Malinow and Malenka 2002). This traffic strongly affects neuronal plasticity (Essmann and others 2008; Hu and others 2007; Malinow and Malenka 2002; Serulle and others 2007). For instance, during LTP and LTD, AMPArs are incorporated and internalized, respectively (Malinow and Malenka 2002). Accordingly, drugs that stimulate incorporation of AMPA receptors could be potential plasticity enhancers. For instance, norepinephrine can induce delivery of AMPAr to the synapse and facilitate LTP (Hu and others 2007). Despite its potential, the facilitation of the incorporation of AMPArs on the membrane has been poorly explored as a therapeutic option.
Potentiation of AMPAr
Instead of affecting trafficking to increase AMPAr number on the synapse, most of the glutamatergic drugs tested as plasticity enhancers are positive allosteric modulators of this receptor (Lynch 2002, 2006). In general, these drugs can slow the channel closure after removal of the agonist (deactivation), which facilitates glutamatergic transmission and increases LTP (Lynch 2002; Rongsheng and others 2005). Positive allosteric modulators of the AMPA receptors have been tested in several conditions. The use of piracetam in Down syndrome has been extremely controversial (Croom 2001; Holmes 1999; Lobaugh and others 2001; Moran and others 2002). In contrast, aniceratam, a newer and more potent AMPAr modulator, has shown promising results in Alzheimer disease (Senin and others 1991; Tsolaki and others 2001) and recovery after traumatic brain injury (Baranova and others 2006). The efficacy of positive allosteric modulators in restoring neuronal plasticity in models of FASD has recently been tested. Piracetam, meclophenoxate, and aniracetam administered after the period of alcohol exposure significantly ameliorated learning deficits in rats that were exposed to alcohol throughout the pregnancy (Vaglenova and others 2008; Vaglenova and Petkov 2001). For instance, alcohol-exposed rats that were treated with 50 mg/kg of aniceratam for 10 days (P18–27) showed an increased number of avoidances when compared to controls in the classical active-avoidance test. Importantly, the behavioral effects of aniracetam are well correlated with a restoration of the electrophysiology properties of the AMPA receptor (Vaglenova and others 2008; Wijayawardhane and others 2007, 2008).
Inhibition of Phosphodiesterases
Phosphodiesterases are enzymes that catalyze the hydrolysis of the 3’ cyclic phosphate bonds of adenosine and/or guanosine 3’, 5’ cyclic monophosphate (Beavo 1995). The rationale of using of phosphodiesterase inhibitors is to increase cAMP/cGMP levels, which would lead to phosphorylation of CREB and other transcription factors such as SRF and ELK-1 (Beavo 1995; Blokland and others 2006; Chai and Tarnawski 2002). In addition, a rise in cAMP/cGMP levels can also improve AMPA function. Recent findings show that both PKA and cGKII (which are activated by cAMP and cGMP, respectively) can phosphorylate AMPArs, promoting its incorporation into the synapse (Serulle and others 2007). The use of phosphodiesterases (PDEs) as a therapeutic target has been the subject of many recent reviews (Blokland and others 2006; Ghavami and others 2006; Lynch 2002; Rose and others 2005; Tully and others 2003). Here we discuss the potential of two types of PDEs for improving plasticity in FASD models.
The PDE1 family is activated by Ca++/calmodulin and exists as several isoforms. Among these isoforms, PDE1A and PDE1B account for more than 90% of total brain PDE1 activity. Although PDE1A is highly expressed in the cerebral cortex and hippocampus, PDE1B is expressed mainly in dopaminergic regions such as the striatum and nucleus accumbens (Kakkar and others 1999). Inhibition of PDE1 leads to an increase in levels of both cAMP and cGMP (Kakkar and others 1999). The alkaloid vinpocetine (vinpocetine-ethyl apovincaminate) is a nonspecific inhibitor of PDE1 activity (Nicholson 1990). Vinpocetine treatment has been shown to facilitate LTP (Molnar and others 1994; Molnar and Gaal 1992), enhance the structural dynamics of dendritical spines (Lendvai and others 2003), improve memory retrieval (DeNoble 1987), and enhance performance on cognitive tests in humans (Hindmarch and others 1991).
The potential of vinpocetine in improving plasticity in FASD was recently tested in the visual cortex of the ferret (Medina and others 2006). In higher mammals (such as ferrets, cats, and primates), the visual cortex presents alternating columns of neurons that are wired preferentially to the right or the left eye. If a unilateral eye lid suture (monocular deprivation) is done during an early period of development, the ocular dominance columns related to the deprived eye shrink, and the columns related to the experienced eye expand (Hubel and Wiesel 1962, 1963; Wiesel and Hubel 1965). This type of plasticity, known as ocular dominance plasticity, is permanently impaired in ferrets that are exposed to alcohol during the third-trimester equivalent of human gestation (Medina and others 2003; Medina and Ramoa 2005). Remarkably, treating alcohol-exposed ferrets with vinpocetine (20 mg/kg, intraperitoneally [IP]) during the monocular deprivation period resulted in a strong ocular dominance shift, which was not seen in animals treated with vehicle (Medina and others 2006). Importantly, the effects of vinpocetine were seen several weeks after the alcohol insult, in a period equivalent to infancy in humans. It would be important to test if inhibition of PDE1 by vinpocetine could restore other plasticity problems such as deficits on learning and memory. In fact, recent findings showed that vinpocetine improves Morris maze performance in rats exposed to alcohol during the third-trimester equivalent of human gestation (Filgueiras and others 2010). Vinpocetine was given during the behavioral test to facilitate the CREB phosphorylation that is often associated with learning (Finkbeiner and others 1997; Lamprecht 2005; Silva and others 1998).
Vinpocetine was also shown to restore the effects of early alcohol exposure on the functional organization of the visual cortex. Neurons in the visual cortex have a preference for stimuli that move in a particular orientation (Hubel and Wiesel 1959, 1962; White and Fitzpatrick 2007). For example, a moving horizontal bar stimulates different groups of neurons than a vertical one. This orientation selectivity is seen in rodents, carnivores, and primates. However, the latter two groups have an additional type of organization: in higher mammals, neurons that share similar orientation tuning cluster together to form orientation selectivity columns (Miller 1994; White and Fitzpatrick 2007). The neuronal orientation tuning and the orientation selectivity columns are considered important components of the visual perception of corners and borders (Livingstone and Hubel 1998; Pasupathy 2006; White and Fitzpatrick 2007), which seems to be affected by early alcohol exposure (Mattson and others 1996; Uecker and Nadel 1996). The impact of third-trimester alcohol exposure on orientation selectivity was investigated in the ferret (Medina and others 2005). It was demonstrated that alcohol exposure during this period dramatically affects orientation tuning of individual neurons as well as the organization of the orientation selectivity columns. These findings may be explained by the deleterious effects of alcohol on the activity-dependent plasticity processes required for the establishment of orientation selectivity (Medina and Krahe 2008; Medina and others 2005). In fact, when alcohol-treated animals are treated with vinpocetine (to enhance neuronal plasticity), orientation tuning at the neuronal level and the organization of orientation selectivity columns are restored (Krahe and others 2009).
In addition to vinpocetine, caffeine is also a PDE1 inhibitor (Kakkar and others 1999) that could have a role in FASD treatment. A recent clinical study showed that perinatal caffeine, which was used to improve the respiratory function, was able to improve survival rate and ameliorate cognitive deficits resulting from prematurity (Schmidt and others 2007; Stevenson 2007). Remarkably, the effects of caffeine on the respiratory function were able to explain only half of the effect of caffeine treatment (Schmidt and others 2007; Stevenson 2007). Because caffeine is also a classical PDE1 inhibitor, it would be interesting to investigate whether the beneficial effects of caffeine could also be related to modulation of cAMP/cGMP levels and activation of CREB.
The PDE4 isoenzymes are cAMP specific and highly expressed in the cerebral cortex and hippocampus (Ghavami and others 2006; Rose and others 2005). The importance of PDE4 for learning and memory has been demonstrated in genetic and pharmacologic studies. Mice lacking the subunit PDE4d show better performance in the Morris water maze and in the radial arm maze (Rose and others 2005). PDE4 inhibition by rolipram ((±)-4-(3-cyclopentyloxy-4-methoxyphenyl)-2-pyrrolidone) has been shown to lower the threshold for inducing hippocampal LTP (Otmakhov and others 2004), restore its duration in aged mice (Barad and others 1998), and restore LTP in a transgenic model of Alzheimer disease (Gong and others 2004). Recently, rolipram was shown to be able to improve object recognition in aged rats (de Lima and others 2008). Despite its potential as a plasticity enhancer, rolipram failed to restore ocular dominance plasticity in the ferret model of FASD (Krahe and others 2010). Although the clinical application of rolipram is complicated by side effects (i.e., emesis), there is an ongoing effort to develop safer PDE4 inhibitors (Dyke and Montana 1999).
Choline Supplementation
Early deprivation of some essential nutrients such as choline can result in long-lasting cognitive problems (Zeisel 2006). Conversely, pre- or perinatal choline supplementation can improve spatial learning and facilitate LTP in animal models (Li and others 2004; Tees and Mohammadi 1999). This effect may be explained by an enhancement of NMDA receptor-mediated population excitatory postsynaptic potentials (pEPSPs) in the hippocampus, which would lower the threshold for LTP (Montoya and Swartzwelder 2000). Moreover, it has been recently demonstrated that choline supplementation is correlated with an increase in ERK (MAPK) and CREB phosphorylation (Mellott and others 2004). However, the specific molecular mechanisms for the effects of choline supplementation in improvement of neuronal plasticity remain poorly understood.
The use of choline supplementation has been very successful in improving plasticity in FASD animal models. In addition to acting as a plasticity enhancer, another rationale for the use of choline supplementation is to counteract the deleterious effects of alcohol on the cholinergic system (Kelly and others 1989; Schambra and others 1990). In a series of studies, Thomas and colleagues (2004, 2007) showed that neonatal or postnatal choline supplementation restores water maze performance and reduces hyperactivity of rats exposed to alcohol during gestation (equivalent to first and second trimesters of human gestation) or during the first weeks of life (equivalent to the third trimester). In addition to its positive effects on learning, it was recently demonstrated that choline supplementation can also mitigate the abnormalities in the development of reflexes and motor coordination seen after early alcohol exposure (Thomas and others 2009).
Further Directions
In addition of drugs that act on the cascade cited in this review (Fig. 3), pharmacological agents that act by different mechanisms may also have potential as plasticity enhancers. In fact, recently, Daniel Savage group showed that a histamine H3 receptor antagonist (ABT-239) was able to improve LTP and learning in a model of FASD (Varaschin et al., 2010; Savage et al., 2010). Although plasticity enhancement by different types of drugs has great potential in restoring brain function in FASD, some challenges should be considered. First, drugs that have poor selectivity may have more the potential to exhibit side effects. Therefore, the dissection of the mechanisms of how a particular drug affects plasticity has an enormous importance. Second, neuronal plasticity is a very complex process that involves making and breaking connections, which in turn are related to LTP and LTD type of phenomena. Because LTP and LTD rely on mechanisms that could be opposites (i.e., increase or decrease of AMPAr in the synaptic membrane; Malinow and Malenka 2002), improvement of the former may affect the latter and vice versa. To overcome these challenges, it is necessary to have a better understanding of the molecular mechanisms of FASD and the development of more selective drugs. This effort, which is currently being carried out by the scientific community, could lead to successful neuronal plasticity restoration in FASD and a significant improvement of brain function in this condition.
Acknowledgments
Financial Disclosure/Funding
The author(s) disclosed receipt of the following financial support for the research and/or authorship of this article: this work was supported by NIH (NIAAA) grant AA-13023 to A.E.M.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interests with respect to the authorship and/or publication of this article.
References
- Abel EL. An update on incidence of FAS: FAS is not an equal opportunity birth defect. Neurotoxicol Teratol. 1995;17:437–43. doi: 10.1016/0892-0362(95)00005-c. [DOI] [PubMed] [Google Scholar]
- Alt A, Nisenbaum ES, Bleakman D, Witkin JM. A role for AMPA receptors in mood disorders. Biochem Pharmacol. 2006;71:1273–88. doi: 10.1016/j.bcp.2005.12.022. [DOI] [PubMed] [Google Scholar]
- Asanuma M, Nishibayashi S, Iwata E, Kondo Y, Nakanishi T, Vargas MG, et al. Alterations of cAMP response element-binding activity in the aged rat brain in response to administration of rolipram, a cAMP-specific phosphodiesterase inhibitor. Brain Res Mol Brain Res. 1996;41:210–5. doi: 10.1016/0169-328x(96)00098-8. [DOI] [PubMed] [Google Scholar]
- Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–9. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- Barad M, Bourtchouladze R, Winder DG, Golan H, Kandel E. Rolipram, a type IV-specific phosphodiesterase inhibitor, facilitates the establishment of long-lasting long-term potentiation and improves memory. Proc Natl Acad Sci U S A. 1998;95:15020–5. doi: 10.1073/pnas.95.25.15020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranova AI, Whiting MD, Hamm RJ. Delayed, post-injury treatment with aniracetam improves cognitive performance after traumatic brain injury in rats. J Neurotrauma. 2006;23:1233–40. doi: 10.1089/neu.2006.23.1233. [DOI] [PubMed] [Google Scholar]
- Barria A, Muller D, Derkach V, Griffith LC, Soderling TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 1997;276:2042–5. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- Beavo JA. Cyclic nucleotide phosphodiesterases: functional implications of multiple isoforms. Physiol Rev. 1995;75:725–48. doi: 10.1152/physrev.1995.75.4.725. [DOI] [PubMed] [Google Scholar]
- Bellinger FP, Davidson MS, Bedi KS, Wilce PA. Neonatal ethanol exposure reduces AMPA but not NMDA receptor levels in the rat neocortex. Brain Res Dev Brain Res. 2002;136:77–84. doi: 10.1016/s0165-3806(02)00363-2. [DOI] [PubMed] [Google Scholar]
- Berardi N, Pizzorusso T, Ratto GM, Maffei L. Molecular basis of plasticity in the visual cortex. Trends Neurosci. 2003;26:369–78. doi: 10.1016/S0166-2236(03)00168-1. [DOI] [PubMed] [Google Scholar]
- Bito H, Deisseroth K, Tsien RW. CREB phosphorylation and dephosphorylation: a Ca++ and stimulus duration dependent switch for hippocampal gene expression. Cell. 1996;87:1203–14. doi: 10.1016/s0092-8674(00)81816-4. [DOI] [PubMed] [Google Scholar]
- Black MD. Therapeutic potential of positive AMPA modulators and their relationship to AMPA receptor subunits: a review of preclinical data. Psychopharmacology (Berl) 2005;179:154–63. doi: 10.1007/s00213-004-2065-6. [DOI] [PubMed] [Google Scholar]
- Blokland A, Schreiber R, Prickaerts J. Improving memory: a role for phosphodiesterases. Curr Pharm Des. 2006;12:2511–23. doi: 10.2174/138161206777698855. [DOI] [PubMed] [Google Scholar]
- Bourne HR, Nicoll R. Molecular machines integrate coincident synaptic signals. Cell. 1993;72:65–75. doi: 10.1016/s0092-8674(05)80029-7. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention. Frequent alcohol consumption among women of childbearing age: behavioral risk factor surveillance system. JAMA. 1994;271:1820–1. [PubMed] [Google Scholar]
- Chai J, Tarnawski AS. Serum response factor: discovery, biochemistry, biological roles and implications for tissue injury healing. J Physiol Pharmacol. 2002;53:147–57. [PubMed] [Google Scholar]
- Chappell TD, Margret CP, Waters RS. Long-term effects of prenatal alcohol exposure on the size of the whisker representation in juvenile and adult rat barrel cortex. Alcohol. 2007;41:239–51. doi: 10.1016/j.alcohol.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements KM, Girard TA, Ellard CG, Wainwright PE. Short-term memory impairment and reduced hippocampal c-Fos expression in an animal model of fetal alcohol syndrome. Alcohol Clin Exp Res. 2005;29:1049–59. doi: 10.1097/01.alc.0000171040.82077.e. [DOI] [PubMed] [Google Scholar]
- Contestabile A. Regulation of transcription factors by nitric oxide in neurons and in neural-derived tumor cells. Prog Neurobiol. 2008;84:317–28. doi: 10.1016/j.pneurobio.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Cook CS, Nowotny AZ, Sulik KK. Fetal alcohol syndrome: eye malformations in a mouse model. Arch Ophthalmol. 1987;105:1576–81. doi: 10.1001/archopht.1987.01060110122045. [DOI] [PubMed] [Google Scholar]
- Costa ET, Savage DD, Valenzuela CF. A review of the effects of prenatal or early postnatal ethanol exposure on brain ligand-gated ion channels. Alcohol Clin Exp Res. 2000;24:706–15. [PubMed] [Google Scholar]
- Croom J. Piracetam study: poorly designed and misinterpreted. Arch Pediatr Adolesc Med. 2001;155:1176–8. [PubMed] [Google Scholar]
- Curtis J, Finkbeiner S. Sending signals from the synapse to the nucleus: possible roles for CaMK, Ras/ERK, and SAPK pathways in the regulation of synaptic plasticity and neuronal growth. J Neurosci Res. 1999;58:88–95. [PubMed] [Google Scholar]
- de Lima MN, Presti-Torres J, Garcia VA, Guimaraes MR, Scalco FS, Roesler R, et al. Amelioration of recognition memory impairment associated with iron loading or aging by the type 4–specific phosphodiesterase inhibitor rolipram in rats. Neuropharmacology. 2008;55:788–92. doi: 10.1016/j.neuropharm.2008.06.025. [DOI] [PubMed] [Google Scholar]
- DeNoble VJ. Vinpocetine enhances retrieval of a step-through passive avoidance response in rats. Pharmacol Biochem Behav. 1987;26:183–6. doi: 10.1016/0091-3057(87)90552-1. [DOI] [PubMed] [Google Scholar]
- Dong J, Sulik KK, Chen SY. Nrf2-mediated transcriptional induction of antioxidant response in mouse embryos exposed to ethanol in vivo: implications for the prevention of fetal alcohol spectrum disorders. Antioxid Redox Signal. 2008;10:2023–33. doi: 10.1089/ars.2007.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyke HJ, Montana JG. The therapeutic potential of PDE4 inhibitors. Expert Opin Investig Drugs. 1999;8:1301–25. doi: 10.1517/13543784.8.9.1301. [DOI] [PubMed] [Google Scholar]
- Essmann CL, Martinez E, Geiger JC, Zimmer M, Traut MH, Stein V, et al. Serine phosphorylation of eph-rinB2 regulates trafficking of synaptic AMPA receptors. Nat Neurosci. 2008;11:1035–43. doi: 10.1038/nn.2171. [DOI] [PubMed] [Google Scholar]
- Etkin A, Alarcon JM, Weisberg SP, Touzani K, Huang YY, Nordheim A, et al. A role in learning for SRF: deletion in the adult forebrain disrupts LTD and the formation of an immediate memory of a novel context. Neuron. 2006;50:127–43. doi: 10.1016/j.neuron.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Fagiolini M, Fritschy JM, Low K, Mohler H, Rudolph U, Hensch TK. Specific GABAA circuits for visual cortical plasticity. Science. 2004;303:1681–3. doi: 10.1126/science.1091032. [DOI] [PubMed] [Google Scholar]
- Filgueiras CC, Krahe TE, Medina AE. Phosphodiesterase type 1 inhibition improves learning in rats exposed to alcohol during the third trimester equivalent of human gestation. Neurosci Lett. 2010;473:202–7. doi: 10.1016/j.neulet.2010.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME. CREB: a major mediator of neuronal neurotrophin responses. Neuron. 1997;19:1031–47. doi: 10.1016/s0896-6273(00)80395-5. [DOI] [PubMed] [Google Scholar]
- Floyd R, O’Connor M, Sokol RJ, Bertrand J, Cordero J. Recognition and prevention of fetal alcohol syndrome. Obstet Gynecol. 2005;106:1059–64. doi: 10.1097/01.AOG.0000181822.91205.6f. [DOI] [PubMed] [Google Scholar]
- Frank DA, Greenberg ME. CREB: a mediator of long-term memory from mollusks to mammals. Cell. 1994;79:5–8. doi: 10.1016/0092-8674(94)90394-8. [DOI] [PubMed] [Google Scholar]
- Ghavami A, Hirst WD, Novak TJ. Selective phosphodiesterase (PDE)–4 inhibitors: a novel approach to treating memory deficit? Drugs R D. 2006;7:63–71. doi: 10.2165/00126839-200607020-00001. [DOI] [PubMed] [Google Scholar]
- Girard TA, Xing HC, Ward GR, Wainwright PE. Early postnatal ethanol exposure has long-term effects on the performance of male rats in a delayed matching-to-place task in the Morris water maze. Alcohol Clin Exp Res. 2000;24:300–6. [PubMed] [Google Scholar]
- Gong B, Vitolo OV, Trinchese F, Liu S, Shelanski M, Arancio O. Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J Clin Invest. 2004;114:1624–34. doi: 10.1172/JCI22831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodlett CR, Johnson TB. Neonatal binge ethanol exposure using intubation: timing and dose effects on place learning. Neurotoxicol Teratol. 1997;19:435–46. doi: 10.1016/s0892-0362(97)00062-7. [DOI] [PubMed] [Google Scholar]
- Gottesfeld Z, Garcia C, Lingham R, Chronister R. Prenatal ethanol exposure impairs lesion-induced plasticity in a dopaminergic synapse after maturity. Neuroscience. 1989;29:715–23. doi: 10.1016/0306-4522(89)90143-7. [DOI] [PubMed] [Google Scholar]
- Guerri C. Neuroanatomical and neurophysiological mechanisms involved in central nervous system dysfunctions induced by prenatal alcohol exposure. Alcohol Clin Exp Res. 1998;22:304–12. doi: 10.1111/j.1530-0277.1998.tb03653.x. [DOI] [PubMed] [Google Scholar]
- Hamilton DA, Akers KG, Rice JP, Johnson TE, Candelaria-Cook FT, Maes LI, et al. Prenatal exposure to moderate levels of ethanol alters social behavior in adult rats: relationship to structural plasticity and immediate early gene expression in frontal cortex. Behav Brain Res. 2010a;207:290–304. doi: 10.1016/j.bbr.2009.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton DA, Candelaria-Cook FT, Akers KG, Rice JP, Maes LI, Rosenberg M, et al. Patterns of social-experience-related c-fos and Arc expression in the frontal cortices of rats exposed to saccharin or moderate levels of ethanol during prenatal brain development. Behav Brain Res. 2010b;214:66–74. doi: 10.1016/j.bbr.2010.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton DA, Kodituwakku P, Sutherland RJ, Savage DD. Children with fetal alcohol syndrome are impaired at place learning but not cued-navigation in a virtual Morris water task. Behav Brain Res. 2003;143:85–94. doi: 10.1016/s0166-4328(03)00028-7. [DOI] [PubMed] [Google Scholar]
- Hebb DO. The organization of behavior. New York: John Wiley; 1949. [Google Scholar]
- Hindmarch I, Fuchs HH, Erzigkeit H. Efficacy and tolerance of vinpocetine in ambulant patients suffering from mild to moderate organic psychosyndromes. Int Clin Psychopharmacol. 1991;6:31–43. doi: 10.1097/00004850-199100610-00005. [DOI] [PubMed] [Google Scholar]
- Holmes LB. Concern about piracetam treatment for children with Down syndrome. Pediatrics. 1999;103:1078–9. doi: 10.1542/peds.103.5.1078-a. [DOI] [PubMed] [Google Scholar]
- Hu H, Real E, Takamiya K, Kang MG, Ledoux J, Huganir RL, et al. Emotion enhances learning via norepinephrine regulation of AMPA-receptor trafficking. Cell. 2007;131:160–73. doi: 10.1016/j.cell.2007.09.017. [DOI] [PubMed] [Google Scholar]
- Hubel DH, Wiesel TN. Receptive fields of single neurones in the cat’s striate cortex. J Physiol. 1959;148:574–91. doi: 10.1113/jphysiol.1959.sp006308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubel DH, Wiesel TN. Receptive fields, binocular interaction and functional architecture in the cat’s visual cortex. J Physiol. 1962;160:106–54. doi: 10.1113/jphysiol.1962.sp006837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubel DH, Wiesel TN. Shape and arrangement of columns in cat’s striate cortex. J Physiol. 1963;165:559–68. doi: 10.1113/jphysiol.1963.sp007079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes PD, Kim YN, Randall PK, Leslie SW. Effect of prenatal ethanol exposure on the developmental profile of the NMDA receptor subunits in rat forebrain and hippocampus. Alcohol Clin Exp Res. 1998;22:1255–61. [PubMed] [Google Scholar]
- Ieraci A, Herrera DG. Nicotinamide protects against ethanol-induced apoptotic neurodegeneration in the developing mouse brain. PLoS Med. 2006;3:0547–57. doi: 10.1371/journal.pmed.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Koch C, Genz K, et al. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;287:1056–60. doi: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Kitabayashi R, Funatsu M, Izumi M, Yuede C, Hartman RE, et al. A single day of ethanol exposure during development has persistent effects on bi-directional plasticity, N-methyl-D-aspartate receptor function and ethanol sensitivity. Neuroscience. 2005;136:269–79. doi: 10.1016/j.neuroscience.2005.07.015. [DOI] [PubMed] [Google Scholar]
- Johnson TB, Stanton ME, Goodlett CR, Cudd TA. Eye-blink classical conditioning in the preweanling lamb. Behav Neurosci. 2008;122:722–9. doi: 10.1037/0735-7044.122.3.722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston MV. Brain plasticity in paediatric neurology. Eur J Paediatr Neurol. 2003;7:105–13. doi: 10.1016/s1090-3798(03)00039-4. [DOI] [PubMed] [Google Scholar]
- Johnston MV. Clinical disorders of brain plasticity. Brain Dev. 2004;26:73–80. doi: 10.1016/S0387-7604(03)00102-5. [DOI] [PubMed] [Google Scholar]
- Jones KL, Smith DW. Recognition of the fetal alcohol syndrome in early infancy. Lancet. 1973;2:999–1001. doi: 10.1016/s0140-6736(73)91092-1. [DOI] [PubMed] [Google Scholar]
- Josselyn SA, Nguyen PV. CREB, synapses and memory disorders: past progress and future challenges. Curr Dug Targets CNS Neurol Disord. 2005;4:481–97. doi: 10.2174/156800705774322058. [DOI] [PubMed] [Google Scholar]
- Kakkar R, Raju RVS, Sharma RK. Calmodulin-dependent cyclic nucleotide phosphodiesterase (PDE1) Cell Mol Life Sci. 1999;55:1164–86. doi: 10.1007/s000180050364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–8. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- Keifer J, Zheng ZK, Zhu D. MAPK signaling pathways mediate AMPA receptor trafficking in an in vitro model of classical conditioning. J Neurophysiol. 2007;97:2067–74. doi: 10.1152/jn.01154.2006. [DOI] [PubMed] [Google Scholar]
- Kelley DJ, Davidson RJ, Elliott JL, Lahvis GP, Yin JC, Bhattacharyya A. The cyclic AMP cascade is altered in the fragile X nervous system. PLos ONE. 2007;2:e931. doi: 10.1371/journal.pone.0000931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly SJ, Black AC, Jr, West JR. Changes in muscarinic cholinergic receptors in the hippocampus of rats exposed to ethyl alcohol during the brain growth spurt. J Pharmacol Exp Ther. 1989;249:798–804. [PubMed] [Google Scholar]
- Klug MG, Burde L. Fetal alcohol syndrome prevention: annual and cumulative cost savings. Neurotoxicol Teratol. 2003;25:763–5. doi: 10.1016/j.ntt.2003.07.012. [DOI] [PubMed] [Google Scholar]
- Kornhauser JM, Cowan CW, Shaywitz AJ, Dolmetsch RE, Griffith EC, Hu LS, et al. CREB transcriptional activity in neurons is regulated by multiple, calcium-specific phosphorylation events. Neuron. 2002;34:221–33. doi: 10.1016/s0896-6273(02)00655-4. [DOI] [PubMed] [Google Scholar]
- Krahe TE, Paul AP, Medina AE. Phosphodiesterase type 4 inhibition does not restore ocular dominance plasticity in a ferret model of fetal alcohol spectrum disorders. Alcohol Clin Exp Res. 2010;34:1–6. doi: 10.1111/j.1530-0277.2009.01114.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krahe TE, Wang W, Medina AE. Phosphodiesterase Inhibition increases CREB phosphorylation and restores orientation selectivity in a model of fetal alcohol spectrum disorders. PLoS One. 2009;4:e6643. doi: 10.1371/journal.pone.0006643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamprecht R. CREB: a message to remember. Cell Mol Life Sci. 2005;55:554–63. doi: 10.1007/s000180050314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavaur J, Bernard F, Trifilieff P, Pascoli V, Kappes V, Pages C, et al. A TAT-DEF-Elk-1 peptide regulates the cytonuclear trafficking of Elk-1 and controls cytoskeleton dynamics. J Neurosci. 2008;27:14448–58. doi: 10.1523/JNEUROSCI.2279-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemoine P, Harrouseau H, Borteyru JP, Menuet JC. Les enfants de parents alcoholiques: Anomalies observees a propos de 127 cas. Ouest Med. 1968;8:476–82. [Google Scholar]
- Lendvai B, Zelles T, Rozsa B, Vizi ES. A vinca alkaloid enhances morphological dynamics of dendritic spines of neocortical layer 2/3 pyramidal cells. Brain Res Bull. 2003;59:257–60. doi: 10.1016/s0361-9230(02)00873-0. [DOI] [PubMed] [Google Scholar]
- Li Q, Guo-Ross S, Lewis DV, Turner D, White AM, Wilson WA, et al. Dietary prenatal choline supplementation alters postnatal hippocampal structure and function. J Neurophysiol. 2004;91:1545–55. doi: 10.1152/jn.00785.2003. [DOI] [PubMed] [Google Scholar]
- Livingstone M, Hubel DH. Segregation of form, color, movement, and depth: anatomy, physiology, and perception. Science. 1998;240:740–9. doi: 10.1126/science.3283936. [DOI] [PubMed] [Google Scholar]
- Lobaugh NJ, Karaskov V, Rombough V, Rovet J, Bryson S, Greenbaum R, et al. Piracetam therapy does not enhance cognitive functioning in children with Down syndrome. Arch Pediatr Adolesc Med. 2001;155:442–8. doi: 10.1001/archpedi.155.4.442. [DOI] [PubMed] [Google Scholar]
- Luo J, Miller MW. Growth factor-mediated neural proliferation: target of ethanol toxicity. Brain Res Brain Res Rev. 1998;27:157–67. doi: 10.1016/s0165-0173(98)00009-5. [DOI] [PubMed] [Google Scholar]
- Lynch G. Memory enhancement: the search for mechanism-based drugs. Nat Neurosci. 2002;5:1035–8. doi: 10.1038/nn935. [DOI] [PubMed] [Google Scholar]
- Lynch G. Glutamate-based therapeutic approaches: ampakines. Curr Opin Pharmacol. 2006;6:82–8. doi: 10.1016/j.coph.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–26. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Mameli M, Zamudio PA, Carta M, Valenzuela CF. Developmentally regulated actions of alcohol on hippocampal glutamatergic transmission. J Neurosci. 2005;25:8027–36. doi: 10.1523/JNEUROSCI.2434-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margret CP, Chappell TD, Li CX, Jan TA, Matta SG, Elberger AJ, et al. Prenatal alcohol exposure (PAE) reduces the size of the forepaw representation in forepaw barrel subfield (FBS) cortex in neonatal rats: relationship between periphery and central representation. Exp Brain Res. 2006;172:387–96. doi: 10.1007/s00221-005-0339-9. [DOI] [PubMed] [Google Scholar]
- Marino MD, Aksenov MY, Kelly SJ. Vitamin E protects against alcohol-induced cell loss and oxidative stress in the neonatal rat hippocampus. Int J Dev Neurosci. 2004;22:363–77. doi: 10.1016/j.ijdevneu.2004.04.005. [DOI] [PubMed] [Google Scholar]
- Mattew SJ, Manji HK, Charney DS. Novel drugs and therapeutic targets for severe mood disorders. Neuropsychopharmacology. 2008;33:2080–92. doi: 10.1038/sj.npp.1301652. [DOI] [PubMed] [Google Scholar]
- Mattson SN, Gramling L, Delis DC, Jones KL, Riley EP. Global-local processing in children prenatally exposed to alcohol. Child Neuropsychol. 1996;2:165–75. [Google Scholar]
- Mattson SN, Riley EP. A review of the neurobehavioral deficits in children with fetal alcohol syndrome or prenatal exposure to alcohol. Alcohol Clin Exp Res. 1998;22:279–94. doi: 10.1111/j.1530-0277.1998.tb03651.x. [DOI] [PubMed] [Google Scholar]
- May P, Gossage J, Kalberg W, Robinson L, Buckley D, Manning M, et al. Prevalence and epidemiologic characteristics of FASD from various research methods with an emphasis on recent in-school studies. Dev Disabil Res Rev. 2009;15:176–92. doi: 10.1002/ddrr.68. [DOI] [PubMed] [Google Scholar]
- May PA, Gossage JP. Estimating the prevalence of fetal alcohol syndrome: a summary. Alcohol Res Health. 2001;25:159–67. [PMC free article] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 1984;309:261–3. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- Medina AE, Krahe TE. Neocortical plasticity deficits in fetal alcohol spectrum disorders: lessons from barrel and visual cortex. J Neurosci Res. 2008;86:256–63. doi: 10.1002/jnr.21447. [DOI] [PubMed] [Google Scholar]
- Medina AE, Krahe TE, Coppola DM, Ramoa AS. Neonatal alcohol exposure induces long-lasting impairment of visual cortical plasticity in ferrets. J Neurosci. 2003;23:10002–12. doi: 10.1523/JNEUROSCI.23-31-10002.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina AE, Krahe TE, Ramoa AS. Early alcohol exposure induces persistent alteration of cortical columnar organization and reduced orientation selectivity in the visual cortex. J Neurophysiol. 2005;93:1317–25. doi: 10.1152/jn.00714.2004. [DOI] [PubMed] [Google Scholar]
- Medina AE, Krahe TE, Ramoa AS. Restoration of neuronal plasticity by a phosphodiesterase type 1 inhibitor in a model of fetal alcohol exposure. J Neurosci. 2006;26:1057–60. doi: 10.1523/JNEUROSCI.4177-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina AE, Ramoa AS. Early alcohol exposure impairs ocular dominance plasticity throughout the critical period. Brain Res Dev Brain Res. 2005;157:107–11. doi: 10.1016/j.devbrainres.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Mellott TJ, Williams CL, Meck WH, Blusztajn JK. Prenatal choline supplementation advances hippocampal development and enhances MAPK and CREB activation. FASEB J. 2004;18:545–7. doi: 10.1096/fj.03-0877fje. [DOI] [PubMed] [Google Scholar]
- Miller KD. A model for the development of simple cell receptive fields and the ordered arrangement of orientation columns through activity-dependent competition between ON- and OFF-center inputs. J Neurosci. 1994;14:409–41. doi: 10.1523/JNEUROSCI.14-01-00409.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MW. Effect of early exposure to ethanol on the protein and DNA contents of specific brain regions in the rat. Brain Res. 1996;734:286–94. [PubMed] [Google Scholar]
- Miller MW, Luo J. Effects of ethanol and transforming growth factor beta (TGF beta) on neuronal proliferation and nCAM expression. Alcohol Clin Exp Res. 2002;26:1281–5. doi: 10.1097/01.ALC.0000026836.38681.58. [DOI] [PubMed] [Google Scholar]
- Miller MW, Robertson S. Prenatal exposure to ethanol alters the postnatal development and transformation of radial glia to astrocytes in the cortex. J Comp Neurol. 1993;337:253–66. doi: 10.1002/cne.903370206. [DOI] [PubMed] [Google Scholar]
- Mohler H. Molecular regulation of cognitive functions and developmental plasticity: impact of GABAA receptors. J Neurochem. 2007;102:1–12. doi: 10.1111/j.1471-4159.2007.04454.x. [DOI] [PubMed] [Google Scholar]
- Molnar P, Gaal L. Effect of different subtypes of cognition enhancers on long-term potentiation in the rat dentate gyrus in vivo. Eur J Pharmacol. 1992;215:17–22. doi: 10.1016/0014-2999(92)90602-z. [DOI] [PubMed] [Google Scholar]
- Molnar P, Gaal L, Horvath C. The impairment of long-term potentiation in rats with medial septal lesion and its restoration by cognition enhancers. Neurobiology. 1994;2:255–66. [PubMed] [Google Scholar]
- Montoya D, Swartzwelder HS. Prenatal choline supplementation alters hippocampal N-methyl-D-aspartate receptor-mediated neurotransmission in adult rats. Neurosci Lett. 2000;296:85–8. doi: 10.1016/s0304-3940(00)01660-8. [DOI] [PubMed] [Google Scholar]
- Moran TH, Capone GT, Knipp S, Davisson MT, Reeves RH, Gearhart JD. The effects of piracetam on cognitive performance in a mouse model of Down’s syndrome. Physiol Behav. 2002;77:403–9. doi: 10.1016/s0031-9384(02)00873-9. [DOI] [PubMed] [Google Scholar]
- Murawski N, Stanton ME. Variants of contextual fear conditioning are differentially impaired in the juvenile rat by binge ethanol exposure on postnatal days 4–9. Behav Brain Res. 2010;212:133–42. doi: 10.1016/j.bbr.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahara AH, Handa RJ. Fetal alcohol exposure alters the induction of immediate early gene mRNA in the rat pre-frontal cortex after an alternation task. Alcohol Clin Exp Res. 1995;19:1389–97. doi: 10.1111/j.1530-0277.1995.tb00997.x. [DOI] [PubMed] [Google Scholar]
- Nicholson CD. Pharmacology of nootropics and metabolically active compounds in relation to their use in dementia. Psychopharmacology (Berl) 1990;101:147–59. doi: 10.1007/BF02244119. [DOI] [PubMed] [Google Scholar]
- Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurons. Nature. 1984;307:462–5. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- O’Connor M, Paley B. Psychiatric conditions associated with prenatal alcohol exposure. Dev Disabil Res Rev. 2009;15:225–34. doi: 10.1002/ddrr.74. [DOI] [PubMed] [Google Scholar]
- Otmakhov N, Khibnick L, Otmakhova N, Carpenter S, Riahi S, Asrican B, et al. Forskolin-induced LTP in the CA1 hippocampal region is NMDA receptor dependent. J Neurophysiol. 2004;91:1955–62. doi: 10.1152/jn.00941.2003. [DOI] [PubMed] [Google Scholar]
- Pandey SC, Roy A, Mittal N. Effects of chronic ethanol intake and its withdrawal on the expression and phosphorylation of the CREB gene transcription factor in rat cortex. J Pharmacol Exp Ther. 2001;296:857–68. [PubMed] [Google Scholar]
- Pasupathy A. Neural basis of shape representation in the primate brain. Prog Brain Res. 2006;154:293–313. doi: 10.1016/S0079-6123(06)54016-6. [DOI] [PubMed] [Google Scholar]
- Pilz RB, Broderick KE. Role of cyclic GMP in gene regulation. Front Biosci. 2005;10:1239–68. doi: 10.2741/1616. [DOI] [PubMed] [Google Scholar]
- Platenik J, Kuramoto N, Yoneda Y. Molecular mechanisms associated with long-term consolidation of the NMDA signals. Life Sci. 2000;67:335–64. doi: 10.1016/s0024-3205(00)00632-9. [DOI] [PubMed] [Google Scholar]
- Powrozec TA, Zhou FC. Effects of prenatal alcohol exposure on the development of the vibrissal somatosensory cortical barrel network. Brain Res Dev Brain Res. 2005;155:135–46. doi: 10.1016/j.devbrainres.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Puglia MP, Valenzuela CF. Repeated third trimester-equivalent ethanol exposure inhibits long-term potentiation in the hippocampal CA1 region of neonatal rats. Alcohol. 2010;44:283–90. doi: 10.1016/j.alcohol.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rema V, Ebner FF. Effect of enriched environment rearing on impairments in cortical excitability and plasticity after prenatal alcohol exposure. J Neurosci. 1999;19:10993–11006. doi: 10.1523/JNEUROSCI.19-24-10993.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson DP, Byrnes ML, Brien JF, Reynolds JN, Dringenberg HC. Impaired acquisition in the water maze and hippocampal long-term potentiation after chronic prenatal ethanol exposure in the guinea-pig. Eur J Neurosci. 2002;16:1593–8. doi: 10.1046/j.1460-9568.2002.02214.x. [DOI] [PubMed] [Google Scholar]
- Rongsheng J, Clark S, Weeks AM, Dudman JT, Gouaux E, Partin KM. Mechanism of positive allosteric modulators acting on AMPA receptors. J Neurosci. 2005;25:9027–36. doi: 10.1523/JNEUROSCI.2567-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose GM, Hopper A, De Vivo M, Tehim A. Phosphodiesterase inhibitors for cognitive enhancement. Curr Pharm Des. 2005;11:3329–34. doi: 10.2174/138161205774370799. [DOI] [PubMed] [Google Scholar]
- Saito M, Mao RF, Wang R, Vadasz C, Saito M. Effects of gangliosides on ethanol-induced neurodegeneration in the developing mouse brain. Alcohol Clin Exp Res. 2007;31:665–74. doi: 10.1111/j.1530-0277.2007.00351.x. [DOI] [PubMed] [Google Scholar]
- Savage DD, Queen SA, Sanchez CF, Paxton LL, Mahoney JC, Goodlett CR, et al. Prenatal ethanol exposure during the last third of gestation in rat reduces hippocampal NMDA agonist binding site density in 45-day-old offspring. Alcohol. 1992;9:37–41. doi: 10.1016/0741-8329(92)90007-w. [DOI] [PubMed] [Google Scholar]
- Savage DD, Rosenberg M, Wolff C, Akers K, El-Emawy A, Staples M, et al. Effects of a novel cognition-enhancing agent on fetal ethanol induced learning deficits. Alcohol Clin Exp Res. 2010;34:1793–802. doi: 10.1111/j.1530-0277.2010.01266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schambra UB, Lauder JM, Petrusz P, Sulik KK. Development of neurotransmitter systems in the mouse embryo following acute ethanol exposure: a histological and immunocytochemical study. Int J Dev Neurosci. 1990;8:507–22. doi: 10.1016/0736-5748(90)90043-2. [DOI] [PubMed] [Google Scholar]
- Schmidt B, Roberts RS, Davis P, Doyle LW, Barrington KJ, Ohlsson A, et al. Long-term effects of caffeine therapy for apnea of prematurity. N Engl J Med. 2007;357:1893–902. doi: 10.1056/NEJMoa073679. [DOI] [PubMed] [Google Scholar]
- Senin U, Abate G, Fieschi C, Gori G, Guala A, Marini G, et al. Aniceratam (Ro 13-5057) in the treatment of senile dementia of Alzheimer type (SDAT): results of a placebo controlled multicentre clinical study. Eur Neuropsychopharmacol. 1991;1:511–7. doi: 10.1016/0924-977x(91)90004-e. [DOI] [PubMed] [Google Scholar]
- Serulle Y, Zhang S, Ninan I, Puzzo D, McCarthy M, Khatri L, et al. A GluR1-cGKII interaction regulates AMPA receptor trafficking. Neuron. 2007;56:670–88. doi: 10.1016/j.neuron.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servais L, Hourez R, Bearzatto B, Gall D, Schiffmann SN, Cheron G. Purkinje cell dysfunction and alteration of long-term synaptic plasticity in fetal alcohol syndrome. Proc Natl Acad Sci U S A. 2007;104:9858–63. doi: 10.1073/pnas.0607037104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegenthaler JA, Miller MW. Transforming growth factor beta1 modulates cell migration in rat cortex: effects of ethanol. Cerebral Cortex. 2004;14:791–802. doi: 10.1093/cercor/bhh039. [DOI] [PubMed] [Google Scholar]
- Siegenthaler JA, Miller MW. Ethanol disrupts cell cycle regulation in developing rat cortex interaction with transforming growth factor beta1. J Neurochem. 2005;95:902–12. doi: 10.1111/j.1471-4159.2005.03461.x. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Kogan JH, Frankland PW, Kida S. CREB and memory. Annu Rev Neurosci. 1998;21:127–48. doi: 10.1146/annurev.neuro.21.1.127. [DOI] [PubMed] [Google Scholar]
- Stanton ME, Goodlett CR. Neonatal ethanol exposure impairs eyeblink conditioning in weanling rats. Alcohol Clin Exp Res. 1998;22:270–5. [PubMed] [Google Scholar]
- Staubli U, Rogers G, Lynch G. Facilitation of glutamate receptors enhances memory. Proc Natl Acad Sci U S A. 1994;91:777–81. doi: 10.1073/pnas.91.2.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stent GS. A physiological mechanism for Hebb’s postulate of learning. Proc Natl Acad Sci U S A. 1973;70:997–1001. doi: 10.1073/pnas.70.4.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson DK. On the caffeination of prematurity. N Engl J Med. 2007;357:1967–8. doi: 10.1056/NEJMe078200. [DOI] [PubMed] [Google Scholar]
- Sulik KK, Johnston MC. Sequence of developmental alterations following acute ethanol exposure in mice: craniofacial features of the fetal alcohol syndrome. Am J Anat. 1983;166:257–69. doi: 10.1002/aja.1001660303. [DOI] [PubMed] [Google Scholar]
- Sulik KK, Johnston MC, Webb MA. Fetal alcohol syndrome: embryogenesis in a mouse model. Science. 1981;214:936–8. doi: 10.1126/science.6795717. [DOI] [PubMed] [Google Scholar]
- Sutherland RJ, McDonald RJ, Savage DD. Prenatal exposure to moderate levels of ethanol can have long-lasting effects on hippocampal synaptic plasticity in adult offspring. Hippocampus. 1997;7:232–8. doi: 10.1002/(SICI)1098-1063(1997)7:2<232::AID-HIPO9>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Zhou H, Neumaier JF, Pham TA. Opposing functions of CREB and MKK1 synergistically regulate the geometry of dendritic spines in visual cortex. J Comp Neurol. 2007;503:605–17. doi: 10.1002/cne.21424. [DOI] [PubMed] [Google Scholar]
- Tees R, Mohammadi E. The effects of neonatal choline dietary supplementation on adult spatial and configural learning and memory in rats. Dev Psychobiol. 1999;35:226–40. [PubMed] [Google Scholar]
- Thomas JD, Abou E, Dominguez H. Prenatal choline supplementation mitigates the adverse effects of prenatal alcohol exposure on development in rats. Neurotoxicol Teratol. 2009;31:311. doi: 10.1016/j.ntt.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JD, Biane JS, O’Bryan KA, O’Neill TM, Dominguez HD. Choline supplementation following third-trimester-equivalent alcohol exposure attenuates behavioral alterations in rats. Behav Neurosci. 2007;121:120–30. doi: 10.1037/0735-7044.121.1.120. [DOI] [PubMed] [Google Scholar]
- Thomas JD, Garrison M, O’Neill TM. Perinatal choline supplementation attenuates behavioral alterations associated with neonatal alcohol exposure in rats. Neurotoxicol Teratol. 2004;26:35–45. doi: 10.1016/j.ntt.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Toso L, Poqqi SH, Abebe D, Roberson R, Dunlap V, Park J, et al. N-methyl-D-aspartate subunit expression during mouse development altered by in utero alcohol exposure. Am J Obstet Gynecol. 2005;193:1534–9. doi: 10.1016/j.ajog.2005.02.105. [DOI] [PubMed] [Google Scholar]
- Tsolaki M, Pantazi T, Kazis A. Efficacy of acetylcholinesterase inhibitors versus nootropics in Alzheimer’s disease: a retrospective, longitudinal study. J Int Med Res. 2001;29:28–36. doi: 10.1177/147323000102900105. [DOI] [PubMed] [Google Scholar]
- Tully T, Bourtchouladze R, Scott R, Tallman J. Targeting the CREB pathway for memory enhancers. Nat Rev Drug Discov. 2003;2:267–77. doi: 10.1038/nrd1061. [DOI] [PubMed] [Google Scholar]
- Uecker A, Nadel L. Spatial locations gone awry: object and spatial memory deficits in children with fetal alcohol syndrome. Neuropsychologia. 1996;34:209–23. doi: 10.1016/0028-3932(95)00096-8. [DOI] [PubMed] [Google Scholar]
- Vaglenova J, Pandiella N, Wijayawardhane N, Vaithianathan T, Birru S, Breese C, et al. Aniracetam reversed learning and memory deficits following prenatal ethanol exposure by modulating functions of synaptic AMPA receptors. Neuropsychopharmacology. 2008;33:1071–83. doi: 10.1038/sj.npp.1301496. [DOI] [PubMed] [Google Scholar]
- Vaglenova J, Petkov VV. Can nootropic drugs be effective against the impact of ethanol teratogenicity on cognitive performance? Eur Neuropsychopharmacol. 2001;11:33–40. doi: 10.1016/s0924-977x(00)00129-2. [DOI] [PubMed] [Google Scholar]
- Valenzuela CF, Partridge LD, Mameli M, Meyer DA. Modulation of glutamatergic transmission by sulfated steroids: role in fetal alcohol spectrum disorder. Brain Res Brain Res Rev. 2008;57:506–19. doi: 10.1016/j.brainresrev.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varaschin R, Akers KG, Rosenberg M, Hamilton DA, Savage DD. Effects of the cognition-enhancing agent ABT-239 on fetal ethanol-induced deficits in dentate gyrus synaptic plasticity. J Pharmacol Exp Ther. 2010;334:191–198. doi: 10.1124/jpet.109.165027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltereit R, Weller M. Signaling from cAMP/PKA to MAPK and synaptic plasticity. Mol Neurobiol. 2003;27:99–106. doi: 10.1385/MN:27:1:99. [DOI] [PubMed] [Google Scholar]
- Webster WS, Walsh DA, Lipson AH, McEwen SE. Teratogenesis after acute alcohol exposure in inbred and outbred mice. Neurobehav Toxicol. 1980;2:227–34. [Google Scholar]
- West JR, Dewey S, Cassel M. Prenatal ethanol exposure alters the post-lesion reorganization (sprouting) of acetylcholinesterase staining in the dentate gyrus of adult rats. Brain Res. 1984;314:83–95. doi: 10.1016/0165-3806(84)90178-0. [DOI] [PubMed] [Google Scholar]
- White LE, Fitzpatrick D. Vision and cortical map development. Neuron. 2007;56:327–38. doi: 10.1016/j.neuron.2007.10.011. [DOI] [PubMed] [Google Scholar]
- Wiesel TN, Hubel DH. Comparison of the effects of unilateral and bilateral eye closure on cortical unit responses in kittens. J Neurophysiol. 1965;28:1029–40. doi: 10.1152/jn.1965.28.6.1029. [DOI] [PubMed] [Google Scholar]
- Wijayawardhane N, Shonesy BC, Vaglenova J, Vaithianathan T, Carpenter M, Breese CR, et al. Postnatal aniracetam treatment improves prenatal ethanol induced attenuation of AMPA receptor-mediated synaptic transmission. Neurobiol Dis. 2007;26:696–706. doi: 10.1016/j.nbd.2007.03.009. [DOI] [PubMed] [Google Scholar]
- Wijayawardhane N, Shonesy BC, Vaithianathan T, Pandiella N, Vaglenova J, Breese CR, et al. Ameliorating effects of preadolescent aniracetam treatment on prenatal ethanol-induced impairment in AMPA receptor activity. Neurobiol Dis. 2008;29:81–91. doi: 10.1016/j.nbd.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Yang X, Horn K, Baraban JM, Wand GS. Chronic ethanol administration decreases phosphorylation of cyclic AMP response element-binding protein in granule cells of rat cerebellum. J Neurochem. 1988;70:223–32. doi: 10.1046/j.1471-4159.1998.70010224.x. [DOI] [PubMed] [Google Scholar]
- Yang X, Horn K, Wand GS. Chronic ethanol exposure impairs phosphorylation of CREB and CRE-binding activity in rat striatum. Alcohol Clin Exp Res. 1998;22:382–90. [PubMed] [Google Scholar]
- Young C, Klocke BJ, Tenkova T, Choi J, Labruyere J, Qin YQ, et al. Ethanol-induced neuronal apoptosis in vivo requires BAX in the developing mouse brain. Cell Death Differ. 2003;10:1148–55. doi: 10.1038/sj.cdd.4401277. [DOI] [PubMed] [Google Scholar]
- Young C, Rothe KA, Klocke BJ, West T, Holtzman DM, Labruyere J, et al. Role of caspase-3 in ethanol-induced developmental neurodegeneration. Neurobiol Dis. 2005;20:608–14. doi: 10.1016/j.nbd.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Youngentob S, Glendinning J. Fetal ethanol exposure increases ethanol intake by making it smell and taste better. Proc Natl Acad Sci U S A. 2009;106:5359–64. doi: 10.1073/pnas.0809804106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel S. The fetal origins of memory: the role of dietary choline in optimal brain development. J Pediatr. 2006;149:s131–6. doi: 10.1016/j.jpeds.2006.06.065. [DOI] [PMC free article] [PubMed] [Google Scholar]