

Abstract
Metabolic imaging has great clinical potential in cancer because perturbations of metabolism are common hallmarks of malignant cellular transformation. Novel imaging strategies focused on glutamine could provide a valuable complement to 18fluoro-dexoyglucose positron emission tomography (18FDG-PET), because glutamine complements glucose in the metabolic platforms that support tumor growth at the cellular level. Furthermore, recent work has demonstrated that distinct aspects of glutamine metabolism are under the control of oncogenes and tumor suppressors. It is plausible that imaging glutamine metabolism could predict both the presence of specific transforming mutations in the tumor and the sensitivity to therapeutic agents designed to target glutamine utilization. Here we review the essential aspects of glutamine metabolism in cancer cells and discuss opportunities for imaging in cancer patients.
Molecular imaging in cancer has the potential to non-invasively illuminate biological properties of a tumor in order to individualize treatment and improve the accuracy of prognosis counseling. While conventional anatomical imaging offers crucial information about tumor size and location, it provides little insight into the molecular characteristics of the tumor. Imaging key aspects of tumor metabolism adds an informative dimension to existing modalities because metabolism reports many clinically relevant aspects of tumor biology, including histological grade, aggressiveness, and effects of the microenvironment. Warburg’s seminal work in cancer metabolism in the 1920s, in particular his observation that some tumors have high rates of glucose uptake relative to normal tissue, paved the way for 18FDG-PET, now the most commonly used form of metabolic imaging. This technique capitalizes on the fact that enhanced glucose uptake is a common effect of many of the mutations that lead to malignant transformation (1). It is used in the clinic to pinpoint the location and distribution of tumor tissue, to determine therapeutic response, and to monitor for recurrence.
Despite the success of 18FDG-PET in clinical oncology, the information it provides about tumor metabolism is actually quite limited. PET provides no information about the fate of the tracer after it enters the tumor, so the user is blind to subtleties of intracellular tumor glucose handling. Furthermore, tumors do not consume glucose as their sole nutrient, and their utilization of other substrates is also exploited with PET (2,3). For example, analogs of methionine or tyrosine labeled with 18F or 11C have been used successfully, and their concentration within tumors probably reflects increased surface expression of amino acid transporters in cancer cells (4). Among the other nutrients consumed by tumors, glutamine is the most versatile and probably the most rapidly consumed (5). Glutamine is the most abundant amino acid in plasma and occupies a unique niche in intermediary metabolism by providing a major inter-organ shuttle for both nitrogen and carbon (6). Here we discuss the reasons why glutamine is a crucial nutrient for the maintenance and growth of tumors, and why glutamine metabolism is an appealing target for new molecular imaging strategies in cancer.
Glutamine metabolism in cancer cells
Glutamine’s function as a source of both nitrogen and carbon makes it a crucial nutrient during cell proliferation (Fig. 1). Glutamine contains amino and amido nitrogens, which are either transferred to metabolic intermediates in the synthesis of nucleic acids, proteins, and hexosamines, or released as ammonia. Although a large fraction of both of these groups are ultimately secreted from rapidly dividing cancer cells, proliferation cannot occur unless some of this nitrogen is retained for the formation of complex molecules (7). In simple models of cell growth, the role as a nitrogen donor appears to explain some of the growth-retardant effects of glutamine deprivation (8,9). Hexosamines, particularly UDP-linked N-acetylglucosamine (UDP-GlcNAc) are used in post-translational protein modifications and are required for glutamine-dependent cell growth and proliferation (10). Hexosamine biosynthesis integrates glucose and glutamine metabolism because the rate-limiting step is the addition of the γ-amino group of glutamine to a hexose sugar by the enzyme glutamine fructose-6-phosphate amidotransferase (GFAT). In hematopoietic cells, GlcNAc and N-linked glycosylation of the IL-3 receptor α-subunit were required for IL-3 to stimulate glutamine uptake and cell growth (11). Since glutamine is also required for de novo GlcNAc biosynthesis, the findings suggest a feed-forward mechanism whereby ongoing glutamine uptake propagates growth signals during cell proliferation.
Figure 1.
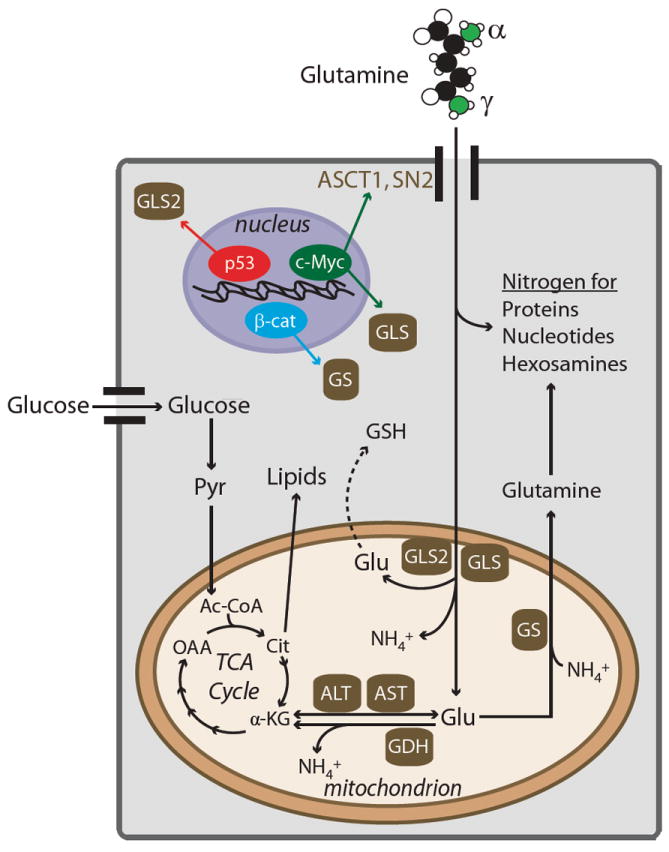
Glutamine metabolism in cancer cells and its regulation by oncogenes and tumor suppressors. After glutamine is imported into the cell through surface transporters such as ASCT1 or SN2, the majority of it either donates nitrogen to macromolecules, or is deamidated by glutaminases, which remove the γ-nitrogen to form glutamate (Glu). Glu can be used for glutathione (GSH) biosynthesis, or processed further in the mitochondria, where removal of the α-nitrogen by aminotransferases or glutamate dehydrogenase (GDH) produces α-ketoglutarate (α-KG). Expression of glutaminases (GLS, GLS2) is upregulated by c-Myc and p53, respectively. Expression of glutamine synthetase (GS) is driven by β-catenin (β-cat). Abbreviations, Ac-CoA, acetyl-CoA; ALT, alanine aminotransferase; AST, aspartate aminotransferase; Cit, citrate; OAA, oxaloacetate; Pyr, pyruvate; TCA, tricarboxylic acid.
Loss of glutamine’s amino/amido groups produces a carbon skeleton (α-ketoglutarate) that is also extensively metabolized. Some cancer cells produce more than 50% of their ATP by oxidizing glutamine-derived α-ketoglutarate in the mitochondria (12). Furthermore, the delivery of glutamine carbon to the tricarboxylic acid (TCA) cycle facilitates growth by maintaining function of the cycle even as intermediates are removed to supply biosynthetic pathways like lipid synthesis (Fig. 1). This process, termed anaplerosis, can be viewed as the sum of all metabolic pathways that produce oxaloacetate without first passing through acetyl-CoA. In some cancer cell lines, a high fraction of anaplerosis is supplied by glutamine, despite the fact that many other nutrients including glucose can be used for anaplerosis (7). Glutamine’s dominant role as an anaplerotic precursor may relate both to its abundance and to the importance of nitrogen-donating reactions that culminate in formation of α-ketoglutarate.
Glutamine also helps mitigate the effects of reactive oxygen species (ROS), which are formed as a result of the high-flux metabolic state of rapid cell growth. After deamidation of glutamine, the resulting glutamate can condense with cysteine to form the dipeptide precursor of glutathione (GSH), a major antioxidant. Mammalian cells can activate expression of genes involved in glutamine catabolism during ROS-induced stress. For example, cells exposed to hydrogen peroxide or ROS-inducing chemotherapeutics activate expression of GLS2, which encodes one of the glutaminases that convert glutamine to glutamate (13). This correlates with enhanced GSH abundance. Stress-induced GLS2 expression is regulated by the tumor suppressor p53, and over-expressing GLS2 reduces colony formation in tumor cells (13). Thus GLS2 may function in a tumor suppression pathway.
Other changes in glutamine metabolism accompany malignant transformation, and these effects depend on the molecular drivers of transformation. Enhanced activity of the oncogenic transcription factor c-Myc drives glutamine metabolism by increasing the expression of the surface transporters ASCT2 and SN2, and of the kidney-type glutaminase encoded by the gene GLS (14, 15). Importantly, cells with enhanced c-Myc were exquisitely sensitive to glutamine withdrawal (14,16). By contrast, other tumor cells seem to increase their capacity to produce glutamine from glutamate. This reaction requires the condensation of glutamate and ammonia by the enzyme glutamine synthetase (GS). Expression of GS correlates with activating mutations in β-catenin in mice and humans (17). Enhanced β-catenin activity is observed in numerous human cancers, including basal cell carcinomas, hepatomas and colorectal cancers. Although the contribution of GS to tumor initiation or progression is unknown, it is possible that this enzyme alleviates glutamine dependence and allows cells to generate a pool of glutamine for biosynthetic reactions or other purposes.
Over the years there have been a number of efforts to inhibit glutamine catabolism in tumors, including in humans. Non-selective inhibitors like 6-diazo-5-oxo-L-norleucine (DON), an amino acid analog that inhibits GFAT, glutaminase, and other glutamine-dependent reactions were in clinical trials as early as the 1950s but found to be ineffective and/or toxic. More recently, a few reports have described agents intended to target glutaminase selectively. One of these, a benzo[a]phenanthridinone compound, was identified in a screen for small molecule inhibitors of Rho-GTPase-mediated transformation. The most active of these compounds (#968) was subsequently found to be a glutaminase inhibitor, and it inhibited the growth of glutaminase-expressing lymphoma cells in mouse xenograft experiments (18). An allosteric glutaminase inhibitor, bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES), was studied in the context of the R132H mutation in isocitrate dehydrogenase-1 (IDH1) (19). This mutation, found frequently in low-grade gliomas, causes the enzyme to convert α-ketoglutarate into 2-hydroxyglutaric acid, an “onco-metabolite” suspected of contributing to tumorigenesis (20). BPTES suppressed α-ketoglutarate levels in astrocytes and preferentially inhibited the growth of cells expressing mutant IDH1 (19). The success of these preliminary studies is stimulating industry-sponsored programs to identify new glutaminase inhibitors with even higher specificity and potency.
Opportunities in glutamine-based imaging
Because many of the metabolic functions supplied by glutamine are independent of those supplied by glucose, imaging glutamine metabolism would provide a window into different aspects of tumor biology than 18FDG-PET. One can imagine several scenarios where glutamine-based PET would complement 18FDG and other nutrient analogs. First, some malignancies lack the high rates of glucose uptake necessary for identification by 18FDG-PET; prostate and bronchoalveolar carcinomas, carcinoid tumors, and low-grade lymphomas fall into this category. It is possible that a subset of these tumors uses glutamine as an alternative nutrient and might be more easily identified with a glutamine-based tracer. Second, these agents would be useful in patients subjected to drugs that suppress glucose uptake, such as inhibitors of the PI3K/Akt/mTOR signaling pathway. In culture, cells exposed to these agents require ongoing glutamine utilization to maintain survival when glucose metabolism is compromised (21). Combining 18FDG-PET with glutamine-based PET during therapy might identify tumors in which persistent glutamine metabolism will support cell survival and negatively impact the outcome. Third, the reliability of 18FDG-PET is limited when the tumor is adjacent to normal tissues where 18FDG is concentrated (e.g. the heart and brain). Glutamine consumption is low in these tissues, so glutamine-based PET would provide improved contrast between the tumor and normal tissue. Fourth, it might be possible to predict genetic alterations in the tumors based on glutamine uptake in vivo, and eventually to tailor therapy based on this information. 18FDG-PET is successful as a general imaging strategy in cancer precisely because enhanced glucose uptake is a common consequence of many different mutations. But this limits its ability to predict the presence of a specific mutation. If enhanced glutamine metabolism is restricted to a smaller subset of tumors (e.g. those with c-myc amplification), then glutamine imaging could be quite informative.
In principle, glutamine could be imaged using 11C or 13N as the radioisotope. The very short half life of 13N (10 minutes) might make it feasible to plan combined, same-day imaging studies to detect tumor glutamine uptake – first using 13N-labeled glutamine followed by 18FDG-PET after an acceptable number of 13N half-lives had passed. However, the intermediary metabolism of glutamine poses some challenges to using 13N or 11C because a large fraction of glutamine’s carbon and nitrogen is rapidly exported from glutaminolytic cells (7). The amido (γ) nitrogen, in particular, is liberated as ammonia by glutaminase, then secreted. The complete glutaminolytic pathway results in the transfer of glutamine-derived carbon to lactate, which is also secreted. Tumors with highly active glutamine synthetase might be amenable to 13N-ammonia PET, since the condensation of this tracer into newly-formed glutamine would trap it inside the cell (22). An alternative approach is to label analogs of glutamine with 18F. Panels of γ-fluorinated derivatives of glutamine and glutamate have been prepared and are readily imported into glutaminolytic tumor cells at rates comparable to 18FDG (23, 24).
Magnetic resonance spectroscopy (MRS) can also be used to evaluate glutamine and its metabolites within tumors. At high magnetic fields (e.g. 7T), it is possible to differentiate glutamine from glutamate, glutathione, and γ-amino-butyric acid by 1H MRS in the brain (25). Perturbations of these pools in tumors may signify novel biological properties and add to the value of clinical MRS. However, the ideal form of metabolic imaging would support some estimation of bona fide metabolic flux – that is, observation and quantification of the transfer of carbon/nitrogen from substrate to product within the tissue of interest. Tracer studies to observe the metabolism of 13C- or 15N-labeled glutamine have been used for decades, but in vivo detection of these nuclei is limited by their low abundance and sensitivity. One potential approach is to enhance the sensitivity using inverse detection of spin-coupled protons; this has been used successfully to monitor 15N metabolism in cancer cell lines (26). Another approach, dynamic nuclear polarization (“hyperpolarization”), massively increases the signal by temporarily transferring the spin state of an unpaired electron to a nuclear spin, usually that of 13C. This causes a signal enhancement of 10,000-fold or more (27), making it feasible to detect carbon-carbon transfers in real time. There has been significant interest in using hyperpolarized 13C to image metabolic fluxes in tumors, particularly the transfer of carbon from pyruvate to lactate by lactate dehydrogenase (28). Hyperpolarization has been used to detect the conversion of [5-13C]glutamine to [5-13C]glutamate in cancer cells (29). This single metabolic conversion potentially integrates a number of individual activities required for tumor cell growth, including glutaminase and nitrogen donation steps in the synthesis of nucleotides and hexosamines. Hyperpolarization of 15N has been performed successfully for some nitrogenous compounds (30), and if this could be extended to ammonia or glutamine, there could be several useful applications in cancer imaging.
Conclusions
Glutamine is an extremely versatile nutrient that contributes to many aspects of intermediary metabolism in cancer cells. It is particularly important in the formation of the macromolecules required for cell proliferation and resistance to oxidative stress. Because glutamine metabolism is altered during malignant transformation, imaging strategies that target glutamine should provide a useful window into tumor biology that would complement 18FDG-PET and other current techniques. As we identify more roles for this critical nutrient in tumor biology, it will be advantageous to harness glutamine-based imaging as a research and clinical tool in cancer.
Acknowledgments
RJD is supported by the NIDDK (grant DK072565), NINDS (grant NS0760675), the Cancer Prevention and Research Institute of Texas (grants HIRP100437 and RP101243) and the Welch Foundation (grant I-1733). KNR is supported by a UT-Southwestern Cancer Biology Pre-doctoral fellowship.
References
- 1.Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB. Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev. 2008 Feb;18(1):54–61. doi: 10.1016/j.gde.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Basu S, Alavi A. Molecular imaging (PET) of brain tumors. Neuroimaging Clin N Am. 2009 Nov;19(4):625–646. doi: 10.1016/j.nic.2009.08.012. [DOI] [PubMed] [Google Scholar]
- 3.Yu EY, Muzi M, Hackenbracht JA, et al. C11-acetate and F-18 FDG PET for men with prostate cancer bone metastases: relative findings and response to therapy. Clin Nucl Med. 2011 Mar;36(3):192–198. doi: 10.1097/RLU.0b013e318208f140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu A, Lee D, Shim H. Metabolic positron emission tomography imaging in cancer detection and therapy response. Semin Oncol. 2011 Feb;38(1):55–69. doi: 10.1053/j.seminoncol.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levine AJ, Puzio-Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science. 2010 Dec 3;330(6009):1340–1344. doi: 10.1126/science.1193494. [DOI] [PubMed] [Google Scholar]
- 6.DeBerardinis RJ, Cheng T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010 Jan 21;29(3):313–324. doi: 10.1038/onc.2009.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeBerardinis RJ, Mancuso A, Daikhin E, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007 Dec 4;104(49):19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meng M, Chen S, Lao T, Liang D, Sang N. Nitrogen anabolism underlies the importance of glutaminolysis in proliferating cells. Cell Cycle. 2010 Oct 1;9(19):3921–3932. doi: 10.4161/cc.9.19.13139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gaglio D, Soldati C, Vanoni M, Alberghina L, Chiaradonna F. Glutamine deprivation induces abortive s-phase rescued by deoxyribonucleotides in k-ras transformed fibroblasts. PLoS One. 2009;4(3):e4715. doi: 10.1371/journal.pone.0004715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lagranha CJ, Doi SQ, Pithon-Curi TC, Curi R, Sellitti DF. Glutamine enhances glucose-induced mesangial cell proliferation. Amino Acids. 2008 May;34(4):683–685. doi: 10.1007/s00726-007-0002-9. [DOI] [PubMed] [Google Scholar]
- 11.Wellen KE, Lu C, Mancuso A, et al. The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev. 2010 Dec 15;24(24):2784–2799. doi: 10.1101/gad.1985910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reitzer LJ, Wice BM, Kennell D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J Biol Chem. 1979 Apr 25;254(8):2669–2676. [PubMed] [Google Scholar]
- 13.Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci U S A. 2010 Apr 20;107(16):7455–7460. doi: 10.1073/pnas.1001006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wise DR, DeBerardinis RJ, Mancuso A, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. 2008 Dec 2;105(48):18782–18787. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao P, Tchernyshyov I, Chang TC, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009 Apr 9;458(7239):762–765. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol. 2007 Jul 2;178(1):93–105. doi: 10.1083/jcb.200703099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cadoret A, Ovejero C, Terris B, et al. New targets of beta-catenin signaling in the liver are involved in the glutamine metabolism. Oncogene. 2002 Nov 28;21(54):8293–8301. doi: 10.1038/sj.onc.1206118. [DOI] [PubMed] [Google Scholar]
- 18.Wang JB, Erickson JW, Fuji R, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. 2010 Sep 14;18(3):207–219. doi: 10.1016/j.ccr.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seltzer MJ, Bennett BD, Joshi AD, et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010 Nov 15;70(22):8981–8987. doi: 10.1158/0008-5472.CAN-10-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009 Dec 10;462(7274):739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang C, Sudderth J, Dang T, Bachoo RM, McDonald JG, DeBerardinis RJ. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res. 2009 Oct 15;69(20):7986–7993. doi: 10.1158/0008-5472.CAN-09-2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiangsong Z, Dianchao Y, Anwu T. Dynamic 13N-ammonia PET: a new imaging method to diagnose hypopituitarism. J Nucl Med. 2005 Jan;46(1):44–47. [PubMed] [Google Scholar]
- 23.Qu W, Zha Z, Ploessl K, et al. Synthesis of optically pure 4-fluoro-glutamines as potential metabolic imaging agents for tumors. J Am Chem Soc. 2011 Feb 2;133(4):1122–1133. doi: 10.1021/ja109203d. [DOI] [PubMed] [Google Scholar]
- 24.Krasikova RN, Kuznetsova OF, Fedorova OS, et al. 4-[(18)F]Fluoroglutamic Acid (BAY 85-8050), a New Amino Acid Radiotracer for PET Imaging of Tumors: Synthesis and in Vitro Characterization. J Med Chem. 2010 Dec 3; doi: 10.1021/jm101068q. [DOI] [PubMed] [Google Scholar]
- 25.Choi C, Dimitrov IE, Douglas D, et al. Improvement of resolution for brain coupled metabolites by optimized (1)H MRS at 7T. NMR Biomed. 2010 Nov;23(9):1044–1052. doi: 10.1002/nbm.1529. [DOI] [PubMed] [Google Scholar]
- 26.Street JC, Delort AM, Braddock PS, Brindle KM. A 1H/15N n.m.r. study of nitrogen metabolism in cultured mammalian cells. Biochem J. 1993 Apr 15;291(Pt 2):485–492. doi: 10.1042/bj2910485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Golman K, Ardenkjaer-Larsen JH, Petersson JS, Mansson S, Leunbach I. Molecular imaging with endogenous substances. Proc Natl Acad Sci U S A. 2003 Sep 2;100(18):10435–10439. doi: 10.1073/pnas.1733836100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kurhanewicz J, Vigneron DB, Brindle K, et al. Analysis of cancer metabolism by imaging hyperpolarized nuclei: prospects for translation to clinical research. Neoplasia. 2011 Feb;13(2):81–97. doi: 10.1593/neo.101102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gallagher FA, Kettunen MI, Day SE, Lerche M, Brindle KM. 13C MR spectroscopy measurements of glutaminase activity in human hepatocellular carcinoma cells using hyperpolarized 13C-labeled glutamine. Magn Reson Med. 2008 Aug;60(2):253–257. doi: 10.1002/mrm.21650. [DOI] [PubMed] [Google Scholar]
- 30.Gabellieri C, Reynolds S, Lavie A, Payne GS, Leach MO, Eykyn TR. Therapeutic target metabolism observed using hyperpolarized 15N choline. J Am Chem Soc. 2008 Apr 9;130(14):4598–4599. doi: 10.1021/ja8001293. [DOI] [PubMed] [Google Scholar]