

Abstract
Invariant natural killer T (iNKT) cells play important roles in the immune response. ITK and TXK/RLK are Tec family kinases that are expressed in iNKT cells, with the expression level of ITK about 7-fold higher than that of TXK. Itk−/− mice have reduced iNKT cell frequency and numbers, with defects in development and cytokine secretion, which are exacerbated in the Itk/Txk DKO mice. By contrast, there is no iNKT cell defect in Txk−/− mice. To determine whether ITK and TXK play distinct roles in iNKT cell development and function, we examined mice that over express TXK in T cells to similar levels as Itk. Over expression of TXK rescues the maturation and cytokine secretion of Itk−/− iNKT cells, as well as altered expression of transcription factors T-bet, eomesodermin and PLZF. By contrast, the increased apoptosis observed in Itk−/− splenic iNKT cells is not affected by TXK over expression, likely due to lack of effect on the elevated expression of p53 regulated pro-apoptotic pathways Fas, Bax and Bad in those cells. Supporting this idea, p53−/− and Bax−/− mice have increased splenic iNKT cells. Our results suggest that TXK plays an overlapping role with ITK in iNKT cell development and function, but that ITK also has a unique function in the survival of iNKT cells, likely via a p53 dependent pathway.
Keywords: Non-conventional T cells, Tec kinases, Fas, T cell development
Introduction
NKT cells are a distinct subset of T cells that recognize glycolipid antigen presented by the CD1d glycoprotein. NKT cells are composed of Type 1 NKT cells and Type 2 NKT cells. Type 1 NKT cells, also named as invariant NKT cells (iNKT), express an invariant Vα chain (Vα14Jα18 in mice and Vα24Jα18 in human) together with certain types of Vβ chains and can be detected by α-GalCer loaded CD1d tetramer (1). Upon stimulation, iNKT cells are able to rapidly secrete large amount of multiple cytokines such as IL-4, IFN-γ and IL-17, and play important roles in the pathogenesis of allergic asthma, bacterial infection and cancer (1).
iNKT cells initiate their development from CD4+ CD8+ (DP) T cells and are selected by glycolipid antigen(s) in the context of CD1d molecules expressed on DP T cells. After selection, iNKT cells further develop through four stages to become mature. The developmental stages of iNKT cells are defined based on the expression of their surface markers CD24, CD44 and NK1.1. The most immature phase of iNKT cells (stage 0) is phenotypically characterized as CD24+CD44−NK1.1−, followed in a stepwise manner by CD24 down-regulation (CD24−CD44−NK1.1−: stage 1), upregulation of CD44 (CD44+NK1.1−: stage 2) and NK1.1 (CD44+NK1.1+: stage 3), to become the final mature iNKT cell. In addition, some of the iNKT cells at developmental stage 2 also migrate to the spleen, where they up regulate NK1.1 as a final maturation step (1). The signaling events that are required for the selection and maturation of iNKT cells have been studied extensively. Both TCR and SLAM-SAP-Fyn signaling pathways are required for the selection of iNKT cells (2–5). Transcription factors PLZF, Egr2, NF-κB family members, signaling protein Dock2 and PDK1 and microRNA processor Dicer have all been shown to be important for the maturation of iNKT cells from stage 1 to stage 2 (6–12). Transcription factor T-bet, vitamin D receptor, cytokine IL-15, cell surface receptor CD28 and ICOS, SLP-76, and PTEN are important for the final maturation of iNKT cells from stage 2 to stage 3 (1, 13–22).
ITK and TXK/RLK (referred to as TXK hereafter) are two Tec family tyrosine kinase members expressed in T cells. ITK is composed of five domains, which from N-terminus to C-terminus are PH, TH, SH3, SH2 and kinase domain. ITK is normally found in the cytoplasm of resting T cells, and upon activation, PI-3 kinase is activated, generating PIP3 in the cell membrane. The generation of PIP3 recruits ITK from the cytoplasm to the cell membrane via the PH domain of Itk. The structure of TXK is very similar to ITK, with the exception of the N-terminus. Instead of a PH domain, TXK has a cysteine-string motif that allows for constitutive localization at the plasma membrane. Both ITK and TXK are expressed in iNKT cells, with the expression level of ITK about 7-fold higher than TXK (23). Previous studies have shown that in the absence of Itk, the number of iNKT cells is decreased, development is blocked at developmental stage 2 and cytokine production is defective (23, 24). All of these defects are exacerbated in Itk/Txk DKO mice, however, Txk−/− mice have normal iNKT cell maturation and number (23). Given the similarities as well as differences in the structure of ITK and TXK, we wanted to determine whether TXK is truly functionally redundant to ITK in iNKT cells. However, due to the low expression levels of TXK in T cells, analysis of Txk−/− mice is not as informative. We therefore utilized transgenic mice which over express TXK to similar levels of endogenous ITK in WT mice (Tg(CD2-Txk)Itk−/−) and show here that TXK has overlapping roles as ITK in iNKT cell development, transcription factor expression and cytokine production, but that ITK has a distinct role in regulating survival of iNKT cells.
Material and Methods
Mice
Tg(CD2-Txk)Itk−/− mice were a kind gift from Dr. Paul Love (via Dr. Pamela Schwartzberg (NHGRI/NIH)) and were generated as described previously (25). Wild type (WT), Itk−/−, Txk−/− and Tg(CD2-Txk)Itk−/− mice were on C57BL/6 background and kept in pathogen-free conditions. Bax deficient mice on a C57BL/6 background were from Jackson labs. Mice lacking p53, along with WT controls, were maintained on a 129S6 background (26). All the mice (except p53−/− mice) used were 6–9 wks. of age, while p53−/− mice were 4–5 weeks prior to tumor development observed in these mice. Cells from 4–5 week old Bad−/− mice on a C57BL/6 background were from Dr. Nika Daniel (Dana Farber Cancer/Institute Harvard Medical School), and compared to age matched WT mice. Experiments were approved by the IACUC at the Pennsylvania State University and Cornell University.
Flow Cytometry
Single cell suspensions from tissues collected from the indicated mice were incubated with Fc block (BD biosciences) for 10 minutes, and then were stained with fluorescent antibodies for 30 min on ice. PE-PBS57/CD1d tetramer was kindly provided by the National Institute of Health tetramer core facility. anti-FITC-TCRβ, V500-CD44, APC-Cy7-TCR, PECy5-CD44 and PECy7-CD4 were from BD Biosciences (San Jose, CA); anti-PECy7-NK1.1, FITC-CD44, APC-NK1.1, FITC-CD122, APC-alexa750-TCRβ were from eBioscience (San Diego, CA); anti-PE/Texas Red CD8 and anti-PE/Texas Red CD4 were from Caltag (Burlingame, CA).
In vivo BrdU incorporation assay
WT, Itk−/− and Tg(CD2-Txk)Itk−/− mice were injected intraperitoneally with 1 μg BrdU in 100 μl PBS, followed by drinking water containing 0.8 mg/ml BrdU for 6 days. BrdU-containing drinking water was changed every two days. Cells from thymus, spleen and liver were then isolated and stained with surface antibodies, followed by BrdU staining using APC-BrdU kit from BD biosciences.
Induction of Fas-mediated apoptosis in vitro
Splenocytes were collected from WT, Itk−/− and Tg(CD2-Txk)Itk−/− mice and directly plated in 24 well plates at 2 X106/ml in each well. Cells were treated with or without 5 μg/ml anti-CD95 (Fas) antibody Jo2 (BD Biosciences) for the indicated time to induce Fas mediated apoptosis. Cells were stained for surface markers, washed in PBS and further stained with 1:50 diluted PI/RNase staining buffer (BD Biosciences) and anti-Cy5-Annexin V (BD Biosciences) in Annexin V binding buffer (BD Biosciences) for 15 minutes. Samples were analyzed within 1-hour post Annexin V staining.
Cell sorting and real-time PCR analysis
Thymic iNKT cells (TCRβ+PBS-57/CD1d tetramer+) were sorted from WT, Itk−/− and Tg(CD2-Txk)Itk−/− mice, and naïve conventional CD4+ T cells (TCRβ+CD4+CD44low) were sorted from spleens of WT and Itk−/− mice, using a Cytopeia Influx Cell Sorter (Cytopeia, Seattle, WA). RNA was isolated using RNeasy Mini kit (Qiagen, Valencia, CA), followed by generation of cDNA with You Prime First-Strand beads kit (GE Healthcare, Piscataway, NJ). Quantitative real-time PCR was carried out using Taqman primer/probe sets for Bax, Bad,, Bcl-2, Bcl-6, BclXL, T-bet, eomesodermin, Egr2, Fas, p53 and PLZF with GAPDH as a housekeeping gene (Applied Biosystems, Foster City, CA). Data were analyzed using the comparative threshold cycle 2−ΔΔCT method, normalized to the expression of GAPDH and the values were expressed as fold increase compared to WT iNKT cells, which was set as 1. To compare the expression level of Itk and Txk, qRT-PCR was performed on cDNA from sorted WT or TxkTg/Itk−/− iNKT cells. Signals were adjusted using standards generated from plasmids carrying the cDNA for murine Itk or Txk and normalized to the expression of GAPDH. The values were expressed as fold increase compared to the Itk expression in WT iNKT cells sorted from thymus or spleen, which was set as 1.
α-Galactosyl Ceramide (GalCer) stimulation and intracellular cytokine staining
WT, Itk−/− and Tg(CD2-Txk)Itk−/− mice were injected intraperitoneally with 100 μl PBS containing either 2 μg α-Galactosyl Ceramide (α-GalCer) or 1 μl DMSO. After 2 hrs., splenocytes were collected and cultured in-vitro for 4 hrs. in the presence of 10 μg/ml Brefeldin A. Cells were stained for surface markers, followed by intracellular staining of IL-4 and IFN-γ using BD fixation/permeabilization kit.
Data Analysis
Statistical analyses were determined using GraphPad Prism (GraphPad Software Inc. La Jolla, CA), using Student’s t-test, with a probability value of p≤0.05 considered statistically significant.
Results
Over expression of TXK does not restore the number of iNKT cells in Itk−/− mice
Previous studies have demonstrated that the absence of ITK results in defects in iNKT cell development, which are exacerbated in Itk/Txk DKO mice, suggesting that TXK also has role in iNKT cell development and function (23, 27). However, the number of iNKT cells is unchanged in the absence of TXK alone (23). In addition, we have determined that development of iNKT cells is also normal in the absence of TXK (Supplemental Figure 1A). Since TXK is expressed at about 7 fold lower levels that ITK in iNKT cells ((23) and Supplemental Figure 1B), this normal iNKT cell number and development in Txk−/− mice may be due to the redundant functions of ITK and TXK in iNKT cells. Given the exacerbation of iNKT cell defects in the Itk/Txk DKO mice, we examined whether increased expression of TXK is able to rescue defects in iNKT cells in the absence of Itk. To do this, we used Tg(CD2-Txk)Itk−/− mice, which over express TXK specifically in T cells to levels that are similar to endogenous ITK in T cells ((23, 25, 28) and Supplemental Figure 1B).
Flow cytometric analysis revealed that in the absence of Itk, the percentage and number of iNKT cells in the thymus and periphery were significantly decreased (Fig. 1A, B & C). Over expression of TXK restored the percentage of iNKT cells in the thymus of Itk−/− mice to WT levels, although the numbers of these cells were not restored due to decreased total thymocyte number in the Tg(CD2-Txk)Itk−/− mice (Supplemental Figure 2, note that the Txk transgene fails to restore thymic cellularity in Itk−/− mice). In contrast, the frequency and number of iNKT cells in the spleen of Itk−/− mice were not restored by over expressing TXK (Fig. 1A, B, C). The percentage and number of hepatic iNKT cells in Tg(CD2-Txk)Itk−/− mice were increased compared to Itk−/− mice, but remained lower than those in WT mice. This result demonstrate that over expression of TXK can restore the percentage of iNKT cells in the thymus of Itk−/− mice, but not the numbers, or percentages in the periphery of these mice.
Figure 1. Over expression of TXK rescues frequency and maturation but not number of iNKT cells in the absence of Itk.

A) Flow cytometric analysis of iNKT cells from thymus, spleen and liver of WT, Itk−/− and Tg(CD2-Txk)Itk−/− mice. Determination of the percentage (B) and number (C) of iNKT cells from thymus, spleen and liver. For thymus and spleen samples, n>9, for liver samples, n=4. p*<0.05, p**<0.01. (D) Thymocytes and (E) splenocytes from WT, Itk−/− and Tg(CD2-Txk)Itk−/− mice were gated on iNKT cells (tetramer+/TcRβ+) and analyzed for the expression of CD44 and NK1.1 (top graph). The percentage and number of CD44− NK1.1−, CD44+NK1.1− and CD44+NK1.1+ iNKT cells were determined (bottom graph). n>8, *p<0.05, **p<0.01.
Over expression of TXK rescues the maturation of iNKT cells in the absence of Itk
To determine the role of TXK in the development of iNKT cells, we analyzed the expression of CD44 and NK1.1 on thymic iNKT cells of WT, Itk−/− and Tg(CD2-Txk)Itk−/− mice. The majority of WT iNKT cells were CD44+NK1.1+, suggesting that most of them are mature and in developmental stage 3 (Fig. 1D). In contrast, a significantly higher percentage of Itk−/− iNKT cells were CD44+NK1.1−, indicating that iNKT cells are blocked in development stage 2 in the absence of Itk. However, in the thymus of Tg(CD2-Txk)Itk−/− mice, the percentage of CD44+NK1.1+ iNKT cells was significantly increased compared to those in Itk−/− mice, and was similar to WT iNKT cells. This indicates that over expression of TXK can rescue the development of iNKT cells in the thymus in the absence of ITK (Fig. 1D). In addition, since CD44+NK1.1− (developmental stage 2) iNKT cells also migrate to spleen to undergo final maturation, we also assessed the development of iNKT cells in the spleen. Similar to the case in the thymus, over expression of TXK also restored the percentage of CD44+NK1.1+ iNKT cells in Itk−/− mice to similar levels as WT mice (Fig. 1E). By contrast and as noted before, the number of mature iNKT cells was partially rescued, although not to the level of WT mice. These data suggest that pathways regulated by TXK can function similarly to ITK in regulating full maturation of iNKT cells.
Over expression of TXK rescues α-Galactosyl Ceramide induced cytokine secretion by iNKT cells in the absence of Itk
iNKT cells are able to secrete large amounts of IL-4 and IFN-γ in response to α-GalCer stimulation, a response which is defective in the absence of ITK (23, 27). To determine whether TXK can function in the absence of ITK in regulating cytokine secretion in iNKT cells, we injected α-GalCer intraperitoneally into WT, Itk−/− and Tg(CD2-Txk)Itk−/− mice, followed by analysis of IL-4 and IFN-γ production by iNKT cells as determined ex-vivo using intracellular cytokine staining. Compared to WT iNKT cells, Itk−/− iNKT cells produced much less IL-4 and IFN-γ (Fig. 2). By contrast, over expression of TXK restored cytokine production in the Itk−/− iNKT cells to WT levels, indicating that TXK can regulate the signaling pathway downstream of ITK for cytokine production in iNKT cells (Fig. 2).
Figure 2. Over expression of TXK rescues α-GalCer induced cytokine secretion of iNKT cells in the absence of ITK.
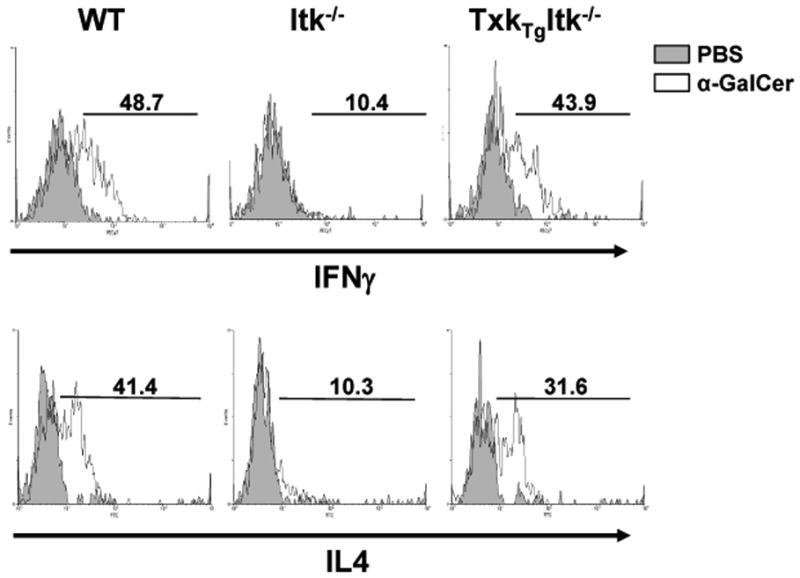
WT, Itk−/− and Tg(CD2-Txk)Itk−/− mice were injected intraperitoneally with either PBS or 2 μg α-GalCer. After 2 hours, splenocytes were collected and cultured in-vitro for 4 h in the presence of 10 μg/ml Brefeldin A. Cells were analyzed by flow cytometry, gated on iNKT cells (tetramer+/TcRβ+) and analyzed for IL-4 and IFN-γ expression. PBS control (shaded), α-GalCer (solid line). Data are representative of two independent experiments.
Over expression of TXK normalizes expression of CD122 and transcription factors in Itk−/− iNKT cells
IL-15 signals and IL-15 receptor CD122 have been shown to be important for the final maturation of iNKT cells, and the expression of CD122 has been suggested to correlate with the maturation of iNKT cells in Itk−/− mice. We have previously shown that a kinase defective mutant of ITK can partly restore the expression of CD122 on iNKT cells in the absence of Itk, suggesting that the signaling pathways that lead to CD122 expression in iNKT cells may be in part, kinase independent (27). To determine if TXK can also restore the expression of CD122 in iNKT cells in the absence of Itk, we determined CD122 expression levels on iNKT cells in the thymus and spleen of the three mouse strains. While the majority of WT thymic and splenic iNKT cells express CD122, the percentage of iNKT cells that express CD122 in Itk−/− mice was dramatically decreased as previously reported (23, 27). In contrast, the percentage of iNKT cells that express CD122 in Tg(CD2-Txk)Itk−/− mice was similar to those in WT mice (Fig. 3A).
Figure 3. Over expression of TXK restores expression of CD122 and transcription factors in Itk−/− iNKT cells.

A) Thymocytes and splenocytes from WT, Itk−/− and Tg(CD2-Txk)Itk−/− mice were gated on iNKT cells (tetramer+/TcRβ+) and stained for CD122. Data are representative of five independent experiments. B) Relative gene expression levels of T-bet, eomesodermin, PLZF and Egr2 in thymic iNKT cells from WT, Itk−/− and Tg(CD2-Txk)Itk−/− mice were determined by real-time quantitative PCR (n>3, *p<0.05 vs. WT iNKT cells; **p<0.05 vs. Itk−/− iNKT cells). C) Relative gene expression levels of T-bet, eomesodermin and PLZF in splenic iNKT cells from WT, Itk−/− and Tg(CD2-Txk)Itk−/− mice were determined by real-time quantitative PCR (n>3, *p<0.05 vs. WT iNKT cells; **p<0.05 vs. Itk−/− iNKT cells).
Several transcription factors have been reported to be important for the development and function of iNKT cells. PLZF and Egr2 are important for the development of NKT cells from stage 1 to stage 2 (6–8), while T-bet is critical for the final maturation of iNKT cells (14, 15). In addition, the expression level of eomesodermin, another member of T-box family of transcription factors, is important for regulating CD122 expression in innate CD8 T cells, although it is not normally expressed in iNKT cells (14, 29). In the absence of Itk, the expression of these transcription factors are deregulated (23, 27). In order to determine the effect of TXK on these transcription factors, their expression levels in thymic and splenic iNKT cells of WT, Itk−/− and Tg(CD2-Txk)Itk−/− mice were analyzed by QRT-PCR. In the absence of Itk, levels of mRNA for T-bet in thymic iNKT cells was decreased, while the levels of mRNA for PLZF, Egr2 and eomesodermin were significantly increased, indicating that signals from ITK is required for proper expression of these transcription factors in thymic iNKT cells (23, 27) and Fig. 3B). Over expression of TXK restored this altered expression of transcription factors in Itk−/− thymic iNKT cells to WT levels (Fig. 3B). In the splenic iNKT cells TXK was able to restore the expression pattern of eomesodermin, but not PLZF (Fig. 3C). These data suggest that similar to ITK, TXK can regulate the pattern of expression of these transcription factors in iNKT cells.
Over expression of TXK does not restore survival of Itk−/− iNKT cells
In order to determine the reason for the inability of TXK to restore iNKT cells numbers in the Tg(CD2-Txk)Itk−/− mice, we examined whether these cells could proliferate similar to those in WT mice. To do this, we analyzed incorporation of BrdU in iNKT cells in the thymus, spleen and liver of these mice. We found that iNKT cells in WT and Tg(CD2-Txk)Itk−/− mice incorporated similar levels of BrdU in these three tissues, while iNKT cells in the thymus of Itk−/− mice had increased BrdU uptake, which reflected the block in iNKT cell development at the NK1.1− stage in these mice, where iNKT cells have higher cycling rates (Fig. 4), (23, 27). This finding suggests that ITK and TXK do not regulate the proliferation of iNKT cells and that the differences in iNKT cells numbers are not due to differences in proliferation of these cells. However, this result indicates that TXK may alleviate the block in differentiation of iNKT cells seen in the absence of Itk, thus normalizing proliferation of thymic iNKT cells.
Figure 4. Over expression of TXK normalizes proliferation of thymic iNKT cells in the absence of ITK.
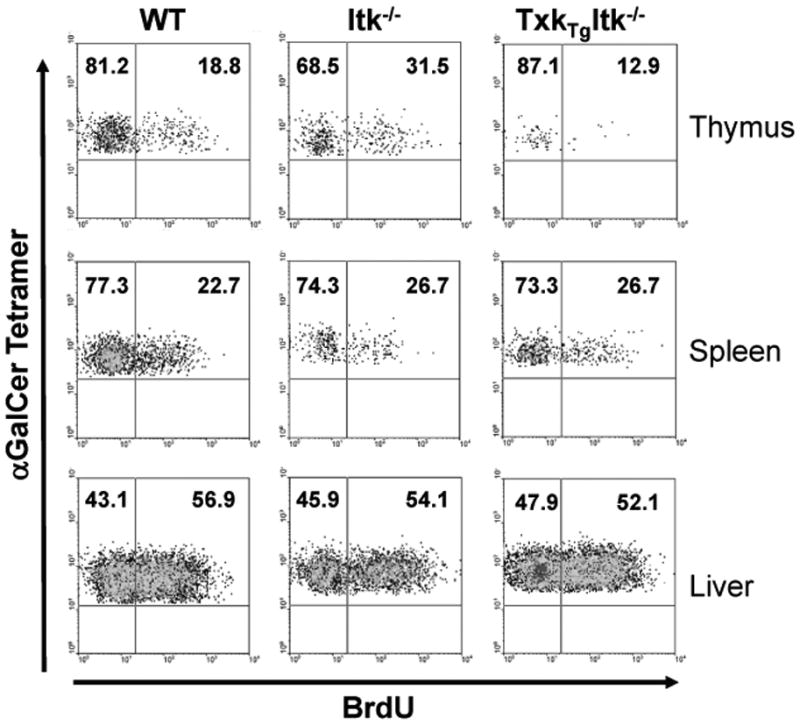
WT, Itk−/− and Tg(CD2-Txk)Itk−/− mice were injected intraperitoneally with 1 μg BrdU and placed on drinking water containing 0.8 mg/ml BrdU for the next 6 days. Cells from the thymus, spleen and liver were gated on iNKT cells (tetramer+/TcRβ+) and analyzed for BrdU uptake. Data are representative of two independent experiments.
Differences in iNKT cells numbers may be due to increased cell death. We therefore determined the level of apoptosis in these cells by staining with Annexin V. Although the percentage of Annexin V+ iNKT cells was similar in the three strains in the thymus, the percentage of those cells in Itk−/− spleens that were Annexin V+ was much higher than that in WT spleens, and over expression of TXK did not alter this increased percentage. This result suggests that apoptosis of iNKT cells is increased in the absence of Itk, and that over expression of TXK does not restore the survival of these cells (Fig. 5A). We further determined whether the expression of molecules that could regulate apoptosis is altered in Itk−/− and Tg(CD2-Txk)Itk−/− iNKT cells and found that expression of FasL was similar in WT, Itk−/− and Tg(CD2-Txk)Itk−/− splenic iNKT cells. In contrast, Fas expression was much higher in the splenic Itk−/− iNKT cells compared to WT iNKT cells, and over expression of TXK did not normalize this pattern (Fig. 5B). We next determined whether the increased expression of Fas on the Itk−/− and Tg(CD2-Txk)Itk−/− splenic iNKT cells reflected increased susceptibility to apoptotic signals in vitro. Splenocytes from WT, Itk−/− and Tg(CD2-Txk)Itk−/− mice were treated with agonistic Fas mAb in vitro, and analyzed for levels of apoptosis. We found that Itk−/− and Tg(CD2-Txk)Itk−/− iNKT cells exhibited increased apoptosis compared to WT iNKT cells in response to Fas triggering, suggesting that reduced numbers of iNKT cells in the periphery of the former strains may be due to increased susceptibility to death signals (Fig. 5C). Similar analysis of the expression levels of receptors for survival cytokines IL-2 and IL-7 revealed no differences between the different strains (Supplemental Figure 3). This inability of the Txk transgene to restore survival was not due to reduced expression of the transgene in the peripheral iNKT cells as the expression levels were similar in both thymic and splenic iNKT cells (Supplemental Figure 1B).
Figure 5. Over expression of TXK fails to restore cell survival of splenic Itk−/− iNKT cells.

A) Thymocytes and splenocytes from WT, Itk−/− and Tg(CD2-Txk)Itk−/− mice were gated on iNKT cells (tetramer+/TcRβ+) and stained for the presence of Annexin V. The percentage of Annexin V+ iNKT cells was determined. n>4, p*<0.05. B) Splenocytes from WT (black line), Itk−/− (red line) and Tg(CD2-Txk)Itk−/− mice (green line) were gated on iNKT cells (tetramer+/TcRβ+) and analyzed for the expression of Fas (left) and FasL (right). MFIs of Fas: WT=2719+/−146.9, Itk−/−=3740+/−195.9, and Tg(CD2-Txk)Itk−/−=3386+/−224.5 (+/−SEM), p<0.05 vs. WT. C) Splenocytes from WT, Itk−/− and Tg(CD2-Txk)Itk−/− mice were treated with Fas agonistic antibody or control for 2 and 8 hours, then iNKT cell apoptosis analyzed for the expression of Annexin V by gating on tetramer+/TcRβ+ cells.
Txk overexpression does not normalize the increased expression of pro-apoptotic genes in Itk−/− splenic iNKT cells
To determine whether the elevated apoptotic response observed in Itk−/− and Tg(CD2-Txk)Itk−/− splenic iNKT cells mice represented a pro-apoptotic state, we analyzed these cells for expression of pro-apoptotic genes Bad, Bax, as well as Fas, anti-apoptotic genes Bcl-2 and BclXL, and p53 and its negative regulator Bcl-6. Comparison of splenic iNKT cells from WT, Itk−/− and Tg(CD2-Txk)Itk−/− mice indicated that the latter mice exhibit higher levels of mRNA for Fas and its downstream pro-apoptotic mediators Bax and Bad, indicating that TXK fails to restore normal levels of Fas and Fas mediated proapoptotic proteins caused by the absence of ITK (Fig. 6A). However, in thymic iNKT cells, increased TXK expression reduced the expression of these genes that are elevated in the absence of Itk, correlating with the lack of enhanced apoptosis of iNKT cells observed in the thymus (Fig. 6B). By contrast, while mRNA for anti-apoptotic gene Bcl-2 was reduced in the absence of ITK in splenic iNKT cells, increased expression of TXK is able to restore the expression of this gene to WT levels, while no difference was observed in BclXL expression (Fig. 6A). The expression of Bcl-6 was reduced in the absence of ITK in splenic iNKT cells, but not in thymic iNKT cells, and was normalized in Tg(CD2-Txk)Itk−/− thymic iNKT cells but not in Tg(CD2-Txk)Itk−/− splenic iNKT cells (Fig. 6A, B). By contrast, p53 expression was induced in the absence of ITK in thymic and splenic iNKT cells, but while its expression was normalized in Tg(CD2-Txk)Itk−/− thymic iNKT cells, this was not the case in splenic Tg(CD2-Txk)Itk−/− iNKT cells (Fig. 6A, B). In addition, Bcl-6 expression was elevated in the absence of ITK in thymic iNKT cells, potentially counteracting the effects of elevated p53, but was not in Itk−/− and Tg(CD2-Txk)Itk−/− splenic iNKT cells. Since Bax, Bad, Fas are all regulated by p53, and Bcl-6 can regulate the activity of p53, these data suggest that ITK, but not TXK, specifically regulates the survival of peripheral iNKT cells by regulating the expression of and function of the Fas pathway, perhaps via a p53 dependent pathway.
Figure 6. Over expression of TXK regulates expression of pro- and anti-apoptotic genes in Itk−/− iNKT cells.

Relative gene expression levels of Fas, Bax, Bad, Bcl-2, BclXL, Bcl-6 and p53 in WT, Itk−/− and Tg(CD2-Txk)Itk−/− iNKT cells from A) spleen or B) thymus were determined by real-time quantitative PCR (*p<0.05 vs. WT iNKT cells; **p<0.05 vs. Itk−/− iNKT cells).
Regulation of percentage and number of splenic iNKT cells by p53 and p53 regulated apoptotic regulators Bax and Bad
The data shown in figure 6 suggest that the p53 pathway may regulate the survival of iNKT cells in the spleen. To determine if this is the case, we analyzed mice lacking p53 and the p53 regulated apoptotic regulators Bax and Bad, for levels of iNKT cells. This analysis revealed that while the absence of p53 did not affect the percentage or numbers of thymic iNKT cells, the percentage and numbers of these cells in the spleen were increased in the absence of p53 (Fig. 7A). Similarly, mice deficient in the p53 regulated pro-apoptotic molecule Bax had increased percentage and number of splenic iNKT cells (Fig. 7B), although Bad deficient mice did not exhibit analogous changes in percentage or numbers of iNKT cells (Fig. 7C). This suggests that the p53 pathway can regulate the survival of splenic iNKT cells, and that the absence of ITK may affect the p53 pathway, thus regulating survival of iNKT cells. The results also indicate that TXK cannot rescue this function of Itk.
Figure 7. p53 and p53 regulated gene Bax regulate the percentage and numbers of splenic iNKT cells.

A) Determination of the percentage and number of iNKT cells from thymus and spleen from WT or p53−/− mice. p*<0.05. B) Determination of the percentage and number of iNKT cells from thymus and spleen from WT or Bax−/− mice. p*<0.05. C) Determination of the percentage and number of iNKT cells from thymus and spleen from WT or Bad−/− mice. p*<0.05.
Discussion
ITK and TXK are both expressed in T cells and ITK has been demonstrated to have a critical role in the development of naïve T cells (30–36). By contrast, a role for TXK has been less clear as Txk single knockouts do not exhibit any obvious phenotype, although this kinase does play a role in T cell development as Itk/Txk double knockouts exhibit a more severe phenotype (32–34). More recent studies examining the role of these kinases in iNKT cell development has revealed that similar to conventional T cells, ITK plays an important role in iNKT cell development, with TXK having a less well-defined role (23, 24, 27, 37). This is likely due to the fact that the expression of TXK in iNKT cells is approximately 7–10 fold less than Itk. Here we have used Tg(CD2-Txk)Itk−/− mice, which have expression of TXK similar to endogenous ITK in iNKT cells and show that at this expression level, TXK rescues defects in the maturation and cytokine production of Itk−/− iNKT cells. This may be due to normalized expression of transcription factor T-bet, Egr2, PLZF and eomesodermin. By contrast, increased expression of TXK does not restore the reduced numbers nor affect the elevated apoptosis of Itk−/− iNKT cells, which may be due to the failure to normalize elevated expression of the death receptor Fas and associated pro-apoptotic molecules. We also show that this may be due to deregulation of a p53 pathway leading to increased apoptosis, and show that lack of p53 or Bax results in increased splenic iNKT cells. These data suggest that ITK and TXK have overlapping roles in the development and function of iNKT cells, however, ITK has a distinct role in the suppression of apoptosis of iNKT cells via a p53 dependent mechanism.
TcR stimulation leads to the recruitment of ITK to the cell membrane and subsequent activation of its kinase activity, due in part to the interaction between the PH domain of ITK and PIP3 lipids at the cell membrane, a Phosphatidyl-inositol-3 kinase dependent event (38). By contrast, TXK lacks a PH domain found in the other Tec kinase family members, and has instead a N-terminal palmitoylation site (39). Among the Tec kinases, this would allow TXK to be uniquely independent of PI3 kinase signaling, and suggest unique functions in activation and downstream signaling for this kinase (38, 39). However, as noted above, Txk single knockouts do not exhibit an obvious phenotype in T cell development, activation or function (40). Indeed, we have previously shown that while the absence of ITK affects naïve T cell differentiation to Th2 cells, increased expression of TXK to levels equivalent to ITK can rescue this Th2 response (28). In addition, increased expression of TXK can also improve positive selection defects observed in the absence of ITK (41). This suggests that TXK can couple to similar intracellular signaling pathways that ITK uses for this function, including the activation of PLCγ1 and increases in intracellular calcium responses (28, 41). It is therefore surprising that when examined in iNKT cells, increased expression of TXK can rescue the function of ITK in iNKT cell development, maturation and function, but cannot restore survival of these cells. This indicates that ITK plays a unique role in regulating iNKT cell survival.
It is clear that TXK makes some contribution to the development of iNKT cells since mice lacking both ITK and TXK exhibit more severe reductions in iNKT cells numbers compared to Itk−/− mice (23). This is largely due to a reduction in maturation from stage 2 to stage 3, which is accompanied by reduced expression of CD122 and T-bet, increased expression of Egr2, PLZF and eomesodermin, and enhanced proliferation of cells at this stage of development. It is noteworthy that T-bet deficiency causes a block in iNKT cells development largely at stage 2, similar to that seen in the absence of Itk, and so this block that is observed in the Itk deficient iNKT cells may be in part a consequence of T-bet deficiency and subsequent reduction in CD122 expression and reduced responses to IL-15 (14, 19, 42). However we have recently reported that there is an ITK kinase domain independent pathway that can partially restore T-bet expression and induce further maturation along the stage 3 pathway, however, full maturation is not restored (27), supporting an important role for T-bet in this process (14). Eomesodermin is not normally expressed in iNKT cells (14), but we observed increased expression in the absence of ITK, and it is possible that aberrant expression of this factor affects the maturation of these cells. While Eomesodermin is related to T-bet and can have overlapping functions (e.g. in CD8+ T cells (14, 29)), it does have unique functions, which may be the case in iNKT cells. Furthermore, while PLZF is important for iNKT cell development (with a block at stage 1) as well as to gain effector function, overexpression of PLZF results in a block in maturation at stage 2 (7, 43). Normalization of expression of these factors, a function of both the ITK kinase independent pathway, as well as TXK, may be critical for further maturation of iNKT cells. Note that the effect of the ITK kinase independent pathway on iNKT cell maturation is dependent on TXK expression, suggesting that TXK collaborates with ITK to regulate iNKT cell maturation. Our current findings show that elevating TXK’s expression can fully rescue maturation and function supports this idea. Given the ability of ITK to partially rescue iNKT cell development in a kinase independent fashion, we speculate that TXK may also be able to do this, since the two kinases interact with similar proteins via their SH2 and SH3 domains (36). However we expect that the kinase activity of both is critical for full maturation of these cells.
Despite the absence of a PH domain, TXK can engage pathways regulated by ITK for conventional T cell positive selection, Th2 differentiation, and now iNKT cell development, maturation and function ((25, 28) and this work). The Tg(CD2-Txk)Itk−/− mice have iNKT cells with normal maturation and function with regards to cytokine secretion, indicating that ITK and TXK share the same or overlapping pathways for the activation of these downstream effects. It is also possible that the reduced cytokine secretion in Itk deficient iNKT cells may be related to immature state of these cells, and that by rescuing maturation, over expression of TXK also restores the ability of these cells to produce these cytokines. However, Itk−/− iNKT cells express similar levels of preformed mRNA for IL-4 and IFNγ as expected in more mature iNKT cells. Moreover, Itk−/− iNKT cells are able to secrete both cytokines after PMA/Ionomycin stimulation, which bypasses ITK and the TcR, suggesting a signaling defect in cytokine secretion in addition to any maturational defects (23, 24, 27, 37). Nevertheless, our findings also reveal that there may be ITK specific signals that result in the activation of pathways that prevent increased apoptosis of iNKT cells, leading to normal numbers of iNKT cells. Our findings suggest that this may be related to p53 pathway activation and expression of Fas and pro-apoptotic pathways in iNKT cells, which is elevated in the absence of Itk, and not normalized when TXK expression is increased. The mechanism by which ITK suppresses the p53 pathway is unclear, however Ras regulated pathways such as PI3 kinase, MEK and JNK pathways have all been shown to suppress p53 activation (44, 45). With a PH domain, ITK interacts both with lipids generated by PI3 kinase, as well as PI3 kinase itself, which TXK may not be able to do since it lacks a PH domain (38, 46). In addition, although TXK can rescue PLCγ1 tyrosine phosphorylation and calcium responses in the absence of ITK (25), ITK may be required for optimal activation of Ras, MEK and/or JNK pathways necessary for suppression of p53 activity in iNKT cells.
Fas is expressed on all iNKT cells, and these cells can be regulated by Fas mediated death (47), and iNKT cells accumulate in older C57BL/6-lpr/lpr mice that lack Fas function, suggesting that this pathway plays a role in the homeostasis of peripheral numbers of these cells (47). Our findings indicate that Fas expression is elevated in the absence of ITK, and that Itk−/− and Tg(CD2-Txk)Itk−/− iNKT cells are more sensitive to Fas induced apoptosis. It is likely that an increase in Fas expression can contribute to the reduction in iNKT cells in Itk−/− and Tg(CD2-Txk)Itk−/− mice by enhancing apoptosis. However the reverse may not apply, i.e., a reduction in Fas may not lead to an increase at this time point, since C57BL/6-lpr/lpr mice do not have higher numbers of iNKT cells at steady state at 6–8 weeks, although it clearly occurs later (47). Perhaps homeostasis of iNKT cells is such that these cells slowly build up in the absence of Fas, but increased Fas can accelerate their demise.
Fas expression has been shown to be regulated by SP1 and inducible by p50/p65 NF-κB transcription factors, GA-binding protein and AP-1 (48, 49). Bax and Bad expression are also elevated in the absence of Itk. Genotoxic damage has also been shown to induce the expression of Fas, Bax and Bad via a p53 dependent mechanism, and Bcl-6 is a negative regulator of p53, such that reduced Bcl-6 levels unleash p53 transcriptional activity (50–53). The absence of ITK compromises the activation of both NF-κB and AP-1 (54), suggesting that elevated Fas expression via these transcription factors is unlikely. Furthermore, in conventional T cells TXK can rescue upstream pathways that lead to activation of these transcription factors (25, 28, 55–57). It is likely that the absence of ITK results in the activation of other pathways, such as the p53 pathway, that would lead to increases in Fas expression, along with other pro-apoptotic factors Bax and Bad, making iNKT cells more susceptible to Fas mediated death and apoptosis. This may partially explain the reduced numbers of iNKT cells in the Itk−/− mouse, as well as the Tg(CD2-Txk)Itk−/− mice, since TXK is unable to normalize the increases in these pro-apoptotic factors. Our finding that p53 expression is elevated in Itk−/− splenic iNKT cells and is not restored by the increased expression of TXK further supports this view. Additional support for a role for p53 in this process includes our finding that p53−/− and Bax−/− mice have increased percentage and numbers of splenic iNKT cells, as would be expected if this pathway regulates the survival of these cells in vivo. By contrast, analysis of the expression of these genes in conventional naïve WT and Itk−/− CD4+ T cells reveals that while there are changes in expression, the pattern is different from that seen in iNKT cells, suggesting that naïve CD4+ T cells behave differently from iNKT cells (Supplemental Figure 4). A number of genes have differential function in iNKT cells vs. conventional T cells, as evidenced by the analysis of components of the TcR pathway or other transcription factors that differentially affect development of these cells (1–16). Thus, pathways activating the p53 pathway may also be differentially regulated in iNKT cells compared to conventional T cells. These differences may be due to the different function of these cells, where conventional T cells generally undergo further differentiation to effector and memory cells, while iNKT cells are already effectors and do not undergo further differentiation. It would be of interest to determine whether the p53 pathway in terminally differentiated innate T cells (such as γδ T cells or other nonconventional T cells), or effector and memory T cells, behaves similar to iNKT cells. Differences in the expression and function of p53 in thymic and splenic iNKT cells may be due to differential signals for maturation of these cells in the thymus vs. the spleen, such that TXK is able to reduce p53 expression in the thymus, but not in the spleen. In addition, the development process for thymic iNKT cells may make them less likely to utilize the p53 pathway, while splenic iNKT cells may be sensitive to this pathway, as we find.
Tec kinases are being considered as targets for the treatments of a number of diseases and selective inhibitors targeting ITK and the related kinase BTK have been developed (58–62). As TXK is related to ITK and is also expressed in T cells, it is of interest to determine if this kinase has overlapping function. Our results suggest that TXK and ITK have overlapping functions in iNKT cell maturation and function, however, targeting ITK may uniquely affect the survival of iNKT cells, which TXK cannot rescue. These findings provide novel insight into the relationship between these two related Tec kinases in iNKT cell development and function.
Supplementary Material
Acknowledgments
We thank members of the August labs for feedback and discussion, Nicole Bem, Susan Magargee, Margaret Potter at Penn State University, and Dr. Rod Getchell, Tina Chew and Amie Wood at Cornell University for technical assistance. We also thank Dr. Nika Daniel and Benjamin Szlyk and Klaudia Polak (Dana-Farber Cancer Institute and Harvard Medical School) for tissues from Bad−/− mice.
Abbreviations used
- α-GalCer
α-Galactosyl Ceramide
- DKO
Double Knock Out
- GATA3
GATA binding protein 3
- iNKT cells
Invariant Natural Killer T cells
- Itk
Interleukin-2 Inducible T cell kinase
- P/I
PMA and Ionomycin
- TcR
T cell Receptor
Footnotes
Supported by NIH grants AI051626 & AI065566 to AA.
References Cited
- 1.Godfrey D, Stankovic S, Baxter A. Raising the NKT cell family. Nat Immunol. 2010;11:197–206. doi: 10.1038/ni.1841. [DOI] [PubMed] [Google Scholar]
- 2.Griewank K, Borowski C, Rietdijk S, Wang N, Julien A, Wei D, Mamchak A, Terhorst C, Bendelac A. Homotypic interactions mediated by Slamf1 and Slamf6 receptors control NKT cell lineage development. Immunity. 2007;27:751–762. doi: 10.1016/j.immuni.2007.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pasquier B, Yin L, Fondanèche M, Relouzat F, Bloch-Queyrat C, Lambert N, Fischer A, de Saint-Basile G, Latour S. Defective NKT cell development in mice and humans lacking the adapter SAP, the X-linked lymphoproliferative syndrome gene product. J Exp Med. 2005;201:695–701. doi: 10.1084/jem.20042432. Epub 2005 Feb 2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eberl G, Lowin-Kropf B, MacDonald H. Cutting edge: NKT cell development is selectively impaired in Fyn- deficient mice. J Immunol. 1999;163:4091–4094. [PubMed] [Google Scholar]
- 5.Nichols K, Hom J, Gong S, Ganguly A, Ma C, Cannons J, Tangye S, Schwartzberg P, Koretzky G, Stein P. Regulation of NKT cell development by SAP, the protein defective in XLP. Nat Med. 2005;11:340–345. doi: 10.1038/nm1189. Mar and (3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Savage A, Constantinides M, Han J, Picard D, Martin E, Li B, Lantz O, Bendelac A. The transcription factor PLZF directs the effector program of the NKT cell lineage. Immunity. 2008;29:391–403. doi: 10.1016/j.immuni.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kovalovsky D, Uche O, Eladad S, Hobbs R, Yi W, Alonzo E, Chua K, Eidson M, Kim H, Im J, Pandolfi P, Sant’Angelo D. The BTB-zinc finger transcriptional regulator PLZF controls the development of invariant natural killer T cell effector functions. Nat Immunol. 2008;9:1055–1064. doi: 10.1038/ni.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lazarevic V, Zullo A, Schweitzer M, Staton T, Gallo E, Crabtree G, Glimcher L. The gene encoding early growth response 2, a target of the transcription factor NFAT, is required for the development and maturation of natural killer T cells. Nat Immunol. 2009;10:306–313. doi: 10.1038/ni.1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kunisaki Y, Tanaka Y, Sanui T, Inayoshi A, Noda M, Nakayama T, Harada M, Taniguchi M, Sasazuki T, Fukui Y. DOCK2 is required in T cell precursors for development of Valpha14 NK T cells. J Immunol. 2006;176:4640–4645. doi: 10.4049/jimmunol.176.8.4640. [DOI] [PubMed] [Google Scholar]
- 10.Elewaut D, Shaikh R, Hammond K, De Winter H, Leishman A, Sidobre S, Turovskaya O, Prigozy T, Ma L, Banks T, Lo D, Ware C, Cheroutre H, Kronenberg M. NIK-dependent RelB activation defines a unique signaling pathway for the development of V alpha 14i NKT cells. J Exp Med. 2003;197:1623–1633. doi: 10.1084/jem.20030141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Finlay D, Kelly A, Clarke R, Sinclair L, Deak M, Alessi D, Cantrell D. Temporal differences in the dependency on phosphoinositide-dependent kinase 1 distinguish the development of invariant Valpha14 NKT cells and conventional T cells. J Immunol. 2010;185:5973–5982. doi: 10.4049/jimmunol.1000827. Epub 2010 Oct 5913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fedeli M, Napolitano A, Wong M, Marcais A, de Lalla C, Colucci F, Merkenschlager M, Dellabona P, Casorati G. Dicer-dependent microRNA pathway controls invariant NKT cell development. J Immunol. 2009;183:2506–2512. doi: 10.4049/jimmunol.0901361. Epub 2009 Jul 2522. [DOI] [PubMed] [Google Scholar]
- 13.Matsuda J, Gapin L, Sidobre S, Kieper W, Tan J, Ceredig R, Surh C, Kronenberg M. Homeostasis of V alpha 14i NKT cells. Nat Immunol. 2002;3:966–974. doi: 10.1038/ni837. [DOI] [PubMed] [Google Scholar]
- 14.Townsend M, Weinmann A, Matsuda J, Salomon R, Farnham P, Biron C, Gapin L, Glimcher L. T-bet regulates the terminal maturation and homeostasis of NK and Valpha14i NKT cells. Immunity. 2004;20:477–494. doi: 10.1016/s1074-7613(04)00076-7. [DOI] [PubMed] [Google Scholar]
- 15.Matsuda J, Zhang Q, Ndonye R, Richardson S, Howell A, Gapin L. T-bet concomitantly controls migration, survival, and effector functions during the development of Valpha14i NKT cells. Blood. 2006;107:2797–2805. doi: 10.1182/blood-2005-08-3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu S, Cantorna M. The vitamin D receptor is required for iNKT cell development. Proc Natl Acad Sci U S A. 2008;105:5207–5212. doi: 10.1073/pnas.0711558105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hayakawa Y, Takeda K, Yagita H, Kaer LV, Saiki I, Okumura K. Differential regulation of Th1 and Th2 functions of NKT cells by CD28 and CD40 costimulatory pathways. J Immunol. 2001;166:6012–6018. doi: 10.4049/jimmunol.166.10.6012. [DOI] [PubMed] [Google Scholar]
- 18.Akbari O, Stock P, Meyer E, Freeman G, Sharpe A, Umetsu D, DeKruyff R. ICOS/ICOSL interaction is required for CD4+ invariant NKT cell function and homeostatic survival. J Immunol. 2008:180. doi: 10.4049/jimmunol.180.8.5448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gordy L, Bezbradica J, Flyak A, Spencer C, Dunkle A, Sun J, Stanic A, Boothby M, He Y, Zhao Z, Van Kaer L, Joyce S. IL-15 Regulates Homeostasis and Terminal Maturation of NKT Cells. J Immunol. 2011;187:6335–6345. doi: 10.4049/jimmunol.1003965. Epub 2011 Nov 6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chung Y, Nurieva R, Esashi E, Wang Y, Zhou D, Gapin L, Dong C. A critical role of costimulation during intrathymic development of invariant NK T cells. J Immunol. 2008;180:2276–2283. doi: 10.4049/jimmunol.180.4.2276. [DOI] [PubMed] [Google Scholar]
- 21.Kishimoto H, Ohteki T, Yajima N, Kawahara K, Natsui M, Kawarasaki S, Hamada K, Horie Y, Kubo Y, Arase S, Taniguchi M, Vanhaesebroeck B, Mak T, Nakano T, Koyasu S, Sasaki T, Suzuki A. The Pten/PI3K pathway governs the homeostasis of Valpha14iNKT cells. Blood. 2007;109:3316–3324. doi: 10.1182/blood-2006-07-038059. [DOI] [PubMed] [Google Scholar]
- 22.Jordan M, Smith J, Burns J, Austin J, Nichols K, Aschenbrenner A, Koretzky G. Complementation in trans of altered thymocyte development in mice expressing mutant forms of the adaptor molecule SLP76. Immunity. 2008;28:359–369. doi: 10.1016/j.immuni.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Felices M, Berg L. The Tec kinases Itk and Rlk regulate NKT cell maturation, cytokine production, and survival. J Immunol. 2008;180:3007–3018. doi: 10.4049/jimmunol.180.5.3007. [DOI] [PubMed] [Google Scholar]
- 24.Au-Yeung B, Fowell D. A key role for Itk in both IFN gamma and IL-4 production by NKT cells. J Immunol. 2007;179:111–119. doi: 10.4049/jimmunol.179.1.111. [DOI] [PubMed] [Google Scholar]
- 25.Sommers C, Rabin R, Grinberg A, Tsay H, Farber J, Love P. A role for the Tec family tyrosine kinase Txk in T cell activation and thymocyte selection. J Exp Med. 1999;190:1427–1438. doi: 10.1084/jem.190.10.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jacks T, Remington L, Williams B, Schmitt E, Halachmi S, Bronson R, Weinberg R. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 27.Qi Q, Xia M, Bai Y, Yu S, Cantorna M, August A. Interleukin-2-inducible T cell kinase (Itk) network edge dependence for the maturation of iNKT cell. J Biol Chem. 2011;286:138–146. doi: 10.1074/jbc.M110.148205. Epub 2010 Oct 2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sahu N, Venegas A, Jankovic D, Mitzner W, Gomez-Rodriguez J, Cannons J, Sommers C, Love P, Sher A, Schwartzberg P, August A. Selective expression rather than specific function of Txk and Itk regulate Th1 and Th2 responses. J Immunol. 2008;181:6125–6131. doi: 10.4049/jimmunol.181.9.6125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Intlekofer A, Takemoto N, Wherry E, Longworth S, Northrup J, Palanivel V, Mullen A, Gasink C, Kaech S, Miller J, Gapin L, Ryan K, Russ A, Lindsten T, Orange J, Goldrath A, Ahmed R, Reiner S. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat Immunol. 2005;6:1236–1244. doi: 10.1038/ni1268. Erratum in: Nat Immunol. 2006 Jan;1237(1231):1113. [DOI] [PubMed] [Google Scholar]
- 30.Berg LJ. Signalling through TEC kinases regulates conventional versus innate CD8(+) T-cell development. Nat Rev Immunol. 2007;7:479–485. doi: 10.1038/nri2091. [DOI] [PubMed] [Google Scholar]
- 31.Hu J, August A. Naive and innate memory phenotype CD4+ T cells have different requirements for active Itk for their development. J Immunol. 2008;180:6544–6552. doi: 10.4049/jimmunol.180.10.6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horai R, Mueller K, Handon R, Cannons J, Anderson S, Kirby M, Schwartzberg P. Requirements for selection of conventional and innate T lymphocyte lineages. Immunity. 2007;27:775–785. doi: 10.1016/j.immuni.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Broussard C, Fleischecker C, Horai R, Chetana M, Venegas A, Sharp L, Hedrick S, Fowlkes B, Schwartzberg P. Altered development of CD8+ T cell lineages in mice deficient for the tec kinases Itk and Rlk. Immunity. 2006;25:93–104. doi: 10.1016/j.immuni.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 34.Atherly L, Lucas J, Felices M, Yin C, Reiner S, Berg L. The Tec family tyrosine kinases Itk and Rlk regulate the development of conventional CD8+ T cells. Immunity. 2006;25:79–91. doi: 10.1016/j.immuni.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 35.Hu J, Sahu N, Walsh E, August A. Memory phenotype CD8+ T cells with innate function selectively develop in the absence of active Itk. Eur J Immunol. 2007;37:2892–2899. doi: 10.1002/eji.200737311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Readinger J, Mueller K, Venegas A, Horai R, Schwartzberg P. Tec kinases regulate T-lymphocyte development and function: new insights into the roles of Itk and Rlk/Txk. Immunol Rev. 2009:228. doi: 10.1111/j.1600-065X.2008.00757.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gadue P, Stein P. NK T cell precursors exhibit differential cytokine regulation and require Itk for efficient maturation. J Immunol. 2002;169:2397–2406. doi: 10.4049/jimmunol.169.5.2397. [DOI] [PubMed] [Google Scholar]
- 38.August A, Sadra A, Dupont B, Hanafusa H. Src-induced activation of inducible T cell kinase (ITK) requires phosphatidylinositol 3-kinase activity and the Pleckstrin homology domain of inducible T cell kinase. Proc Natl Acad Sci USA. 1997;94:11227–11232. doi: 10.1073/pnas.94.21.11227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Debnath J, Chamorro M, Czar M, Schaeffer E, Lenardo M, Varmus H, Schwartzberg P. rlk/TXK encodes two forms of a novel cysteine string tyrosine kinase activated by Src family kinases. Mol Cell Biol. 1999;19:1498–1507. doi: 10.1128/mcb.19.2.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schaeffer E, Debnath J, Yap G, McVicar D, Liao X, Littman D, Sher A, Varmus H, Lenardo M, Schwartzberg P. Requirement for Tec kinases Rlk and Itk in T cell receptor signaling and immunity. Science. 1999;284:638–641. doi: 10.1126/science.284.5414.638. [DOI] [PubMed] [Google Scholar]
- 41.Lucas J, Felices M, Evans J, Berg L. Subtle defects in pre-TCR signaling in the absence of the Tec kinase Itk. J Immunol. 2007;179:7561–7567. doi: 10.4049/jimmunol.179.11.7561. [DOI] [PubMed] [Google Scholar]
- 42.Castillo E, Acero L, Stonier S, Zhou D, Schluns K. Thymic and peripheral microenvironments differentially mediate development and maturation of iNKT cells by IL-15 transpresentation. Blood. 2010;116:2494–2503. doi: 10.1182/blood-2010-03-277103. Epub 2010 Jun 2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raberger J, Schebesta A, Sakaguchi S, Boucheron N, Blomberg K, Berglöf A, Kolbe T, Smith C, Rülicke T, Ellmeier W. The transcriptional regulator PLZF induces the development of CD44 high memory phenotype T cells. Proc Natl Acad Sci U S A. 2008;105:17919–17924. doi: 10.1073/pnas.0805733105. Epub 12008 Nov 17912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hong M, Lai M, Lin Y, Lai M. Antagonism of p53-dependent apoptosis by mitogen signals. Cancer Res. 1999;59:2847–2852. [PubMed] [Google Scholar]
- 45.Das M, Jiang F, Sluss H, Zhang C, Shokat K, Flavell R, Davis R. Suppression of p53-dependent senescence by the JNK signal transduction pathway. Proc Natl Acad Sci U S A. 2007;104:15759–15764. doi: 10.1073/pnas.0707782104. Epub 12007 Sep 15724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu Y, Cuevas B, Gibson S, Khan H, LaPushin R, Imboden J, Mills G. Phosphatidylinositol 3-Kinase Is Required for CD28 But Not CD3 Regulation of the TEC Family Tyrosine Kinase EMT/ITK/TSK: Functional and Physical Interaction of EMT with Phosphatidylinositol 3-Kinase. J Immunol. 1998;161:5404–5412. [PubMed] [Google Scholar]
- 47.Leite-de-Moraes M, Herbelin A, Gouarin C, Koezuka Y, Schneider E, Dy M. Fas/Fas ligand interactions promote activation-induced cell death of NK T lymphocytes. J Immunol. 2000;165:4367–4371. doi: 10.4049/jimmunol.165.8.4367. [DOI] [PubMed] [Google Scholar]
- 48.Chan H, Bartos D, Owen-Schaub L. Activation-dependent transcriptional regulation of the human Fas promoter requires NF-kappaB p50–p65 recruitment. Mol Cell Biol. 1999;19:2098–2108. doi: 10.1128/mcb.19.3.2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li X, Chong A, Wu J, Roebuck K, Kumar A, Parrillo J, Rapp U, Kimberly R, Williams J, Xu X. Transcriptional regulation of Fas gene expression by GA-binding protein and AP-1 in T cell antigen receptor. CD3 complex-stimulated T cells. J Biol Chem. 1999;274:35203–35210. doi: 10.1074/jbc.274.49.35203. [DOI] [PubMed] [Google Scholar]
- 50.Owen-Schaub L, Zhang W, Cusack J, Angelo L, Santee S, Fujiwara T, Roth J, Deisseroth A, Zhang W, Kruzel E, Radinsky R. Wild-type human p53 and a temperature-sensitive mutant induce Fas/APO-1 expression. Mol Cell Biol. 1995;15:3032–3040. doi: 10.1128/mcb.15.6.3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuribayashi K, El-Deiry W. Regulation of programmed cell death by the p53 pathway. Adv Exp Med Biol. 2008;615:201–221. doi: 10.1007/978-1-4020-6554-5_10. [DOI] [PubMed] [Google Scholar]
- 52.Kuribayashi K, Finnberg N, El-Deiry W. Studying p53-dependent cell death in vitro and in vivo. Methods Enzymol. 2008;446:159–173. doi: 10.1016/S0076-6879(08)01609-1. [DOI] [PubMed] [Google Scholar]
- 53.Phan R, Dalla-Favera R. The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature. 2004;432:635–639. doi: 10.1038/nature03147. [DOI] [PubMed] [Google Scholar]
- 54.Schaeffer E, Yap G, Lewis C, Czar M, McVicar D, Cheever A, Sher A, Schwartzberg P. Mutation of Tec family kinases alters T helper cell differentiation. Nat Immunol. 2001;2:1183–1188. doi: 10.1038/ni734. [DOI] [PubMed] [Google Scholar]
- 55.Czar M, Debnath J, Schaeffer E, Lewis C, Schwartzberg P. Biochemical and genetic analyses of the Tec kinases Itk and Rlk/Txk. Biochem Soc Trans. 2001;29:863–867. doi: 10.1042/0300-5127:0290863. [DOI] [PubMed] [Google Scholar]
- 56.Schneider H, Guerette B, Guntermann C, Rudd CE. Resting Lymphocyte Kinase (Rlk/Txk) Targets Lymphoid Adaptor SLP-76 in the Cooperative Activation of Interleukin-2 Transcription in T-cells. J Biol Chem. 2000;275:3835–3840. doi: 10.1074/jbc.275.6.3835. [DOI] [PubMed] [Google Scholar]
- 57.Tomlinson M, Kurosaki T, Berson A, Fujii G, Johnston J, Bolen J. Reconstitution of Btk signaling by the atypical tec family tyrosine kinases Bmx and Txk. J Biol Chem. 1999;274:13577–13585. doi: 10.1074/jbc.274.19.13577. [DOI] [PubMed] [Google Scholar]
- 58.Lo H. Itk inhibitors: a patent review. Expert Opin Ther Pat. 2010;20:459–469. doi: 10.1517/13543771003674409. [DOI] [PubMed] [Google Scholar]
- 59.Sahu N, August A. ITK inhibitors in inflammation and immune-mediated disorders. Curr Top Med Chem. 2009;9:690–703. doi: 10.2174/156802609789044443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Di Paolo J, Huang T, Balazs M, Barbosa J, Barck K, Bravo B, Carano R, Darrow J, Davies D, DeForge L, Diehl L, Ferrando R, Gallion S, Giannetti A, Gribling P, Hurez V, Hymowitz S, Jones R, Kropf J, Lee W, Maciejewski P, Mitchell S, Rong H, Staker B, Whitney J, Yeh S, Young W, Yu C, Zhang J, Reif K, Currie K. Specific Btk inhibition suppresses B cell- and myeloid cell-mediated arthritis. Nat Chem Biol. 2011;7:41–50. doi: 10.1038/nchembio.481. [DOI] [PubMed] [Google Scholar]
- 61.Uckun F, Qazi S. Bruton’s tyrosine kinase as a molecular target in treatment of leukemias and lymphomas as well as inflammatory disorders and autoimmunity. Expert Opin Ther Pat. 2010;20:1457–1470. doi: 10.1517/13543776.2010.517750. [DOI] [PubMed] [Google Scholar]
- 62.Honigberg L, Smith A, Sirisawad M, Verner E, Loury D, Chang B, Li S, Pan Z, Thamm D, Miller R, Buggy J. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci U S A. 2010;107:13075–13080. doi: 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.