

Abstract
An understanding of metabolic pathways based solely on biochemistry textbooks would underestimate the pervasive role of metabolism in essentially every aspect of biology. It is evident from recent work that many human diseases involve abnormal metabolic states – often genetically programmed – that perturb normal physiology and lead to severe tissue dysfunction. Understanding these metabolic outliers is now a crucial frontier in disease-oriented research. This review discusses the broad impact of metabolism in cellular function, how modern concepts of metabolism can inform our understanding of common diseases like cancer, and considers the prospects of developing new metabolic approaches to disease treatment.
Introduction – Metabolism pervades every aspect of biology
Metabolism is broadly defined as the sum of biochemical processes in living organisms that either produce or consume energy. It is a dauntingly large sum: more than 8,700 reactions and 16,000 metabolites are now annotated in the Kyoto Encyclopedia of Genes and Genomes (http://www.genome.jp/kegg/pathway.html). Core metabolism can be simplified to those pathways involving abundant nutrients like carbohydrates, fatty acids and amino acids, essential for energy homeostasis and macromolecular synthesis in humans (Figure 1). Pathways of core metabolism can then be separated conveniently into three classes: those that synthesize simple molecules or polymerize them into more complex macromolecules (anabolism); those that degrade molecules to release energy (catabolism); and those that help eliminate the toxic waste produced by the other classes (waste disposal). These pathways are profoundly important. Stated bluntly, they are the sole source of energy that allows life to resist the urge to degrade into entropy.
Figure 1. An overview of intermediary metabolism.
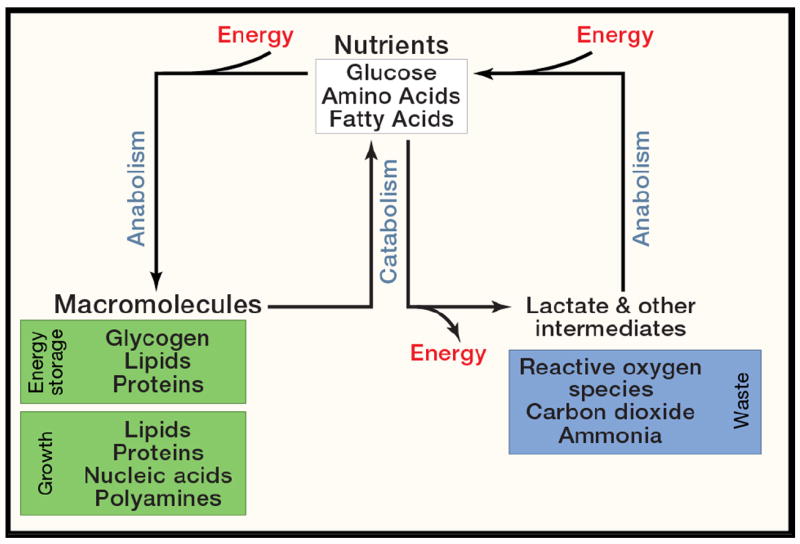
A simplified view of core metabolism, focusing on the use of major nutrients (glucose, amino acids and fatty acids) to produce or store energy, and to grow.
Defining these pathways and understanding their physiological roles have been among the most fruitful pursuits in biological research. The “golden age of biochemistry” (roughly 1920s-1960s) defined most of the metabolic network responsible for nutrient utilization and energy production in humans and other organisms. These included core activities like glycolysis (Embden, Meyerhof and Parnas), respiration (Warburg), the tricarboxylic acid (TCA) and urea cycles (Krebs), glycogen catabolism (Cori and Cori), oxidative phosphorylation (Mitchell), and the supremacy of ATP in energy-transfer reactions (Lipmann). Biochemistry and the analysis of metabolic pathways dominated basic and medically-oriented research during these decades, with some fifteen Nobel Prizes in either Physiology/Medicine or Chemistry awarded for work related to energy balance or core metabolic pathways. By the end of this period it was possible to understand, at the level of enzymatic control, such complex matters as the temporal and organ-specific regulation of fuel preferences (Krebs, 1972).
Research in metabolism has been propelled by the realization that metabolic perturbations accompany common human diseases. This insight predates the formal study of metabolism by many centuries. Almost 2,000 years ago, Celsus knew that rich foods and drink precipitated attacks of gout, and Indian physicians knew that the urine of diabetic patients attracted ants, while normal urine did not (Trowell, 1982). A greater appreciation for the relationship between precise metabolic activities and disease states blossomed during the golden age, but momentum in metabolic research gradually dissipated with the advent of newer areas of biological investigation in the latter half of the 20th century, and perhaps from the suspicion that most of what could be known about intermediary metabolism had already been discovered. The search for the genetic and molecular bases of cancer, diabetes, obesity and neurodegeneration displaced focus from understanding the altered metabolic states in these diseases. Many common diseases are now understood in terms of inherited or somatic mutations that impact gene expression, signal transduction, cellular differentiation and other processes not traditionally viewed in bioenergetic or metabolic terms.
Ironically, ongoing exploration of cell biology and disease has recently stimulated a renaissance of interest in small-molecule metabolism (McKnight, 2010). The last ten years have revealed a host of functions for metabolites and metabolic pathways that could not have been predicted from a conventional understanding of biochemistry. As a result, it is no longer possible to view metabolism merely as a self-regulating network operating independently of other biological systems. Rather, metabolism impacts, or is impacted by, virtually every other cellular process; there is no longer any space in biological research that is totally free from the influence of metabolism. This is perhaps not surprising when one considers that fundamental aspects of energy metabolism are conserved throughout evolution, whereas higher levels of regulation and the complex organization of multi-cellular organisms came much later.
Recent work has identified numerous regulatory mechanisms that either link cell signaling to the orchestration of metabolic pathways, or that enable cells to sense fuel availability and transmit the information through signaling networks (Figure 2). The integration of biochemical pathways in the cellular response to growth factors is a good example of this. In most mammalian cells, growth occurs only when promoted by extracellular ligands. These growth factors stimulate signal transduction pathways such as the phosphoinositol 3’-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) pathway. Activation of this and other pathways alter the phosphorylation states of numerous targets which together coordinate the cellular activities that culminate in cell division. But a successful transition from a resting state to growth can only occur if metabolism is reprogrammed to meet the rising demands of proliferation. Growth factor-induced signaling coordinates these functions, including maintaining a bioenergetic state permissive for growth (Lum et al, 2005). In particular, the PI3K/Akt/mTOR pathway stimulates both a rapid increase in essential nutrient uptake and the proper allocation of these nutrients into catabolic and anabolic pathways to produce energy and macromolecules, respectively (Gibbons et al., 2009). Interruption of any of these metabolic effects renders the growth factor ineffective.
Figure 2. Metabolism helps to implement cell growth programs.
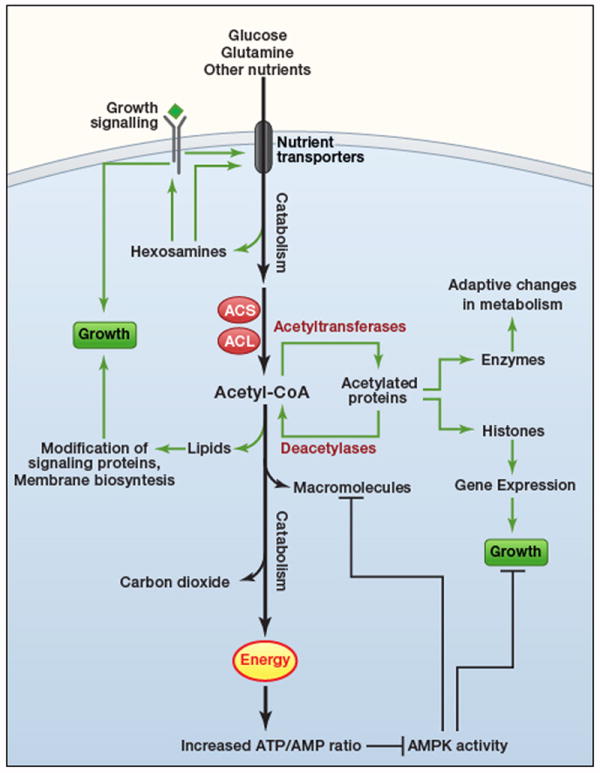
In mammals, cell growth and proliferation are controlled by extracellular factors. These ligands bind to cell surface receptors and initiate signal transduction cascades, stimulating numerous cellular activities to enable growth and replicative division. Proper control of metabolism is required for these effects. One of the proximal effects of growth factor signaling is to increase surface expression of transporters for glucose and other nutrients, which provide energy and metabolic precursors to produce macromolecules. Catabolism of these nutrients (heavy arrows) produces carbon dioxide and energy. If nutrients are present in excess, so that flux through these basic catabolic pathways is satisfied, other pathways stemming from core metabolism are induced to propagate growth signals. Hexosamine biosynthesis reinforces growth signals by enabling cells to maintain surface expression of growth factor receptors and nutrient transporters. Acetyl-CoA generated by acetyl-CoA synthetases (ACS) and ATP-citrate lyase (ACL) provides substrate for the synthesis of lipids and other macromolecules, and for acetylation reactions to regulate gene expression and enzyme function. The favorable energy state during growth factor signaling also suppresses AMPK, thereby permitting cells to engage in energy-consuming biosynthetic pathways and to progress through the cell cycle.
Dynamic mechanisms also sense cellular energy status and regulate the balance between anabolism and catabolism. While the PI3K/Akt/mTOR pathway promotes anabolism and suppresses catabolism, AMP-activated protein kinase (AMPK) does the reverse (Figure 2). This serine-threonine kinase is a “fuel sensor” activated during compromised bioenergetic states such as acute nutrient deprivation and hypoxia (Hardie, 2011). By phosphorylating a number of key targets, AMPK inactivates energy-consuming, growth-promoting pathways like protein and lipid synthesis, and activates catabolism of fatty acids and other fuels. This enables the cell to re-balance energy supply with demand. Interestingly, AMPK also regulates a p53-dependent cell-cycle checkpoint activated by glucose deprivation in cultured cells, thereby limiting growth in energetically unfavorable states (Jones et al., 2005). AMPK also coordinates the expression of stress response genes by localizing to chromatin and phosphorylating histone H2B on serine 36, and this activity facilitates AMPK’s effects on gene expression (Bungard et al., 2010). Therefore AMPK executes a number of activities that allow cells to respond decisively and comprehensively to energy shortage.
Metabolism also affects cell signaling by providing substrates for post-translational modifications that modulate protein trafficking, localization and enzyme activity. The most obvious example is ATP, which provides the substrate for phosphorylation in kinase cascades. But other examples involve metabolites that are produced in more complex pathways stemming from core metabolism. These modifications can signal states of nutrient abundance, because generating sufficient quantities of the requisite metabolite for protein modification requires access to nutrients in excess of the levels needed to run basic bioenergetic programs. For example, both glucose and glutamine are abundant nutrients that are catabolized to produce energy in growth-factor stimulated cells. But they also collaborate in another biochemical pathway to produce hexosamines, which modify nutrient transporters and growth factor receptors, enabling their expression on the cell membrane (Figure 2). Unless glucose is present in adequate amounts to supply flux through the hexosamine pathway, cells lose the ability to respond to growth factor signaling and no longer take up the full complement of nutrients required for growth (Wellen et al., 2010). Similar examples involve the post-translational modification of signaling mediators by fatty acids and other lipid-like molecules, which facilitate membrane localization and/or activation of these proteins.
Acetyl-CoA, a central metabolite at the intersection of carbohydrate, fatty acid and amino acid oxidation, exerts tremendous influence on cell signaling. Acetylation of lysine residues on the N-terminal tails of histone proteins is a major factor in chromatin dynamics and gene expression. The acetyl-CoA groups used to modify histones are predominantly produced by acetyl-CoA synthetase in yeast and ATP-citrate lyase in mammalian cells, both of which enter the nucleus to produce a localized acetyl-CoA pool for this purpose (Figure 2). Loss of function of these enzymes reduces histone acetylation, with global consequences on gene expression (Takahashi et al., 2006; Wellen et al., 2009). Many other cellular proteins besides histones are acetylated, including most of the enzymes in core metabolic pathways (Zhao et al., 2010). In the liver, these modifications appear to regulate metabolic flux in synchrony with the feed/fast cycle. For example, the acetylation status of several key enzymes in gluconeogenesis and the urea cycle correlates with their stability or activity. In both mammals and bacteria, enzyme acetylation fluctuates according to which nutrients are available to the cell (Wang et al., 2010; Zhao et al., 2010). Thus both unicellular and multicellular organisms use metabolite-mediated post-translational modification to match nutrient abundance with the distribution of carbon throughout metabolic networks.
Deacetylation reactions also regulate metabolism (Figure 2). A class of deacetylases, the sirtuins, are nicotinamide adenine dinucleotide (NAD)-dependent deactylases whose targets include histones and metabolic enzymes. Sirtuins are key evolutionarily-conserved factors linking caloric restriction to longevity. Over-expression of sirtuins in model systems ameliorates a variety of age-related phenotypes including cancer, diabetes and neurodegeneration (Guarente, 2011). There is a tremendous amount of interest in identifying potent pharmacological activators of sirtuins to treat or prevent these diseases. Interestingly, several sirtuins localize to the mitochondria, where they deacetylate or otherwise modify metabolic enzymes. It is unknown whether these sirtuins serve to antagonize the effects of as-yet unidentified mitochondrial acetyltransferases or to reverse non-enzymatic acetylation, but evidence indicates their importance in regulating induction of catabolism and waste disposal during the feed/fast cycle. For example, the urea cycle enzyme carbamoyl phosphate synthetase (CPS) is deacetylated by the sirtuin SIRT5 during fasting (Nakagawa et al., 2009). This likely enables the liver to accommodate the increased amino acid degradation and ammonia production stimulated by the fasting state. CPS is one of many enzymes previously thought to be regulated primarily through allosteric mechanisms, but now recognized to be subject to additional levels of control.
Acetyl-CoA levels also regulate higher levels of organization in eukaryotes, particularly fundamental processes such as the commitment to cell growth. When budding yeast are cultured in glucose-limiting conditions, they collectively oscillate through a metabolic cycle that is linked to synchronized cell growth (Tu et al., 2005). A rise in intracellular acetyl-CoA occurs in phase with the induction of growth genes, and a bolus of extracellular nutrients that can be readily converted to acetyl-CoA causes cells to short-circuit the metabolic cycle and enter directly into growth (Cai et al., 2011). Peak levels of acetyl-CoA are accompanied by histone acetylation at specific regions of chromatin containing growth genes. Thus, in these cells, extracellular nutrients stimulate gene expression via an acetyl-CoA-transmitted signal that culminates in a commitment to cell growth/division.
Work in all of these areas emphasizes that metabolism pervades every aspect of biology from the single-cell to whole-organism level. No cellular functions occur independently of metabolism, and a metabolic perturbation at one node has ripple effects that can extend throughout the network and out into other systems. Thus metabolic disturbances have an extremely long reach, and this extends to disease phenotypes.
Genetic variation in human metabolism and its impact on health and disease
Much of the metabolic variation among individuals is genetically defined. Metabolically-derived phenotypes are profoundly important both to human disease and to the history of experimental biology. Indeed, most of the classical experiments in genetics followed phenotypes caused by metabolic mutants. Mendel’s pea seeds were smooth or wrinkled according to whether they expressed wild-type or mutant alleles of a starch-branching enzyme (Bhattacharyya et al., 1990), and T. H. Morgan followed Drosophila eye color phenotypes that reflected altered transport of metabolites required for pigment synthesis (Sullivan and Sullivan, 1975). Ultimately observations like these led to the “one gene-one enzyme” formulation of genetics and phenotypic inheritance (Beadle and Tatum, 1941), a precursor of the central dogma of molecular biology.
Metabolic variation also causes human phenotypes, and the characterization of extreme metabolic perturbations fostered the concept that human diseases could be inherited as simple traits. This idea was formulated by Archibald Garrod in London during the first decade of the 20th Century. Garrod’s discoveries were propelled by an unusual combination of interests: chemistry, urine pigments, and joint diseases (some of these were evidently inherited from his father, who first observed uric acid crystals in the urine of gout patients). Garrod’s breakthrough was the observation that alkaptonuria – an arthritic disorder characterized by massive excretion of pigment-generating homogentisic acid in the urine – recurred in families in an autosomal recessive pattern (Garrod, 1902). Notably, Garrod reported the finding just two years after the rediscovery of Mendel’s pea experiments, to which he referred in the paper. Garrod observed similar heritable metabolic anomalies in albinism, cystinuria and pentosuria, collectively termed these diseases “inborn errors of metabolism (IEMs),” and published his seminal treatise on the subject in 1923. This conclusively established the importance of metabolic pathways in human health and, conversely, the impact of genetically-defined metabolic outliers on disease.
There are now some 500 recognized IEMs (Childs et al., 2001), making this the largest category of heritable human diseases. IEMs involve essentially every known biochemical pathway and organ system, resulting in an extremely broad array of phenotypes spanning clinically silent abnormalities in metabolite abundance, chronic/progressive accumulation of toxic macromolecules, and severe, acutely life-threatening states of bioenergetic catastrophe. The vast majority of IEMs result from recessively inherited loss-of-function mutations in enzymes and transporters, although several are caused by mutations that either stimulate basal enzymatic activity or evoke altogether new functions (Kranendijk et al., 2010; Stanley et al., 1998). Individuals affected with IEMs often display a near-complete loss of normal pathway function, making them the equivalent of human knockouts. Nevertheless, many severe IEMs are compatible with embryonic development and postnatal life, although significant organ-system dysfunction or context-specific pathologies may result. Many IEMs, particularly those involving the core pathways in Figure 1, are treated by modifying the diet so as to reduce exposure to offending nutrients, and these interventions have substantially improved health and lifespan.
IEMs provide a valuable view of “life on the edge” of metabolic dysfunction. Indeed, our overall understanding of human metabolism owes a great deal to the clinical care of IEM patients. Mitochondrial fatty acid oxidation was understood to be an efficient bioenergetic pathway, but its specific role in human energy homeostasis became obvious from the severe, life-threatening hypoketotic hypoglycemia elicited by prolonged fasting in children with diminished function of the pathway (Houten and Wanders, 2010). Similarly, glucose oxidation in the citric acid cycle produces most of the energy for the brain, but the crucial role for pyruvate dehydrogenase (PDH) in brain development is evident in the profound central nervous system dysfunction resulting from mutation of PDH’s catalytic subunit (Robinson et al., 1996). In other examples, the physiological function of an enzyme or pathway was unknown until patients were identified with genetic defects. Peroxisomal enzyme deficiencies fall into this category. Synthesis of plasmalogens and oxidation of fatty acids 24-26 carbons in length occur in the peroxisomes, but this was only realized when abnormal levels of these metabolites were observed in patients with peroxisomal dysfunction (Wanders and Waterham, 2006).
Since the 1960s, population-based efforts have substantially broadened the view of human metabolic diversity beyond severely affected individuals with overt disease. State-operated newborn screening programs quantify levels of diagnostic metabolites in dried blood spots, with the goal to identify pre-symptomatic babies in whom prompt therapy can improve clinical outcome. These programs are descended from the work of Robert Guthrie, who developed a simple, inexpensive assay to detect newborns with phenyketonuria (PKU) in the 1960s. PKU results from genetic deficiencies in phenylalanine (Phe) oxidation and accumulation of toxic Phe-related metabolites that impair cognitive development. Because affected infants do not immediately manifest symptoms, morbidity is avoided by the early initiation of Phe-reduced diets in newborns ascertained through screening. Pilot PKU screening efforts in the United States were extremely successful, and within 10 years full-scale programs were underway throughout the U.S. and Europe. Since the 1990s, improved analytical technologies, particularly high-throughput mass spectrometry, enabled programs to expand dramatically the number of metabolites analyzed. Most programs now detect more than 20 individual disorders from a single blood sample (Chace et al., 2002). These programs have revealed a remarkable degree of phenotypic heterogeneity among individuals with abnormal levels of diagnostic metabolites, many of whom never manifest any clinical symptoms. In some cases, clinically silent “disease” has been detected in the asymptomatic mothers of babies subjected to screening programs, apparently because small amounts of the mother’s metabolites are transferred to the unaffected baby through the placenta, breast milk or other routes (Eichhorst et al., 2010). In other cases, IEMs once thought to be highly penetrant and severe now appear to be clinically silent in most individuals (Alfardan et al., 2010). Overall, metabolite screening in millions of babies has led to a wealth of data about the spectrum of metabolite abundance in humans and its predictive value for disease.
Combining metabolite profiling with genomic studies has identified additional genetic determinants of metabolic diversity and their relationship to disease. Profiling plasma lipids in large populations has uncovered sequence variations that substantially impact lipid phenotypes (Cohen et al., 2005; Teslovich et al., 2010). Other studies have profiled a much broader array of metabolites (acylcarnitines, amino acids, prostaglandins, sphingo- and glycerophospholipids, etc) in hundreds of healthy adults (Gieger et al., 2008; Illig et al., 2010). Treating the abundance of each metabolite or ratios of related metabolites as quantitative traits, the authors used genome-wide association studies to search for polymorphisms that exhibited large effects on metabolite abundance. Surprisingly, these studies identified a number of relatively common alleles that accounted for significant fractions of the variation in metabolite levels, sometimes more than 20% of the total variance (Gieger et al., 2008; Illig et al., 2010). There were several examples in which a polymorphism changed the coding sequence of a metabolic enzyme directly related to the variable metabolite. Other polymorphisms had previously been shown to associate with cardiovascular disorders, gout and type-2 diabetes, all of which are known to involve changes in core metabolic pathways (Suhre et al., 2011). However, there were also associations with drug toxicity, Crohn’s disease and other conditions not conventionally thought to be related to intermediary metabolism (Suhre et al., 2011). These studies prove that a wide range of human metabolic diversity is genetically programmed, and they emphasize the intimate connections between metabolism and health.
Cancer as a paradigm of genetically-defined metabolic abnormalities
Cancer is a prime example of a common human disease with genetically-defined, pathological metabolic perturbations. Altered cellular metabolism is a hallmark of cancer, contributing to malignant transformation and to the initiation, growth and maintenance of tumors (Hanahan and Weinberg, 2011). Although the recent renaissance in metabolism research, particularly work in basic regulation of core metabolic pathways, owes much to cancer cell biology (Bensaad et al., 2006; Christofk et al., 2008; Gao et al., 2009; Matoba et al., 2006; Vander Heiden et al., 2010), the principle of perturbed metabolism in tumors is very old, dating almost to the era of early work in chemical carcinogens and viruses as cancer-promoting agents. Otto Warburg performed the first rigorous work in cancer metabolism in the early 1920s, studying the behavior of tissue slices ex vivo using manometric techniques developed in his own laboratory (Koppenol et al., 2011). Warburg observed that carcinoma slices from rats and humans consumed much more glucose and secreted much more lactate than normal tissue, even when presented with enough oxygen to metabolize glucose completely to CO2. This was interpreted as a fundamental change in the way glucose metabolism is regulated in cancer cells (Warburg, 1956). Among Warburg’s many seminal contributions to biochemistry (he won the Nobel Prize in 1931 for work on respiration), he is best remembered and most frequently cited for that observation, now called the Warburg effect.
After Warburg, generations of cancer biologists and biochemists have refined his hypothesis and attempted to provide mechanistic explanations for it – but basically these studies have confirmed the central observation that many tumors can out-compete surrounding tissue for glucose. This trait explains the success of 18fluoro-2-deoxyglucose positron emission tomography (FDG-PET) to image tumors of many histological types. Other work has identified additional metabolic characteristics of tumor tissue, most consistently the tendency to metabolize glutamine and to synthesize fatty acids, both of which promote tumor growth in experimental models (DeBerardinis and Cheng, 2010; Swinnen et al., 2006). One common hypothesis to unify these metabolic pathways is that aggressive tumor growth requires a restructuring of metabolism to meet the bioenergetic and biosynthetic demands of rapid cell growth and to protect the cells against stresses induced by a harsh microenvironment (Deberardinis et al., 2008; Semenza, 2010; Shanware et al., 2011). Metabolic flux studies in cancer cells have validated this model, emphasizing the integration of these three core pathways (DeBerardinis et al., 2007). Thus, cancer is a paradigm for how perturbed metabolism at the cellular level contributes to disease.
What drives metabolic reprogramming in tumor cells? As in IEMs, the metabolic idiosyncrasies of tumors are genetically defined, resulting from the same mutations that promote malignancy. However, in IEMs, germline mutations elicit wholesale changes in metabolism that tend to reduce overall fitness. Cancer mutations are in general acquired somatically and associated with metabolic effects that appear to increase fitness and growth at the cellular level. Two different classes of mutations can reprogram metabolism in tumors. First, many human oncogenes and tumor suppressors regulate glucose metabolism (Figure 3A) (DeBerardinis, 2008; Jones and Thompson, 2009). Tumor-promoting mutations in these genes tend to converge on a metabolic phenotype of enhanced glycolysis and energy production, thereby contributing to self-sufficiency of growth and evasion of growth-suppressive signals. The PI3K/Akt/mTOR pathway is essential to this process because it regulates both nutrient uptake and the allocation of carbon and nitrogen into biosynthetic pathways (Zoncu et al., 2011). Cell growth downstream of this pathway requires the induction of de novo lipid synthesis via mTOR, and this effect is due in part to increased nuclear localization of sterol regulatory element-binding protein (SREBP), a basic helix-loop-helix leucine zipper transcription factor that activates expression of a suite of lipogenic enzymes (Porstmann et al., 2008). Recent work demonstrated that mTOR’s influence on SREBP involves phosphorylation of Lipin-1, a phosphatidic acid phosphatase. mTOR-dependent Lipin-1 phosphorylation causes it to be retained in the cytosol, whereas mTOR inhibition led to the dephosphorylation of Lipin-1 and its translocation to the nucleus, where it was involved with depletion of nuclear SREBP (Peterson et al., 2011). Thus reprogramming metabolism into a platform that promotes cell growth and proliferation is an essential component of growth-factor signaling pathways and of malignancy resulting from constitutive activation of these pathways.
Figure 3. Cancer cell metabolism.

(A) Cancer cells rely primarily on glucose and glutamine to supply intermediary metabolism. Several metabolite pools fed by these nutrients and thought to be essential for tumor cell growth are highlighted in yellow. Uptake and catabolism of glucose and glutamine is regulated by oncogenic signaling. Suspected metabolic tumor suppressors (red) and oncogenes (green) control the abundance of a handful of key metabolites (bold) that regulate additional signaling functions as described in the text. These signaling activities likely contribute to malignant transformation or the propagation of growth signals within transformed cells. Thus, in addition to their traditional roles in metabolism, FH, SDH and the 2-HG dehydrogenases serve to suppress levels of pro-oncogenic metabolites. 2SC, S-(2-succinyl)-cysteine.
(B) Detailed view of selected metabolites and enzymes discussed in the text. Abbreviations: Aco, aconitase; IDH, isocitrate dehydrogenase; αKGDH, α-ketoglutarate dehydrogenase; SCS, succinyl-CoA synthetase; SDH, succinate dehydrogenase; FH, fumarate hydratase; mut IDH1/IDH2, mutant isocitrate dehydrogenase 1 or 2; (D)-2HG DH; (D)-2-hydroxyglutaric acid dehydrogenase.
Second, metabolic reprogramming can occur as the direct effect of enzyme mutations. This was first observed in a subset of patients with IEMs who displayed an increased risk of cancer, particularly hepatocellular carcinoma (HCC). Many of these diseases either involve the accumulation of mutagens or cause cirrhosis, an independent risk factor for HCC (Erez et al., 2011). In these cases the connection between metabolism and malignancy is probably indirect. But other disorders produce a metabolic state that mimics oncogene activation. Glucose-6-phosphatase (G6Pase) deficiency in Glycogen Storage Disease Type 1a is a risk factor for hepatocellular adenomas and HCC, but cirrhosis is not a prominent feature. G6Pase catalyzes the final common step in glycogenolysis and gluconeogenesis, and G6Pase deficiency results in extreme fasting intolerance and hypoglycemia. The failure to generate glucose from either of these two pathways results in a large flux into other pathways supplied by glucose-6-phosphate in the liver, including glycolysis, lipid synthesis and nucleotide metabolism. Along with massive accumulation of glycogen and fat in the liver, G6Pase-deficienct patients develop severe elevations of lactic acid, lipids and uric acid in the bloodstream. The pathophysiology of tumorigenesis is unclear and it is unknown how the redirection of glucose metabolism affects hepatocellular differentiation. But it is noteworthy that G6Pase deficiency mimics the Warburg effect and other oncogene-dependent metabolic phenomena, with the shunting of glucose-6-phosphate into lactate and other metabolite pools.
Recently it has become apparent that cancer is also associated with metabolic mutations confined to the tumor. In these diseases, the metabolic enzymes behave genetically as oncogenes or tumor suppressors (Figure 3A) (Frezza et al., 2011a; Thompson, 2009). These forms of cancer are a unique opportunity to determine the cell-intrinsic consequences of metabolic perturbations. The first such examples were mutations in TCA cycle enzymes in familial cancer syndromes. Mutations in succinate dehydrogenase (SDH), an oxidoreductase complex that functions in both the ETC and TCA cycle, were identified in dominantly-inherited familial paraganglioma (Baysal, 2008; Baysal et al., 2000). Loss-of-function mutations have been identified in all four subunits of the SDH complex (Baysal, 2008; Burnichon et al., 2010), and in SDH5, which encodes a protein involved in incorporation of SDH’s flavin adenine dinucleotide cofactor (Hao et al., 2009). Mutations in the TCA cycle enzyme fumarate hydratase (FH) were identified in familial syndromes characterized by susceptibility to renal cell cancer and leiomyomatosis (smooth muscle tumors of the uterus and skin) (Tomlinson et al., 2002). In families with SDH- or FH-deficient tumors, affected individuals inherit one mutation and their tumors display loss of the wild-type allele. Thus, both SDH and FH are tumor suppressors.
The mechanisms connecting dysfunctional SDH/FH to malignancy are probably multi-factorial (Figure 3A). Strong evidence implicates dysfunctional cell signaling stimulated by succinate and/or fumarate, which accumulate to high levels in the tumors. Both metabolites have been demonstrated to interfere with processes potentially involved in tumor suppression, including c-Jun-mediated apoptosis in pheochromocytoma cells (Lee et al., 2005) and AMPK signaling (Tong et al., 2011). Furthermore, both succinate and fumarate aberrantly increase the function of hypoxia inducible factors (HIFs). These dimeric transcription factors orchestrate the metabolic effects of hypoxia, including the increased expression of glucose transporters and glycolytic enzymes. When cells have adequate access to oxygen, HIF transcriptional activity is constrained through the constitutive degradation of its α-subunits (HIF-1α and HIF-2α). This requires post-translational modification by prolyl hydroxylases (PHDs), α-ketoglutarate-dependent enzymes that generate succinate as an end product (Figure 3B). Both succinate and fumarate are competitive PHD inhibitors, and the massive accumulation of these metabolites in cells lacking SDH or FH activity elicit a hypoxic response even under oxygen-replete condition (Isaacs et al., 2005; Selak et al., 2005). These effects chronically poise SDH- and FH-deficient cells for glycolysis, regardless of whether or not oxygen is available. Accumulation of reactive oxygen species, an independent trigger for HIF stabilization, may contribute to this process in some cases of defective SDH or FH (Guzy et al., 2008; Sudarshan et al., 2009). However both HIF-1α and HIF-2α are dispensable for the formation of hyperplastic renal cysts in mice lacking expression of FH in the kidney (Adam et al., 2011), suggesting additional consequences of these enzyme deficiencies.
Interestingly, because of its electrophilic properties, high levels of fumarate can modify cysteine residues through a process termed succination. This process converts sulfhydryl groups on cysteine to S-(2-succinyl)-cysteine (2SC), in some cases interfering with protein function (Alderson et al., 2006). Cells lacking FH were shown to have high levels of succination on Kelch-like ECH-associated protein 1 (KEAP1), an electrophile sensor and negative regulator of the transcription factor Nuclear Factor E2-related factor 2 (Nrf2) (Adam et al., 2011; Ooi et al., 2011). Nrf2 induces expression of a suite of genes involved in defense against reactive oxygen species, perhaps enabling malignant FH-deficient cells to tolerate high levels of exogenous or endogenous oxidants. Another possibility is that Nrf2 activation produces metabolic advantages for FH-deficient cells. One of Nrf2’s targets is the gene encoding heme oxygenase-1 (HMOX1), an enzyme involved in heme degradation. The pathway of heme synthesis from glutamine followed by degradation via HMOX1 is enhanced in FH-deficient cells, and silencing HMOX1 prevents these cells from forming colonies (Frezza et al., 2011b). Although it is still unclear how best to exploit any of these effects for cancer therapy, these findings collectively demonstrate that dysfunctional mitochondrial metabolism and elevations of dicarboxylic intermediates of the TCA cycle promote aberrant signaling in tumor cells.
Other enzyme mutations may function as oncogenes. Genomic sequencing of gliomas (Parsons et al., 2008; Yan et al., 2009) and acute myelogenous leukemia (Mardis et al., 2009) identified mutations in two isoforms of NADP+-dependent isocitrate dehydrogenase (IDH1 and IDH2). These enzymes normally oxidize isocitrate to α-ketoglutarate, with NADP+ reduced to NADPH in the process. IDH1 and IDH2 mutations in cancer are somatically acquired, present on only one allele, and confined to the enzyme’s active site. Thus, unlike FH and SDH, it was difficult to make a case for IDH1 and IDH2 as tumor suppressors. The data were more consistent with a gain-of-function referable to the altered enzymatic active site. This suspicion was confirmed by metabolomics: tumor tissue and cell lines expressing mutant IDH1 or IDH2 produced large quantities of a metabolite, (D)-2-hydroxyglutarate (2-HG), that is vanishingly scarce when only the wild-type enzymes were expressed (Dang et al., 2009). This metabolite is produced from the NADPH-dependent reduction of α-ketoglutarate to 2-HG (Fig. 3A,B), a neomorphic enzyme activity that occurs very efficiently when both mutant and wild-type alleles are expressed together (Pietrak et al., 2011). The presence of 2-HG in these tumors is compelling because it mirrors one of the best-established connections between an IEM and cancer. Children with (L)-2-hydroxyglutaric aciduria, an autosomal recessive condition caused by deficiency of (L)-2-HG dehydrogenase, which converts 2-HG to α-ketoglutarate, accumulate the (L)-isomer of 2-HG in all body fluids. A large fraction of children with this disease have developed brain tumors or other types of cancer (Moroni et al., 2004). Thus, active-site mutations in IDH1 and IDH2 seem to confer oncogenic properties to the enzyme, and production of the oncometabolite 2-HG may be the crucial factor that tips susceptible populations of cells into a transformed state.
The mechanisms responsible for this phenomenon are now the subject of intense study. Surprisingly, even large accumulations of 2-HG do not cause wholesale perturbations of metabolite levels in IDH1-mutant tumors (Dang et al., 2009). Recent data suggest that 2-HG exerts its effects by influencing some of the more than 50 mammalian dioxygenases that use α-ketoglutarate as a substrate (Fig. 3A,B). These enzymes regulate a number of crucial processes including methylation of histones and DNA. Indeed, high concentrations of 2-HG inhibited the function of α-ketoglutarate-dependent histone demethylases and 5-methylcytosine hydroxylases in vitro, and primary IDH1- or IDH2-mutant tumors showed evidence of extensively altered histone and DNA methylation in vivo (Figueroa et al., 2010; Xu et al., 2011). Furthermore, although mutations in IDH1/2 and in the 5-methylcytosine hydroxylase TET2 occur frequently in acute myelogenous leukemia, the mutations were found to be mutually exclusive, suggesting that the two different types of mutation exert redundant effects on the cell (Figueroa et al., 2010). Because widespread methylation changes of histones and DNA could broadly influence epigenetics, IDH1/2 mutations have the potential to exert tremendous effects on cellular function and differentiation (Figure 3A). Furthermore, if this epigenetic state is maintained by ongoing 2-HG effects, it may be possible to reverse some of the effects by inhibiting mutant IDH1/2 enzyme activity or by increasing the availability of α-ketoglutarate to re-stimulate dioxygenase function. It should be emphasized that the presence of IDH1/2 mutations in gliomas is associated with both lower histological grade and a slower rate of disease progression, suggesting the possibility that inhibiting mutant IDH function may prevent progression to its most malignant form, glioblastoma multiforme (Parsons et al., 2008; Yan et al., 2009).
The surprising involvement of IDH1, IDH2, SDH and FH in cancer has prompted efforts to identify other metabolic genes whose alteration at the genomic level causes or facilitates tumorigenesis. This work recently uncovered phosphoglycerate dehydrogenase (PHGDH), an enzyme required to produce the amino acids serine and glycine from glucose (Figure 3A). Serine and glycine are crucial intermediates in a variety of biosynthetic processes including the production of nucleotides, proteins, glutathione, creatine, and methylated DNA. High levels of PHGDH protein expression relative to normal tissue correlates with disease severity in breast cancer patients (Pollari et al., 2011). Interestingly, like traditional oncogenes, the region of chromosome 1p12 encoding PHGDH is amplified in a significant fraction of tumors, and cell lines derived from these tumors tend to be highly dependent on PHGDH expression (Locasale et al., 2011; Possemato et al., 2011). It remains to be established whether PHGDH is a bona fide oncogene, but over-expressing PHGDH was sufficient to promote anchorage independence in breast epithelial cells and to disturb cell polarity, phenotypes taken to be consistent with malignancy (Locasale et al., 2011). Therefore high levels of PHGDH define another class of genetically-defined metabolic outliers with vulnerabilities that could perhaps be exploited therapeutically in cancer.
Prospects, frontiers and applications
Where do we go from here? Armed with a broader view of metabolism’s influence, it should become increasingly feasible to reverse disease states caused by genetically-determined metabolic dysfunction. One area that should benefit from this new information is the treatment of children with IEMs. Although dietary modifications and other advancements have greatly reduced morbidity and mortality in some of these diseases, the treatments are often invasive and do not fully protect patients from occasional states of severe metabolic decompensation, which can result in permanent disability. It is possible that agents developed to manipulate metabolism in cancer will find applications in other diseases that share some of the same metabolic features, like dysregulated production of lactic acid or altered TCA cycle function. For example, recent work demonstrates that tumor cells with mutations in the TCA cycle or ETC (Mullen et al., 2011), or with hypoxia-induced suppression of oxidative metabolism (Metallo et al., 2011; Scott et al., 2011; Wise et al., 2011) use reductive carboxylation of α-KG to produce citrate and other precursors during growth. In this reaction, NADPH-dependent isoforms of IDH act in reverse with respect to their conventional role as oxidative decarboxylases (Fig. 3B). It is possible that a similar pathway contributes to disease in patients with fixed defects in mitochondrial metabolism, or with perinatal asphyxia, stroke, cardiac ischemia and other conditions involving pathological hypoxia.
Furthermore, our understanding of the pathophysiology of IEMs still draws heavily from the traditional view of intermediary metabolism that focuses primarily on catabolism. The metabolic regulation of macromolecular synthesis, chromatin dynamics and cell renewal is just beginning to be considered, and the interplay between metabolite accumulation, cell signaling and other processes highly relevant to these diseases should be investigated further. For example, the biology of IDH1/2-mutant tumors may provide insight into the effects of global 2-HG accumulation in patients with (L)- or (D)-2-hydroxyglutaric aciduria. This point was emphasized by the recent observation that up to half of patients with (D)-2-hydroxyglutaric aciduria contain heterozygous active-site mutations in IDH2 (Kranendijk et al., 2010). It is unknown whether these patients or patients with other genetic forms of 2-HG aciduria have any of the epigenetic effects observed in IDH1/2-mutant tumors.
Conversely, it should be possible to bring nearly 100 years of clinical experience with IEMs to bear on cancer and other diseases. There is significant interest in developing agents to inhibit the Warburg effect in tumors, but because glycolysis is nearly ubiquitous among human tissues, there is concern about the toxicity of such agents. The extremely large number of well-characterized IEMs in almost every known metabolic pathway may provide a guide as to which types of toxicity to anticipate for some of these targets. SLC2A1 is a HIF-1 transcriptional target and encodes the predominant glucose transporter in cancer cells, GLUT1. GLUT1 is also the major transporter at the blood-brain barrier and is responsible for supplying glucose to the brain. GLUT1 deficiency has a wide phenotypic spectrum but its most common form involves severe central nervous system manifestations including seizures, mental retardation and poor growth of the postnatal brain. Glucose levels in the cerebrospinal fluid may be less than half of normal. It is unknown whether the increased glucose dependence of tumors relative to differentiated tissue will support an acceptable therapeutic window in cancer treatment, but clearly extreme caution would be indicated in attempts to inhibit GLUT1. Other glycolytic IEMs have less severe manifestations. LDHA is a transcriptional target of HIF-1 and c-Myc. It encodes the isoform of lactate dehydrogenase observed in most tumors and is responsible for robust lactate synthesis in cancer cell lines. Severe LDHA deficiency is also a well-characterized human disease, Glycogen Storage Disease Type XI. This disorder causes exercise intolerance, cramps and occasional myoglobinuria in adolescents and adults, but is not associated with severe dysfunction of major homeostatic organs. Perhaps inhibition of LDHA would be tolerated during intermittent cancer therapy. Ongoing population-based studies in metabolomics and genomics should uncover the full breadth of metabolic diversity in healthy humans, and this information may help predict how to target metabolic pathways safely in cancer.
Altered metabolic states in disease also present a tremendous opportunity to develop novel methods in diagnostic imaging. Several platforms for metabolic imaging are already in clinical practice, particularly FDG-PET. But with an ever-expanding knowledge of the interplay between metabolism and disease, it will become more feasible to expand the repertoire of non-invasive techniques to detect diseased tissue and monitor its response to therapy. A number of efforts in this regard are already underway and have a good chance for translation to human patients in the near future. The importance of additional nutrients besides glucose to tumor metabolism has prompted the development of several new PET agents for cancer, including glutamate and glutamine (Koglin et al., 2011; Qu et al., 2011). These agents would add another level of molecular detail to help assess individual tumors, and may improve the detection of tumors invisible on FDG-PET. Magnetic resonance spectroscopy (MRS) at 1.5-Tesla has been used for decades to produce semi-quantitative, non-invasive measurements of abundant metabolite pools in cancer (Glunde and Bhujwalla, 2011). Recently, higher-field human MRI systems have drastically improved the resolution of 1H spectra and made it possible to detect many additional metabolites in human tumors. Successful quantification of significant metabolites like glycine and 2-HG has already been reported for human gliomas (Choi, 2011a; Choi et al., 2011b), and additional metabolites will be added to the list as their relevance in cancer, IEMs, or other diseases is more firmly established. It is also now possible to image metabolites labeled with 13C and to monitor specific enzyme activities in vivo by tracking the transfer of the label from substrate to product. This method requires that polarization of the 13C nuclear spin state be enhanced by transferring to it the high spin polarization of an unpaired electron from a free radical, a process known as hyperpolarization of the 13C nucleus. This produces a transient gain in sensitivity of 13C detection by 10,000-fold or more. Hyperpolarization has been used successfully to quantify oncogene-driven metabolic activities in experimental tumors (Hu et al., 2011). Although still an investigational method, metabolic imaging using hyperpolarization of 13C and other stable isotopes has considerable translational potential in human disease because of its high sensitivity, integration with MRI, lack of the need for radioactive tracers, and in particular its ability to probe the flux of specific enzymatic reactions in vivo (Kurhanewicz et al., 2011).
In fact one of the primary challenges now in human metabolism research – and one of the next frontiers in systems biology – is to analyze the impact of disease on metabolic flux in vivo; that is, to use sensitive and efficient methods to measure the transfer of carbon, nitrogen, etc. along metabolic pathways in live subjects, with and without disease. While methods to catalog and quantify small molecules in biological fluids (metabolomics) are highly informative, they do not present a complete view of metabolism and in some cases can produce misleading results. Newborn screening programs provide a valuable example of this issue. An elevated level of a metabolite in these screening tests almost always reflects decreased rather than increased flux of the pathway, similar to the effect of a traffic light on the accumulation of automobiles (Figure 4A). Furthermore, because metabolism involves so many intersecting pathways, it may be impossible to infer which pathway(s) are affected when abnormal levels of a metabolite are observed. For example, it is unknown which extracellular nutrients are metabolized to 2-HG in IDH1/2-mutant tumors in vivo. One way to address these issues is to combine metabolic pathway analysis with metabolomic studies. Such studies would involve introducing isotopically-labeled nutrients into an animal model or a human patient, and then harvesting metabolites of interest from the blood, urine, breath, or tissue samples (Figure 4B). The total abundance of these metabolites would be measured using a metabolomics platform, then a subset of the most informative metabolites would be studied further by mass spectrometry and/or NMR spectroscopy to determine the abundance and position of isotopic label within each molecule. Similar approaches focusing on a handful of metabolites have already been used successfully in humans to measure gluconeogenesis and the urea cycle in vivo, and to compare metabolism between lung tumors and surrounding tissue (Fan et al., 2009; Landau et al., 1996; Yudkoff et al., 2010). Combined metabolomics-metabolic flux studies, despite their technical challenges, have tremendous value because they could produce a quantitative and comprehensive readout of the variation in metabolic pathway activity, leading to a deeper understanding of metabolic individuality and the biological basis of disease in humans.
Figure 4. Metabolic flux analysis.
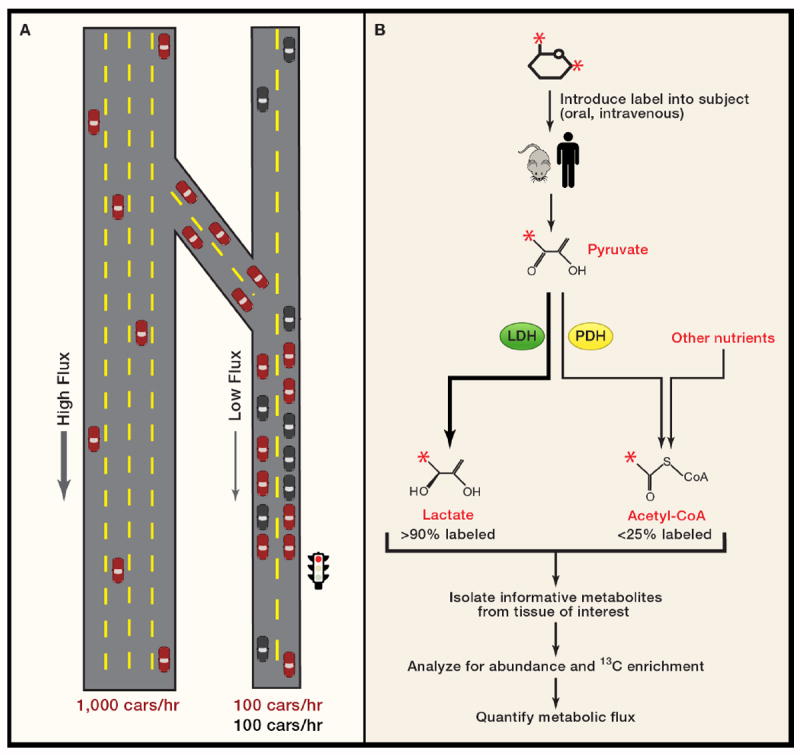
(A) Analysis of metabolism is similar in principle to the analysis of traffic patterns, with many of the same uncertainties. The high “flux” on a four-lane highway leads to a low density of cars, all of which travel unimpeded southward. Upon exiting the highway, drivers experience reduced flux because of the traffic light, akin to mutation or under-expression of a metabolic enzyme. This causes an increased density of cars north of the light. Flux downstream of the block is unimpeded. Note that red cars on the two-lane road also merge with black cars, leading to a reduced fraction of red cars downstream of the intersection. The sum effect of these factors on overall flux is demonstrated by counting the cars that pass the checkered flags. On the highway, 1000 cars, all red, pass in one hour. On the two-lane road, only 200 cars pass, and only half are red.
(B) Simple schematic of metabolic flux analysis. Glucose labeled with 13C at positions 1 and 6 (red asterisks) is given via injection or oral administration to a subject, which metabolizes it. After a period of time, tissue or body fluids are sampled to determine the abundance of various metabolites, the fraction of the metabolite that contains 13C, and the position(s) of 13C within the molecule. Data are acquired using mass spectrometry or NMR spectroscopy. Mathematical models are then applied to translate the data into metabolic flux. In this example, labeling of lactate and acetyl-CoA are examined. The pathways producing these two metabolites diverge at pyruvate. LDH, a highly active enzyme, rapidly converts pyruvate to lactate, resulting in a very high enrichment in the lactate pool in a short time. Meanwhile, two factors conspire to reduce enrichment in acetyl-CoA. First, this pathway involves PDH, a highly regulated and less active enzyme. Second, entry of carbon from unlabeled nutrients contributes to the acetyl-CoA pool, reducing the fraction of acetyl-CoA molecules containing 13C from glucose.
Conclusions
Interest in intermediary metabolism and the rapid improvement of analytical technologies needed to study it have never been higher. Major new findings in disease-oriented metabolic research are being reported on a weekly basis, reinforcing the concept that metabolism pervades every area of biology and pathology. The next decade promises to see continued progress in understanding the metabolic basis of common human diseases, and it is becoming increasingly feasible that some of this knowledge will be translated into novel diagnostic and therapeutic approaches, particularly in cancer.
Acknowledgments
We apologize to colleagues whose work could not be cited owing to space limitations. We thank members of the DeBerardinis laboratory for reading the manuscript critically. R.J.D. is supported by grants from the NIH (1R01 CA157996), the Cancer Prevention and Research Institute of Texas (HIRP100437 and RP101243), the Robert A. Welch Foundation (I-1733) and the Damon-Runyon Cancer Research Foundation. C.B.T. is supported by grants from SU2C, NIH and NCI.
References
- Adam J, Hatipoglu E, O’Flaherty L, Ternette N, Sahgal N, Lockstone H, Baban D, Nye E, Stamp GW, Wolhuter K, et al. Renal Cyst Formation in Fh1-Deficient Mice Is Independent of the Hif/Phd Pathway: Roles for Fumarate in KEAP1 Succination and Nrf2 Signaling. Cancer Cell. 2011;20:524–537. doi: 10.1016/j.ccr.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alderson NL, Wang Y, Blatnik M, Frizzell N, Walla MD, Lyons TJ, Alt N, Carson JA, Nagai R, Thorpe SR, et al. S-(2-Succinyl)cysteine: a novel chemical modification of tissue proteins by a Krebs cycle intermediate. Arch Biochem Biophys. 2006;450:1–8. doi: 10.1016/j.abb.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Alfardan J, Mohsen AW, Copeland S, Ellison J, Keppen-Davis L, Rohrbach M, Powell BR, Gillis J, Matern D, Kant J, et al. Characterization of new ACADSB gene sequence mutations and clinical implications in patients with 2-methylbutyrylglycinuria identified by newborn screening. Mol Genet Metab. 2010;100:333–338. doi: 10.1016/j.ymgme.2010.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baysal BE. Clinical and molecular progress in hereditary paraganglioma. J Med Genet. 2008;45:689–694. doi: 10.1136/jmg.2008.058560. [DOI] [PubMed] [Google Scholar]
- Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PE, Rubinstein WS, Myers EN, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848–851. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- Beadle GW, Tatum EL. Genetic Control of Biochemical Reactions in Neurospora. Proc Natl Acad Sci U S A. 1941;27:499–506. doi: 10.1073/pnas.27.11.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya MK, Smith AM, Ellis TH, Hedley C, Martin C. The wrinkled-seed character of pea described by Mendel is caused by a transposon-like insertion in a gene encoding starch-branching enzyme. Cell. 1990;60:115–122. doi: 10.1016/0092-8674(90)90721-p. [DOI] [PubMed] [Google Scholar]
- Bungard D, Fuerth BJ, Zeng PY, Faubert B, Maas NL, Viollet B, Carling D, Thompson CB, Jones RG, Berger SL. Signaling kinase AMPK activates stress-promoted transcription via histone H2B phosphorylation. Science. 2010;329:1201–1205. doi: 10.1126/science.1191241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnichon N, Briere JJ, Libe R, Vescovo L, Riviere J, Tissier F, Jouanno E, Jeunemaitre X, Benit P, Tzagoloff A, et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet. 2010;19:3011–3020. doi: 10.1093/hmg/ddq206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Sutter BM, Li B, Tu BP. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol Cell. 2011;42:426–437. doi: 10.1016/j.molcel.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chace DH, Kalas TA, Naylor EW. The application of tandem mass spectrometry to neonatal screening for inherited disorders of intermediary metabolism. Annu Rev Genomics Hum Genet. 2002;3:17–45. doi: 10.1146/annurev.genom.3.022502.103213. [DOI] [PubMed] [Google Scholar]
- Childs B, Valle D, Jimenez-Sanchez G. The Inborn Error and Biochemical Individuality. In: Sciver CS, Beaudet AL, Sly WS, Valle D, editors. The Metabolic & Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001. pp. 155–166. [Google Scholar]
- Choi C, et al. Noninvasive detection of 2-hydroxyglutarate in human gliomas in vivo by J difference editing at 3.0 T. Nature Medicine. 2011a In press. [Google Scholar]
- Choi C, Ganji SK, DeBerardinis RJ, Dimitrov IE, Pascual JM, Bachoo R, Mickey BE, Malloy CR, Maher EA. Measurement of glycine in the human brain in vivo by 1H-MRS at 3 T: application in brain tumors. Magn Reson Med. 2011b;66:609–618. doi: 10.1002/mrm.22857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature. 2008;452:181–186. doi: 10.1038/nature06667. [DOI] [PubMed] [Google Scholar]
- Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet. 2005;37:161–165. doi: 10.1038/ng1509. [DOI] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ. Is cancer a disease of abnormal cellular metabolism? New angles on an old idea. Genet Med. 2008;10:767–777. doi: 10.1097/GIM.0b013e31818b0d9b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Cheng T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010;29:313–324. doi: 10.1038/onc.2009.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007;104:19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB. Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev. 2008;18:54–61. doi: 10.1016/j.gde.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichhorst J, Alcorn J, Lepage J, Etter M, Antonishyn NA, Fitterer B, Birch DA, Agopsowicz KL, Ruthnum L, Greenberg CR, et al. Elevated neonatal 3-OH isovalerylcarnitine due to breast milk sources in maternal 3-MCC deficiency. Mol Genet Metab. 2010;101:84–86. doi: 10.1016/j.ymgme.2010.05.002. [DOI] [PubMed] [Google Scholar]
- Erez A, Shchelochkov OA, Plon SE, Scaglia F, Lee B. Insights into the pathogenesis and treatment of cancer from inborn errors of metabolism. Am J Hum Genet. 2011;88:402–421. doi: 10.1016/j.ajhg.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan TW, Lane AN, Higashi RM, Farag MA, Gao H, Bousamra M, Miller DM. Altered regulation of metabolic pathways in human lung cancer discerned by (13)C stable isotope-resolved metabolomics (SIRM) Mol Cancer. 2009;8:41. doi: 10.1186/1476-4598-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C, Pollard PJ, Gottlieb E. Inborn and acquired metabolic defects in cancer. J Mol Med (Berl) 2011a;89:213–220. doi: 10.1007/s00109-011-0728-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C, Zheng L, Folger O, Rajagopalan KN, MacKenzie ED, Jerby L, Micaroni M, Chaneton B, Adam J, Hedley A, et al. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature. 2011b;477:225–228. doi: 10.1038/nature10363. [DOI] [PubMed] [Google Scholar]
- Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762–765. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrod AE. The incidence of alkaptonuria: A study in chemical individuality. Lancet. 1902;ii:1616–1620. [PMC free article] [PubMed] [Google Scholar]
- Gibbons JJ, Abraham RT, Yu K. Mammalian target of rapamycin: discovery of rapamycin reveals a signaling pathway important for normal and cancer cell growth. Semin Oncol. 2009;36(Suppl 3):S3–S17. doi: 10.1053/j.seminoncol.2009.10.011. [DOI] [PubMed] [Google Scholar]
- Gieger C, Geistlinger L, Altmaier E, Hrabe de Angelis M, Kronenberg F, Meitinger T, Mewes HW, Wichmann HE, Weinberger KM, Adamski J, et al. Genetics meets metabolomics: a genome-wide association study of metabolite profiles in human serum. PLoS Genet. 2008;4:e1000282. doi: 10.1371/journal.pgen.1000282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glunde K, Bhujwalla ZM. Metabolic tumor imaging using magnetic resonance spectroscopy. Semin Oncol. 2011;38:26–41. doi: 10.1053/j.seminoncol.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarente L, Franklin H. Epstein Lecture: Sirtuins, aging, and medicine. N Engl J Med. 2011;364:2235–2244. doi: 10.1056/NEJMra1100831. [DOI] [PubMed] [Google Scholar]
- Guzy RD, Sharma B, Bell E, Chandel NS, Schumacker PT. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol Cell Biol. 2008;28:718–731. doi: 10.1128/MCB.01338-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hao HX, Khalimonchuk O, Schraders M, Dephoure N, Bayley JP, Kunst H, Devilee P, Cremers CW, Schiffman JD, Bentz BG, et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science. 2009;325:1139–1142. doi: 10.1126/science.1175689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG. AMP-activated protein kinase--an energy sensor that regulates all aspects of cell function. Genes Dev. 2011;25:1895–1908. doi: 10.1101/gad.17420111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houten SM, Wanders RJ. A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation. J Inherit Metab Dis. 2010;33:469–477. doi: 10.1007/s10545-010-9061-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, Balakrishnan A, Bok RA, Anderton B, Larson PE, Nelson SJ, Kurhanewicz J, Vigneron DB, Goga A. 13C-pyruvate imaging reveals alterations in glycolysis that precede c-Myc-induced tumor formation and regression. Cell Metab. 2011;14:131–142. doi: 10.1016/j.cmet.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illig T, Gieger C, Zhai G, Romisch-Margl W, Wang-Sattler R, Prehn C, Altmaier E, Kastenmuller G, Kato BS, Mewes HW, et al. A genome-wide perspective of genetic variation in human metabolism. Nat Genet. 2010;42:137–141. doi: 10.1038/ng.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacs JS, Jung YJ, Mole DR, Lee S, Torres-Cabala C, Chung YL, Merino M, Trepel J, Zbar B, Toro J, et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell. 2005;8:143–153. doi: 10.1016/j.ccr.2005.06.017. [DOI] [PubMed] [Google Scholar]
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009;23:537–548. doi: 10.1101/gad.1756509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koglin N, Mueller A, Berndt M, Schmitt-Willich H, Toschi L, Stephens AW, Gekeler V, Friebe M, Dinkelborg LM. Specific PET Imaging of xC- Transporter Activity Using a 18F-Labeled Glutamate Derivative Reveals a Dominant Pathway in Tumor Metabolism. Clin Cancer Res. 2011;17:6000–6011. doi: 10.1158/1078-0432.CCR-11-0687. [DOI] [PubMed] [Google Scholar]
- Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- Kranendijk M, Struys EA, van Schaftingen E, Gibson KM, Kanhai WA, van der Knaap MS, Amiel J, Buist NR, Das AM, de Klerk JB, et al. IDH2 mutations in patients with D-2-hydroxyglutaric aciduria. Science. 2010;330:336. doi: 10.1126/science.1192632. [DOI] [PubMed] [Google Scholar]
- Krebs HA. Some aspects of the regulation of fuel supply in omnivorous animals. Adv Enzyme Regul. 1972;10:397–420. doi: 10.1016/0065-2571(72)90025-8. [DOI] [PubMed] [Google Scholar]
- Kurhanewicz J, Vigneron DB, Brindle K, Chekmenev EY, Comment A, Cunningham CH, Deberardinis RJ, Green GG, Leach MO, Rajan SS, et al. Analysis of cancer metabolism by imaging hyperpolarized nuclei: prospects for translation to clinical research. Neoplasia. 2011;13:81–97. doi: 10.1593/neo.101102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau BR, Wahren J, Chandramouli V, Schumann WC, Ekberg K, Kalhan SC. Contributions of gluconeogenesis to glucose production in the fasted state. J Clin Invest. 1996;98:378–385. doi: 10.1172/JCI118803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Nakamura E, Yang H, Wei W, Linggi MS, Sajan MP, Farese RV, Freeman RS, Carter BD, Kaelin WG, Jr, et al. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: developmental culling and cancer. Cancer Cell. 2005;8:155–167. doi: 10.1016/j.ccr.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, Heffron G, Metallo CM, Muranen T, Sharfi H, et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet. 2011;43:869–874. doi: 10.1038/ng.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–248. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM. p53 regulates mitochondrial respiration. Science. 2006;312:1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- McKnight SL. On getting there from here. Science. 2010;330:1338–1339. doi: 10.1126/science.1199908. [DOI] [PubMed] [Google Scholar]
- Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, Kelleher JK, Vander Heiden MG, Iliopoulos O, Stephanopoulos G. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2011 doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroni I, Bugiani M, D’Incerti L, Maccagnano C, Rimoldi M, Bissola L, Pollo B, Finocchiaro G, Uziel G. L-2-hydroxyglutaric aciduria and brain malignant tumors: a predisposing condition? Neurology. 2004;62:1882–1884. doi: 10.1212/01.wnl.0000125335.21381.87. [DOI] [PubMed] [Google Scholar]
- Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ. Reductive carboxylation supports growth in tumor cells with defective mitochondria. Nature. 2011 doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Lomb DJ, Haigis MC, Guarente L. SIRT5 Deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell. 2009;137:560–570. doi: 10.1016/j.cell.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi A, Wong JC, Petillo D, Roossien D, Perrier-Trudova V, Whitten D, Min BW, Tan MH, Zhang Z, Yang XJ, et al. An Antioxidant Response Phenotype Shared between Hereditary and Sporadic Type 2 Papillary Renal Cell Carcinoma. Cancer Cell. 2011;20:511–523. doi: 10.1016/j.ccr.2011.08.024. [DOI] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, Guertin DA, Madden KL, Carpenter AE, Finck BN, et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146:408–420. doi: 10.1016/j.cell.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrak B, Zhao H, Qi H, Quinn C, Gao E, Boyer JG, Concha N, Brown K, Duraiswami C, Wooster R, et al. A tale of two subunits: how the neomorphic R132H IDH1 mutation enhances production of alphaHG. Biochemistry. 2011;50:4804–4812. doi: 10.1021/bi200499m. [DOI] [PubMed] [Google Scholar]
- Pollari S, Kakonen SM, Edgren H, Wolf M, Kohonen P, Sara H, Guise T, Nees M, Kallioniemi O. Enhanced serine production by bone metastatic breast cancer cells stimulates osteoclastogenesis. Breast Cancer Res Treat. 2011;125:421–430. doi: 10.1007/s10549-010-0848-5. [DOI] [PubMed] [Google Scholar]
- Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, Griffiths JR, Chung YL, Schulze A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008;8:224–236. doi: 10.1016/j.cmet.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, Sethumadhavan S, Woo HK, Jang HG, Jha AK, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346–350. doi: 10.1038/nature10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu W, Zha Z, Ploessl K, Lieberman BP, Zhu L, Wise DR, Thompson CB, Kung HF. Synthesis of optically pure 4-fluoro-glutamines as potential metabolic imaging agents for tumors. J Am Chem Soc. 2011;133:1122–1133. doi: 10.1021/ja109203d. [DOI] [PubMed] [Google Scholar]
- Robinson BH, MacKay N, Chun K, Ling M. Disorders of pyruvate carboxylase and the pyruvate dehydrogenase complex. J Inherit Metab Dis. 1996;19:452–462. doi: 10.1007/BF01799106. [DOI] [PubMed] [Google Scholar]
- Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7:77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. 2010;29:625–634. doi: 10.1038/onc.2009.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Regulation of Metabolism by Hypoxia-Inducible Factor 1. Cold Spring Harb Symp Quant Biol. 2011 doi: 10.1101/sqb.2011.76.010678. [DOI] [PubMed] [Google Scholar]
- Shanware NP, Mullen AR, DeBerardinis RJ, Abraham RT. Glutamine: pleiotropic roles in tumor growth and stress resistance. J Mol Med (Berl) 2011;89:229–236. doi: 10.1007/s00109-011-0731-9. [DOI] [PubMed] [Google Scholar]
- Scott DA, Richardson AD, Filipp FV, Knutzen CA, Chiang GG, Ronai ZA, Osterman AL, Smith JW. Comparative metabolic flux profiling of melanoma cell lines: beyond the Warburg effect. J Biol Chem. 2011;286:42626–42634. doi: 10.1074/jbc.M111.282046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley CA, Lieu YK, Hsu BY, Burlina AB, Greenberg CR, Hopwood NJ, Perlman K, Rich BH, Zammarchi E, Poncz M. Hyperinsulinism and hyperammonemia in infants with regulatory mutations of the glutamate dehydrogenase gene. N Engl J Med. 1998;338:1352–1357. doi: 10.1056/NEJM199805073381904. [DOI] [PubMed] [Google Scholar]
- Sudarshan S, Sourbier C, Kong HS, Block K, Valera Romero VA, Yang Y, Galindo C, Mollapour M, Scroggins B, Goode N, et al. Fumarate hydratase deficiency in renal cancer induces glycolytic addiction and hypoxia-inducible transcription factor 1alpha stabilization by glucose-dependent generation of reactive oxygen species. Mol Cell Biol. 2009;29:4080–4090. doi: 10.1128/MCB.00483-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhre K, Shin SY, Petersen AK, Mohney RP, Meredith D, Wagele B, Altmaier E, Deloukas P, Erdmann J, Grundberg E, et al. Human metabolic individuality in biomedical and pharmaceutical research. Nature. 2011;477:54–60. doi: 10.1038/nature10354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan DT, Sullivan MC. Transport defects as the physiological basis for eye color mutants of Drosophila melanogaster. Biochem Genet. 1975;13:603–613. doi: 10.1007/BF00484918. [DOI] [PubMed] [Google Scholar]
- Swinnen JV, Brusselmans K, Verhoeven G. Increased lipogenesis in cancer cells: new players, novel targets. Curr Opin Clin Nutr Metab Care. 2006;9:358–365. doi: 10.1097/01.mco.0000232894.28674.30. [DOI] [PubMed] [Google Scholar]
- Takahashi H, McCaffery JM, Irizarry RA, Boeke JD. Nucleocytosolic acetyl-coenzyme a synthetase is required for histone acetylation and global transcription. Mol Cell. 2006;23:207–217. doi: 10.1016/j.molcel.2006.05.040. [DOI] [PubMed] [Google Scholar]
- Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson CB. Metabolic enzymes as oncogenes or tumor suppressors. N Engl J Med. 2009;360:813–815. doi: 10.1056/NEJMe0810213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, Leigh I, Gorman P, Lamlum H, Rahman S, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002;30:406–410. doi: 10.1038/ng849. [DOI] [PubMed] [Google Scholar]
- Tong WH, Sourbier C, Kovtunovych G, Jeong SY, Vira M, Ghosh M, Romero VV, Sougrat R, Vaulont S, Viollet B, et al. The Glycolytic Shift in Fumarate-Hydratase-Deficient Kidney Cancer Lowers AMPK Levels, Increases Anabolic Propensities and Lowers Cellular Iron Levels. Cancer Cell. 2011;20:315–327. doi: 10.1016/j.ccr.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trowell HC. Ants distinguish diabetes mellitus from diabetes insipidus. Br Med J (Clin Res Ed) 1982;285:217. doi: 10.1136/bmj.285.6336.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu BP, Kudlicki A, Rowicka M, McKnight SL. Logic of the yeast metabolic cycle: temporal compartmentalization of cellular processes. Science. 2005;310:1152–1158. doi: 10.1126/science.1120499. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Locasale JW, Swanson KD, Sharfi H, Heffron GJ, Amador-Noguez D, Christofk HR, Wagner G, Rabinowitz JD, Asara JM, et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science. 2010;329:1492–1499. doi: 10.1126/science.1188015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanders RJ, Waterham HR. Biochemistry of mammalian peroxisomes revisited. Annu Rev Biochem. 2006;75:295–332. doi: 10.1146/annurev.biochem.74.082803.133329. [DOI] [PubMed] [Google Scholar]
- Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, Zhao W, Yao Y, et al. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science. 2010;327:1004–1007. doi: 10.1126/science.1179687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Lu C, Mancuso A, Lemons JM, Ryczko M, Dennis JW, Rabinowitz JD, Coller HA, Thompson CB. The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev. 2010;24:2784–2799. doi: 10.1101/gad.1985910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise DR, Ward PS, Shay JE, Cross JR, Gruber JJ, Sachdeva UM, Platt JM, Dematteo RG, Simon MC, Thompson CB. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of a-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci U S A. 2011;108:19611–19616. doi: 10.1073/pnas.1117773108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Xiao MT, Liu LX, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yudkoff M, Ah Mew N, Daikhin Y, Horyn O, Nissim I, Payan I, Tuchman M. Measuring in vivo ureagenesis with stable isotopes. Mol Genet Metab. 2010;100(Suppl 1):S37–41. doi: 10.1016/j.ymgme.2010.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, et al. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]