

Abstract
Skeletal muscle toxicity is the primary adverse effect for statins. In this review, we summarize current knowledge regarding the genetic and non-genetic determinants of risk for statin induced myopathy. Many genetic factors were initially identified through candidate-gene association studies (CGAS) limited to pharmacokinetic (PK) targets. Through genome-wide association studies (GWAS), it has become clear that SLCO1B1 is among the strongest PK predictors of myopathy risk. GWAS have also expanded our understanding of pharmacodynamic (PD) candidate genes, including RYR2. It is anticipated that deep re-sequencing efforts will define new loci with rare variants that contribute as well, and sophisticated computational approaches will be needed to characterize gene-gene (GxG) and gene-environment (GxE) interactions. Beyond environment, race is a critical covariate, and its influence is only partly explained by geographic differences in the frequency of known PD and PK variants. As such, admixture analyses will be essential to a full understanding of statin-induced myopathy.
Keywords: statin, myopathy, pharmacokinetic, pharmacodynamics, candidate gene studies, genome-wide studies, ancestry, admixture, covariate, gene-gene interactions
Statins are among the most commonly prescribed drugs in the industrialized world [1]. They inhibit HMG Coenzyme A reductase (HMGCR), the rate-limiting enzyme in cholesterol biosynthesis (Figure 1) [2, 3], and they reduce the frequency of coronary artery disease in patients at risk [4]. There are seven statins currently available within the class. The first statin to be approved by the U.S. Food and Drug Administration was lovastatin in 1987, followed by simvastatin, 1988; pravastatin, 1991; fluvastatin, 1994; atorvastatin, 1997; rosuvastatin, 2003; and pitavastatin, 2009 [5, 6]. One additional agent, cerivastatin, was released (in 1998) and subsequently withdrawn from the market due to a markedly increased frequency of muscle toxicity. Between 1998 and 2001, forty reported cases of muscle toxicity due to cerivastatin use were reported to be fatal [7].
Figure 1.
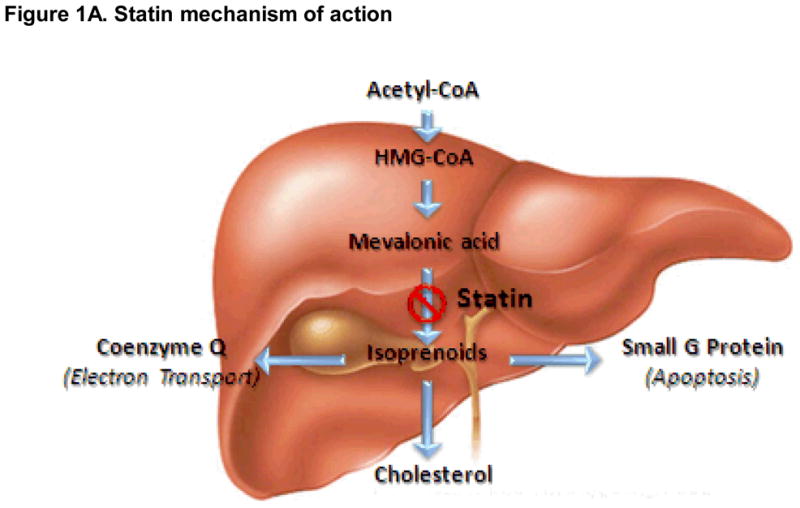

Figure 1A. Pharmacodynamic (PD) factors influencing risk for statin-induced myopathy. Simplified view of key candidate gene products influencing statin mechanism of action. Arrows represent primary direction of each biosynthetic reaction.
Figure 1B. Interactive network for Candidate Pharmacodynamic (PD) Genes. Genes with red nodes represent Hub genes in our analysis; others are generated through the network analysis from Ingenuity Pathways Knowledge Base (http://www.ingenuity.com). Edges are displayed with labels that describe the nature of the relationship between the nodes. All edges are supported by at least one reference from the literature, or from canonical information stored in the Ingenuity Pathways Knowledge Base. The lines between genes represent known interactions, with solid lines representing direct interactions and dashed lines representing indirect interactions. Nodes are displayed using various shapes that represent the functional class of the gene product (see http://www.ingenuity.com).
In general, statins are considered safe [8]. Statin-related adverse drug reactions (ADRs) include neurocognitive complaints, musculoskeletal complaints, and very rarely nephrotoxicity or hepatotoxicity [9]. Musculoskeletal problems represent the most common form of statin intolerance. Over the past two decades, statin use has expanded dramatically, and statin-induced muscle toxicity is becoming more fully characterized [6, 10–12]. The clinical presentation of this ADR varies widely, from mild myalgias (focal or diffuse) to rhabdomyolysis (severe skeletal muscle damage accompanied by acute kidney injury) [13]. In the context of clinical practice, statin-induced muscle toxicity is diagnosed based upon two variables: muscle pain and circulating levels of a relatively non-specific muscle enzyme, creatine kinase (CK). CK level is often used as a marker for severity for statin-related muscle damage [9, 14]. Because CK levels can occasionally be elevated in the absence of pain, accurate risk prediction models are needed for preventing rhabdomyolysis [15].
Clinical Determinants of Risk
The true frequency of statin-related muscle damage is difficult to quantify. Randomized clinical trials often underestimate the frequency of this ADR because patients with symptoms of intolerance are typically excluded during the run-in period [16–18]. Frequency estimates derived from databases maintained by regulatory agencies also tend to underestimate the problem because such event-reporting is voluntary [19]. Thus, large observational databases linked to electronic medical records may represent the most accurate way of quantifying statin-induced muscle toxicity within the community [20]. Mild myalgias related to statin use are quite common, reported to occur at frequencies ranging from 1% to 10% [11]. Myalgias accompanied by elevation in serum CK level occur at a much lower frequency [21, 22]. The Health Improvement Network (THIN) and MediPlus databases report an annual incidence approaching 700 per million exposed per year, for intermediate myotoxicity [23]. Rhabdomyolysis represents the most severe and potentially lethal form of this ADR [9, 14], and many clinicians consider an elevation in serum CK level >50-fold upper limit of normal (~10,000 Units per liter) both necessary and sufficient to make the diagnosis. Graham and colleagues surveyed more than 250,000 statin-exposed patients, and reported rhabdomyolysis rates of 0.000044 events per person-year [24]. Similar rates have been observed for more than 100,000 first-time statin users followed in the United Kingdom over a course of 20 months [25]. In general, the frequency of severe muscle damage is low in the context of monotherapy (i.e., when statins are used in the absence of other medications).
Risk and severity increase, however, in the presence of co-medications known to influence statin disposition [9, 22, 26]. Examples include medications altering the expression or activity of CYP3A4/5, UGT1A1, ABCB1, and OATP1B1 (gene name SLCO1B1). Each of these determinants of statin myopathy risk is discussed further below, within the context of pharmacokinetic candidate genes. Additional clinical factors influencing the severity of statin-induced myopathy include advanced age, small body mass index, female gender, metabolic co-morbidities, and vigorous physical exercise [20, 27–31] (Table 1). Age is a particularly strong contributor. By studying the medical records of more than 250,000 patients, Schech et al. reported that statin users ≥65 years of age have four times the risk of hospitalization due to rhabdomyolysis than younger statin users [32]. Some of this difference can be attributed to age-related changes in body composition, and increased frequency of relevant co-morbidities (e.g., chronic liver disease or chronic kidney disease). Female gender also appears to increase risk, approximately 2-fold, perhaps due to smaller vascular volumes and reduced muscle mass resulting in greater tissue drug exposure per statin dose [33].
Table 1.
Clinical Determinants of Statin-Induced Myopathy.
| Clinical Determinants | form of toxicity | magnitude of effect (odds ratio [95% confidence interval]) | significant (p-value) | references | case/control | statin |
|---|---|---|---|---|---|---|
| Age | rhabdomyolysis | 4.36 [1.5, 14.1] | [32] | 252460 new users | ||
| [117] | ||||||
| [118] | ||||||
| Race | rhabdomyolysis | [119] | ||||
| Asian | two fold greater plasma exposure in Asian than in European | [35] | rosuvastatin | |||
| FDA Public Health Advisory on Crestor (rosuvastatin). Media release, 2005 | ||||||
| African American | incidence of CK elevation | higher in African American than white | <0.001 | [120] | lovastatin | |
| [121] | ||||||
| Female gender | 3.1 [1.9–4.9] | <0.001 | [122] | 76 | atorvastatin, rosuvastatin | |
| discontinuation, myalgia, or CK >3x ULN | <0.01 | [96] | ||||
| Clicnial commorbidities | ||||||
| History of muscle pain with another lipid lowering therapy | 10.12 | <0.0001 | [123] | 7924 (cohort) | ||
| Unexplained cramps | 4.14 | <0.0001 | ||||
| History of elevated CK | 2.04 | <0.0001 | ||||
| Family history of muscular symptoms with lipid lowering therapy | 1.89 | 0.017 | ||||
| Hypothyroidism | 1.71 | 0.017 | ||||
| McArdle disease | Myopathy | [78] | 136/116 | Multiple statins | ||
| Malignant hyperthemiaa | Myopathy | |||||
| Diabetes mellitus | 0.01 | [124] | 338 | |||
| Ongoing infection | 0.005 | |||||
| Renal disease | 0.002 | |||||
| Hyperuricaemia | 0.02 | |||||
| Alcohol overconsumption | 0.004 | |||||
| Trauma | 0.002 | |||||
| Intense physical activity | [125] | 22 total | ||||
| [27] | ||||||
| Concomitant medications | ||||||
| CYP3A4 inhibitor (with simvastatin) | [126] | simvastatin | ||||
| Inhibitors of CYP3A4 and SLCO1B1, including traconazole, ritonavir, verapamil and diltiazem | [42] | |||||
| Antiretroviral drug and statin (inhibitor of CYP) | [127] | |||||
| Gemfibrozil and pravastatin | [128] | pravastatin | ||||
| Gemfibrozil and simvastatin | [46] | |||||
| Fibrates | [118] | |||||
McClure and colleagues have shown that dose is also a very strong predictor of risk; the incidence rate for myotoxicity was roughly 10-fold greater in patients on high-dose statin therapy (i.e., defined as a dose equivalent to 40 mg of lovastatin daily or greater) [22] This observation has been replicated in an independent clinical practice-based cohort [27]. From the records of nearly 2,000,000 unique individuals served by a single comprehensive system of care, 213 validated cases of statin-induced muscle toxicity were enrolled in a population-based study of genetic risk determinants (http://www.pharmgkb.org/contributors/pgrn/parc_profile.jsp). Within this observational cohort, the relationship between simvastatin dose and severity of myotoxicity was dose-dependent [20, 27, 34]. In 2011, the U.S. Food and Drug Administration (FDA) announced a new safety recommendation for high-dose simvastatin. FDA recommended that the 80 mg dose of simvastatin should be limited to patients “…who have been taking this dose for >12 months and have not experience any muscle toxicity” (www.fda.org).
The FDA has also recommended limiting the dose of some statins based upon major continental race (FDA Public Health Advisory on rosuvastatin; Media release March 2, 2005; http://www.accessdata.fda.gov/drugsatfda_docs/label/2005/21366slr005lbl.pdf). This recommendation was based upon (1) the observation that patients of Asian ancestry exhibit 2-fold increase in AUC for rosuvastatin, compared to patients of European ancestry, following single dose exposure [35], and (2) the observation that patients of Asian ancestry have greater lipid lowering efficacy at lower doses of rosuvastatin, compared to patients of European ancestry [36]. After reviewing the evidence from these and similar studies, the FDA concluded that Asian Americans were one of three important groups with an elevated risk/benefit ratio (the others were patients on immune suppression and patients with severe kidney failure). The FDA now recommends limiting patients of Asian ancestry to a 5 mg starting dose for rosuvastatin.
Figure 2.


Figure 2A. Pharmacokinetic (PK) factors influencing risk for statin-induced myopathy. Simplified view of key candidate gene products influencing the absorption, distribution, metabolism and elimination of statins. Arrows represent primary direction of each biotransformation reaction.
Figure 2B. Interactive network for Candidate Pharmacokinetic (PK) Genes. Genes with red nodes represent Hub genes in our analysis; others are generated through the network analysis from Ingenuity Pathways Knowledge Base (http://www.ingenuity.com). Edges are displayed with labels that describe the nature of the relationship between the nodes. All edges are supported by at least one reference from the literature, or from canonical information stored in the Ingenuity Pathways Knowledge Base. The lines between genes represent known interactions, with solid lines representing direct interactions and dashed lines representing indirect interactions. Nodes are displayed using various shapes that represent the functional class of the gene product (see http://www.ingenuity.com).
Challenges and solutions
Differences in the overall genetic architecture between major continental races can cause variability within the clinical response to statins [29–32]. As noted below, some of the variability in myopathy risk is due to race-specific differences in the frequency of pharmacodynamic and pharmacokinetic candidate gene variants. However, a large fraction of the race-dependent variance in this trait remains unexplained. Failure to account for population confounding in the characterization of statin-induced myopathy may therefore lead to errors in inferring true association. Practice-based cohorts of diverse ancestry will be essential to our full understanding of myopathy risk in the general population [22, 37].
Within the general community, population confounding is introduced by groups of individuals that have some degree of reproductive isolation from the rest of the population, and for which allele frequencies are likely to be different from the population as a whole [37]. Any given study may therefore be subjected to population confounding due to a) population stratification, or b) admixture. Stratification occurs when cases and controls are unintentionally drawn from two or more population subgroups. Polymorphisms that genetically mark the high-risk subgroup (i.e., found by chance at a higher frequency in that subgroup) can erroneously appear to be associated with the trait [37]. Further, gene variants occurring at a very low frequency are typically not shared across divergent populations because they have either arisen relatively recently or because their frequencies have been influenced by population history (e.g., the out-of-Africa expansion or natural selection)[38]. Admixture is a common form of gene flow between populations. It refers to the process in which two or more genetically and phenotypically diverse populations with different allele frequencies mate and form a new, mixed or ‘hybrid’ population [37]. A classic example of an admixed population in humans is the African-American population. As a result of the genetic admixture, the African-American population contains stretches of DNA as large as 20–30 centiMorgan (cM) that resemble mosaics of chromosomal segments, or ancestral blocks.
Markers with large allele frequency differences between ancestral populations, known as ancestry informative markers (AIMs), can be used to control for population confounding arising due to variations in background ancestry [39]. It will therefore be critical to adequately control for population stratification and admixture when analyzing pharmacogenetic variants [40]. Although three different methods (genomic control, structured association and principle component analysis) are used to correct for confounding, good epidemiology study design is the most efficient way to avoid confounding in population-based cohorts. No amount of adjustment can correct for poor study design where population subgroups for cases and controls do not overlap.
Genetic Determinants of Risk
The degree to which genetic factors contribute to inter-individual variability in myopathy risk has been an active area of investigation for more than a decade. Much of our initial understanding came from candidate gene association studies (CGAS), particularly within the context of pharmacokinetic (PK) targets. In general, candidate gene studies are based on prior knowledge [41]. Because statin disposition varies drug-by-drug, reflecting subtle differences in uptake, oxidation, conjugation, and efflux, models used to predict the impact of PK genes on myopathy risk require flexibility.
Pharmacokinetic candidate genes
The clinical severity of statin-induced muscle toxicity is clearly influenced by variability in enzymes modulating statin disposition (absorption, distribution, metabolism and elimination, ADME) (Figure 2) (Table 2) [26]. While many statins undergo phase I oxidation (atorvastatin, fluvastatin, lovastatin, simvastatin), the impact of phase I oxidation on others (pitavastatin, pravastatin, rosuvastatin) is very limited [42]. Atorvastatin and lovastatin are oxidized primarily by the cytochrome P450 (CYP) 3A4 and 3A5 enzymes. Although the same enzymes are known to be responsible for the metabolism of simvastatin, fluvastatin, and cerivastatin, the oxidation of fluvastatin (and possibly pitavastatin) is influenced by CYP2C9 [43], whereas both simvastatin and cerivastatin metabolites are further oxidized by CYP2C8 [43–46]. Although controversial, the oxidation of simvastatin metabolites may also be influenced by the highly polymorphic CYP2D6 [45, 47–50]. Each of these genes - CY3A4/5, CYP2C8/9, and CYP2D6 – is polymorphic, and variability in phase I drug metabolizing enzyme genes might therefore account for patient-to-patient differences in muscle related ADRs.
Table 2.
Genetic Determinants of Statin-Induced Myopathy
| Genetic Determinants | form of toxicity | magnitude of effect (odds ratio [95% confidence interval]) | significant (p-value) | References | case/control | statin |
|---|---|---|---|---|---|---|
| PK genes | ||||||
| CYP3A5 | myalgia | p=0.025 without gemfibrozil, p=0.01 without gemfibrozil and niacin | [52] | 69/68 | atorvastatin | |
| CYP2C8 | rhabdomyolysis | [129] | 1 | cerivastatin | ||
| rhabdomyolysis | [130] | 1 | cerivastatin | |||
| CYP2D6 | myopathy/myalgia | Discovery O.R. = 3.6; Validation O.R. = 2.7 | Discovery P=0.004; Validation P=0.036;total P=0.001 | [51] | 75/188 | atorvastain |
| ABCB1 | myalgia | <0.05 | [62] | 15/99 | simvastatin | |
| SLCO1B1 | rhabdomyolysis | [129] | 1 | cerivastatin | ||
| myopathy | O.R. = 4.5 [2.6 to 7.7] per copy of the C allele; O.R. = 16.9 [4.7 to 61.1] in CC as compared with TT | [95] | 85/90 | simvastatin | ||
| myopathy | [131] | 1 | pravastatin | |||
| rhabdomyolysis | O.R. = 1.89 [1.40–2.56] (rs4149056) | [102] | 185/732 | cerivastatin | ||
| severe myopathy | 0.042 | [132] | 25/84 | simvastatin | ||
| myopathy | [133] | 1 | pravastatin | |||
| severemyopathy | O.R. = 2.05 (rs4149056) | 0.043 | [97] | 816/1275 | ||
| O.R. = 0.71 (rs2306283) | 0.026 | |||||
| muscular intolerance | O.R. = 2.7 [1.3–4.9] for atorvastatin | <0.001 | [122] | 76 | atorvastatin, rosuvastatin | |
| discontinuation, myalgia, or CK >3x ULN | 0.01 | [96] | 99 | atorvastatin, simvastatin, pravastatin | ||
| PD genes | ||||||
| CPT 2 | myopathy | [78] | 136/116 | multiple statins, including cerivastatin | ||
| COQ2 | myopathy | O.R. = 2.42 [0.99 to 5.89], O.R. = 2.33 [1.13 to 4.81] and O.R. = 2.58 [1.26 to 5.28] for SNP1 and SNP2, as well as diplotypes (two-SNP haplotypes) created from both, respectively. | [80] | 133/158 | multiple statins,including rosuvastatin and atorvastatin | |
| muscular intolerance | O.R. atorvastatin = 3.1 [1.9–6.4]; O.R. rosuvastatin = 2.6 [1.7–4.4] | atorvastatin, p < 0.001; rosuvastatin, p < 0.001 | [122] | 76 | atorvastatin, rosuvastatin | |
| RYR2 | rhabdomyolysis | O.R. = 0.48 [0.36–0.63] (rs2819742) | 1.74E-07 | [102] | 185/732 | cerivastatin |
The potential effect of CYP2D6 polymorphisms on statin intolerance was first explored in a cohort of 88 participants [49]. Frudakis et al. demonstrated that CYP2D6*4 was associated with the frequency of statin induced muscle events (p = 0.001), independent of demographic variables [51]. Kaspera et al. sequenced CYP2C8 in 126 rhabdomyolysis cases, and identified 12 novel single nucleotide polymorphisms (SNPs) with a potential to alter CYP2C8 enzyme function [44]. A common splice variant in CYP3A5 has been associated with the magnitude of CK elevation by our group, specifically within the context of atorvastatin [52]. The strength of this latter association was dependent upon the presence of concomitant medications known to interact with statins through processes other than phase I oxidation (e.g., phase II conjugation) [52].
Many statins and hydroxy-statin derivatives undergo further modification, through UDP-glucuronosyl transferase 1 (UGT1) - dependent processes (Figure 2) [53]. It is therefore likely that genetic variability in the UGT1 enzyme family would contribute to myopathy risk as well. The entire family of UGT1 gene products (UGT1A1-12) is derived from the same locus. Because atorvastatin δ-lactone is associated with toxicity, Riedmaier et al. studied the role of UGTs in atorvastatin lactonization [54]. After analyzing 150 human liver samples, they showed that atorvastatin lactonization is associated with both UGT1A3 immunoreactive protein levels and mRNA levels. Genetic analysis UGT1A3 mRNA and protein levels are altered by the UGT1A3*2 allele, a variant also shown to influence the rate of atorvastatin lactonization. Interestingly, expression level of UGT1A3 mRNA was also positively influenced by the well-defined UGT1A1 variant allele – UGT1A1*28 (p<0.001). This variant has previously been associated with clinical outcome within the context of a number of drug classes [55].
Beyond phase I (oxidative) and phase II (conjugative) statin metabolism, variability in membrane transport also contributes strongly to myopathy risk. The organic anion transporting polypeptide OATP-1B1 (gene name SLCO1B1) is expressed on the sinusoidal membrane of human hepatocytes and facilitates the hepatic uptake of most statins. Other relevant hepatic uptake transporters include OATP1B3, OATP2B1, OATP1A2 and the sodium-dependent taurocholate co-transporting polypeptide, NTCP [56, 57]. Genetic variability in membrane transport clearly influences statin-related clinical outcome (Table 2). Polymorphisms in candidate solute transporter genes are associated with the altered hepatic uptake of simvastatin [58] and pravastatin [59]. Much of this variability can be attributed to two coding variants in SLCO1B1 (Asn130Asp and Val174Ala) [60]. As outlined in a later section, the latter variant has since been shown to be highly informative in determining risk for the development of toxicity to simvastatin.
As shown in Figure 2, other transporters can influence the development and severity of statin-induced muscle toxicity as well. Many statins are substrates for efflux transporters such as multidrug resistance protein MDR1 (gene name ABCB1) or multidrug resistance-associated protein MRP2 (gene name ABCC2) [61]. Located on the canalicular membrane of hepatocytes, these ATP-binding cassette proteins mediate the final step in the hepatobiliary clearance of statins. It therefore seems likely that variability in the activity of these transporters would alter the course of statin related clinical events. Genotype-phenotype association study performed in a cohort of 116 hypercholesterolemic patients has revealed that ABCB1 variants influence the efficacy of simvastatin. The same analysis also revealed that ABCB1 variants (1236T, 2677 non-G and 3435T) were less frequent in patients with adverse muscle effects [62].
Change in the activity of another efflux protein from the same family (gene name ABCG2) further alters the pharmacokinetics of most statins [63, 64]. This is particularly true for atorvastatin and rosuvastatin, two of the most potent drugs in the class [63, 64]. In a recent study of 305 Chinese patients treated with 10 mg rosuvastatin daily, one SNP in ABCG2 (rs2231142) was strongly associated with statin efficacy [65]. Participants carrying a CC genotype at rs2231142 had a 6.9% greater reduction in LDL cholesterol levels compared to those with AA genotypes. Because the frequency of this genotype differs widely by race, it may explain a significant portion of the increased myopathy risk observed in Asians [36, 65]. Other ABCG2 variants may be involved as well [66, 67].
Pharmacodynamic candidate genes
The rate-limiting enzyme in cholesterol biosynthesis is HMGCR. Statins inhibit the activity of this enzyme. It has been demonstrated in multiple studies that genetic variants in the HMGCR gene are important determinants for statin efficacy [68–74]. Although it seems reasonable to assume that those polymorphisms would also alter risk of statin toxicity, Frudakis et al. failed to observe any association between HMGCR variants and statin myopathy in a well-designed case-control study (263 samples) [51]. Thus, alteration in cholesterol biosynthesis alone might not be sufficient to induce myopathy [75]. Inhibition of HMGCR also attenuates the levels of many distal intermediates (Figure 1A) [3]. After the generation of mevalonic acid, the pathway subsequently produces geranyl pyrophosphate (10 carbons), farnesyl pyrophosphate (15 carbons) and geranylgeranyl pyrophosphate (20 carbons). The isoprenoid side chains of these biosynthetic intermediates can transfer farnesyl or geranyl moieties to C-terminal cysteine(s) of target proteins, through a process call “protein prenylation.”
Because prenylation is necessary for synthesizing the side chain within uibiquinone (coenzyme Q10, CoQ10), statins may disturb the integrity of electron transport within the mitochondria (Figure 1A). As such, mitochondrial dysfunction due to altered levels of CoQ10 has been suggested as a potential mechanism for statin myopathy. The role of mitochondrial dysfunction in the pathogenesis of statin-induced myopathy is supported by extensive pathological evidence [76–78]. Vladutiu et al. have demonstrated that 52% of muscle biopsies from patients with statin related myalgias revealed mitochondrial abnormalities, and 31% of these biopsies revealed multiple defects [78]. Further work by the same group identified variants in adenosine monophosphate deaminase (AMPD1), myophosphorylase (PYGM) and carnitine palmitoyltransferase II (CPT2) as contributors to risk [78]. Additional pharmacodynamic variants contributing to myopathy risk have been reviewed by Peters et al. (including subclinical McArdle disease) (Table 2) [79]. Oh et al. genotyped two SNPs in COQ2 (encoding an important enzyme in CoQ10 biosynthesis) in 133 statin-induced myopathy cases and 158 matched controls [80]. Both SNPs were associated with increased risk of statin intolerance, and a haplotype based on these variants yielded an even stronger association (2.5-fold increase in risk) [80]. These observations have led a number of investigators to explore the possibility that statin myopathy could be attenuated by coadministration of oral CoQ10 [81]. The trials, however, have been small, and the results have been disappointing; e.g., Young et al. randomized 44 patients, who had previously failed statins due to muscle pain, to receive simvastatin with either placebo or CoQ10 supplementation (200 mg/day). No difference in myalgia score was observed between the treatment groups. Thus, oral CoQ10 did not improve statin tolerance [82].
Prenylation also influences the balance between myocyte viability and apoptosis Statin-induced apoptosis has been demonstrated in vitro, using myotubes [83], myoblasts [84], and differentiated primary human skeletal muscle cells [85]. This effect can be reproduced by geranyl-geranyl-transferase inhibitors, and rescued by replacement of mevalonic acid [83]. Compelling evidence suggests that statins cause apoptosis in skeletal muscle by disrupting the prenylation of small G proteins like Rho [84], Rab [86], and Rap [83]. For example, statins induce apoptosis at concentrations that suppress the prenylation of Rap1a (a 21kD GTPase) [83], and Itagaki et al. have shown that this process is accompanied by the redistribution of small G proteins in myoblasts [87]. It remains unclear, however, whether the altered prenylation of small G-proteins is necessary and sufficient to produce myopathy in vivo, or whether myocyte apoptosis is first activated by disrupted Ca2+ homeostasis following mitochondrial injury.
Genome-wide studies
Although the candidate gene approach was widely applied in the identification of genes responsible for complex diseases, and evolutionarily important quantitative traits, the utility of this approach is largely limited by its reliance upon a priori knowledge [88]. On the other hand, genome-wide approaches usually proceed without any presuppositions regarding the importance of specific functional features of the traits being investigated. Genome-wide approaches include linkage studies (in families) and genome wide association studies (in unrelated individuals). Both approaches represent unbiased hypothesis-free experiments that hold the potential to identify new biology [89].
Linkage studies represent the earliest type of whole-genome scanning. By constructing pedigrees, early linkage analyses tested for the joint transmission of chromosomal segments and complex phenotypic traits within families. Linkage is the method of choice to identify rare variants with a large impact on disease risk if the trait aggregates in families. The diseases caused by such variants show obvious inheritance patterns and are typically called Mendelian diseases [90]. Although powerful, linkage analyses typically only localize ~10 to 100 cM (centiMorgans) intervals because of the limited number of recombination events within pedigrees [91, 92]. Furthermore, this approach has limited capacity for identifying genes with low penetrance and modest effect size. Thus, the main advantage of family-based studies is that they are not susceptible to false positives from racial admixture and population stratification. Linkage studies have typically not been applied routinely within a pharmacogenomic context, due to the difficulty in identifying families with multiple members exposed to the same drug at the same dose.
Conversely, genome-wide association studies (GWAS) are applied often within the context of pharmacogenomics, for large cohorts of unrelated individuals [93]. GWAS conducted in randomized controlled trials (RCTs) can provide an unbiased survey of the genomic architecture underlying treatment outcome. It is now possible to examine large numbers of polymorphisms, on the order of 100000–1000000, across the entire genome using highly parallel genotyping arrays [94]. In 2008, the SEARCH Collaborative Group applied a 317K SNP scan to 85 cases of incipient myopathy and 90 frequency-matched drug exposed controls, to identify markers of muscle toxicity specifically within the context of high dose simvastatin (80 mg daily) [95]. This was the first published genome-wide association study of statin-induced muscle toxicity. A single variant survived statistical correction for multiple testing: a base substitution in the SLCO1B1 gene [95].
After genomic re-sequencing of SLCO1B1, the putative causative allele (Val174Ala) was re-tested for association in a subset of definite myopathy cases from the original SEARCH study cohort, revealing an odds ratio for myopathy of 4.5 per copy of the variant allele [95]. This association has since been replicated in several independent study populations [95–98]. In the Heart Protection Study (HPS), 24 cases of myopathy were identified in 10,269 participants receiving primary prevention with a lower dose of simvastatin (40 mg daily); 21 were genotyped retrospectively for the variant identified in SEARCH [95], and the relative risk was 2.6 per copy of the variant allele. In a practice-based setting, where the definition of intolerance includes discontinuation of the drug for any reason, the relative risk appears to be closer to 1.5 [96–98]. Efforts are now being made to move this pharmacogenetic association into clinical practice through the application of novel decision-support mechanisms [99, 100]
GWAS using statin-induced myopathy cases may also provide deeper insight into the underlying mechanism of toxicity (i.e., leveraging the genetics to inform the biology) [101, 102]. In 2011, Marciante et al. published a combined CGAS - GWAS using a cohort of 185 confirmed cerivastatin-induced myopathy cases (CK >10xULN with pain) and 732 matched controls [102]. In addition to replicating the well-established SLCO1B1 association for another statin (odds ratio 1.9, p = 0.002), Marciante et al. also leveraged GWAS to identify an association between cerivastatin-induced myopathy and an intronic SNP (rs2819742) in the ryanodine receptor 2 gene (RYR2) (odds ratio 0.48, p = 1.74−07) [102]. Other GWAS cohorts are providing new candidates for statin-related myopathy Muscle specific genes (e.g., gene products modulating Ca2+ flux and excitation-contraction coupling) represent attractive targets for mechanism-based study in vitro.
Challenges and solutions
GWAS have identified numerous common variants associated with complex diseases and provided valuable insights into their fundamental genetic architecture. The main strength of GWAS is an ability to discover truly novel candidate SNPs/genes. However, GWAS are vulnerable to selection bias for “top hits.” [103]. Genes highly relevant in the context of interaction with other important variants will not be detectable at conventional levels of significance [104]. Thus, variants contributing to myopathy risk must be considered within the context of gene-gene (GxG) and gene-environment (GxE) interactions as well. Grouping SNPs into functional units (either genes or groups of genes) might improve the signal for association with a trait. The gene-based approach estimates the combined effect of markers within a gene rather than each marker individually, and is potentially more powerful than individual SNP-based analysis [105].A pathway based approach (interactive sets based on gene ontology, and biological networks based on structural or functional similarity) may facilitate a deeper understanding of findings related to determinants of statin outcome, and account for genetic heterogeneity, as well as improve statistical power to detect relevant genes [106].
Accurate risk prediction models must be flexible enough to simultaneously integrate clinical and genetic factors influencing the absorption, distribution, metabolism, and elimination of statins (i.e., within processes impacting PK), and they must be robust enough to further interpret such determinants in the context of variables influencing mechanism (i.e., across PD and PK processes). One potential approach includes the application of the worlds rapidly growing annotated databases (e.g., GO, gene ontology and gene network analysis). The GO provides structured information on the properties of the products of genes, using three general domains: cellular components (the parts of a cell or its extracellular environment), molecular function (the activities of gene products at the molecular level), and biological process (sets of molecular events with a defined beginning and end) (http://www.geneontology.org/). Clinical responses to statins are complex traits that likely extend from many gene products highly interconnected within networks. For purposes of illustration, we uploaded PD and PK candidate genes into an IPA network analysis (http://www.ingenuity.com/), to uncover the biological and functional interactions among these candidates relevant to statin-related clinical outcome (Figures 1B, 2B). This software application resolves biological and chemical interactions between genes and gene products with functional commonalities. A similar approach was used by Baye et al (2011) to validate recursive partitioning (RP) based gene-gene interactions in childhood asthma [107].
The scientific community is also moving toward whole genome sequencing, in cohorts of increasing sample size, and it is therefore inevitable that risk prediction models for statin-induced myopathy will need to be sufficiently robust to accommodate huge numbers of functional variants that are relatively rare [108]. Most SNP genotyping platforms used to date have only captured polymorphisms at a frequency of 5% or more. Thus, rare variants have been missed in most myopathy GWAS. Rare variants, however, are becoming increasingly catalogued within large populations through efforts like The 1000 Genomes Project (www.1000genomes.org) and next generation sequencing (NGS) [not only to analyze genomic DNA but also as a valuable tool to dissect whole transcriptomes (mRNA, ncRNA, microRNAs) and epigenomes (DNA methylation, protein-DNA interaction)]. While these populations could serve as controls (i.e., statin-exposed, non-myopathic individuals) for sequencing efforts directed at myopathic individuals, sophisticated analytical methodologies will be required [109, 110]. Collapsing methods based on functionality look promising [109]. Using knowledge in annotated databases, functional elements can be weighted by a variety of strategies, such as the regulation of expression through prediction methods that assess transcription factor binding (TF search, ConSite and TRANSFAC), enhancer interactions (VISTA enhancer browser), and mRNA stability (miRBase, TargetScan).
Lastly, these genomic approaches will need to be supported by additional technologies related to rigorous phenotyping (e.g., transcriptomics, proteomics and lipidomics). Transcriptional profiling arrays now consider alternative splicing [111], tissue specific gene expression [112], and evolutionary aspects of gene expression [113]. Laaksonen et al. analyzed the expression of over 46,000 genes in muscle biopsy samples obtained from 6 subjects receiving atorvastatin, 5 receiving simvastatin (1 of 6 cases yielded insufficient RNA), and 6 receiving placebo (limited to males, and frequency-matched according to age) [114]. Simvastatin treatment resulted in expression change in 111 genes - 26 down-regulated and 85 up-regulated. More than twenty biological pathways were affected according to their bioinformatics analysis. The most significant up-regulated genes included ALOX5AP, CCL5, COL3A1 MYL5 and MYBPH. The same muscle biopsy specimens were then also characterized by lipidomics (LC tandem MS), quantifying 132 unique molecular lipid species [114, 115]. Regression of lipidomic data on gene expression data for pathway-based signaling networks confirmed the involvement of lipid-derived signaling pathways (e.g., prostanoid biosynthesis), and suggested a role for Ca2+-dependent pathways capable of modulating excitation contraction coupling and apoptosis (e.g., Phospholipase C, PLC) [114, 115]. Thus, comprehensive approaches linking genomics and transcriptional profiling, with novel phenotyping strategies in the context of larger populations hold the potential to define the genetic architecture of statin-induced myopathy with unprecedented power.
Summary
Like most complex traits, statin-induced muscle toxicity is highly variable in its clinical presentation, and strongly influenced by both GxG and GxE interactions. Although candidate genes studies have revealed a number of variants influencing statin PK, only one locus (SLCO1B1) has been validated using genome-wide technologies. Additional PD variants are emerging from GWAS (RYR2), reinforcing the claim that the field of genomics will inform the underlying biology. While these results move future mechanistic queries in the direction of Ca2+ homeostasis, additional population-based studies are needed to place the findings into context alongside huge numbers of rare variants in cohorts that are diverse enough to facilitate analyses adjusting for admixture.
Future Prospective
As the translational genomics community continues moving toward prospective implementation of ancestry- and gene-based dosing models for a variety of commonly used medications, some groups [79] have argued that statins should be among the first drugs to benefit from automated decision support utilizing these data. The Clinical Pharmacogenomics Implementation Consortium (CPIC) routinely publishes guidelines for such an approach [116]. To date, three such guidelines have been published (for warfarin, clopidogrel, and thiopurines), and it is anticipated that a guideline for gene-based statin dosing will be among the next publications to be posted: http://www.pharmgkb.org/contributors/consortia/cpic_gene-drug_pairs.jsp. As medical centers across diverse systems of care begin adopting this approach, the health care enterprise will soon be in a position to assess impact for pharmacogenomics on quality of care for the most commonly prescribed class of drugs in the industrialized world.
Executive summary.
-
Background
Skeletal muscle toxicity is the most common statin-related ADR.
-
Clinical determinants of risk
age, race, gender, exercise, co-morbidity, co-medication and dose impact risk.
Confounding can occur as a result of population stratification or admixture.
Good epidemiology study design and rigorous phenotyping are the most efficient way to avoid confounding in population-based cohort studies.
-
Genetic determinants of risk
-
The degree to which genetic factors contribute to inter-individual variability in myopathy risk has been an active area of investigation for more than a decade.
-
Candidate gene studies
Pharmacokinetic candidate genes
Pharmacodynamic candidate genes
-
Genome-wide studies
Linkage studies (in families)
Genome wide association studies (in unrelated individuals)
-
-
Emerging advances in the field
-
Technologies related to rigorous genotyping
Whole genome sequencing
Collapsing methods for rare variants
-
Technologies related to rigorous phenotyping
Transcriptomics
Proteomics
Lipidomics
-
Implementation
Electronic medical records
Automated decision support
-
-
References
- 1.Ribeiro RA, Ziegelmann PK, Duncan BB, et al. Impact of statin dose on major cardiovascular events: A mixed treatment comparison meta-analysis involving more than 175,000 patients. Int J Cardiol. 2011 doi: 10.1016/j.ijcard.2011.10.128. [DOI] [PubMed] [Google Scholar]
- 2.Mangravite LM, Wilke RA, Zhang J, Krauss RM. Pharmacogenomics of statin response. Curr Opin Mol Ther. 2008;10(6):555–561. [PubMed] [Google Scholar]
- 3.Wilke RA, Mareedu RK, Moore JH. The Pathway Less Traveled: Moving from Candidate Genes to Candidate Pathways in the Analysis of Genome-Wide Data from Large Scale Pharmacogenetic Association Studies. Curr Pharmacogenomics Person Med. 2008;6(3):150–159. doi: 10.2174/1875692110806030150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bulbulia R, Bowman L, Wallendszus K, et al. Effects on 11-year mortality and morbidity of lowering LDL cholesterol with simvastatin for about 5 years in 20,536 high-risk individuals: a randomised controlled trial. Lancet. 2011;378(9808):2013–2020. doi: 10.1016/S0140-6736(11)61125-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahmad H, Cheng-Lai A. Pitavastatin: a new HMG-CoA reductase inhibitor for the treatment of hypercholesterolemia. Cardiol Rev. 2010;18(5):264–267. doi: 10.1097/CRD.0b013e3181ebdb2f. [DOI] [PubMed] [Google Scholar]
- 6.Tobert JA. Lovastatin and beyond: the history of the HMG-CoA reductase inhibitors. Nat Rev Drug Discov. 2003;2(7):517–526. doi: 10.1038/nrd1112. [DOI] [PubMed] [Google Scholar]
- 7.Ballantyne CM, Corsini A, Davidson MH, et al. Risk for myopathy with statin therapy in high-risk patients. Arch Intern Med. 2003;163(5):553–564. doi: 10.1001/archinte.163.5.553. [DOI] [PubMed] [Google Scholar]
- 8.Kohli P, Cannon CP. Statins and safety: can we finally be reassured? Lancet. 2011;378(9808):1980–1981. doi: 10.1016/S0140-6736(11)61544-4. [DOI] [PubMed] [Google Scholar]
- 9.Mckenney JM, Davidson MH, Jacobson TA, Guyton JR. Final conclusions and recommendations of the National Lipid Association Statin Safety Assessment Task Force. Am J Cardiol. 2006;97(8A):89C–94C. doi: 10.1016/j.amjcard.2006.02.030. [DOI] [PubMed] [Google Scholar]
- 10.Wilke RA, Dolan ME. Genetics and variable drug response. JAMA. 2011;306(3):306–307. doi: 10.1001/jama.2011.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thompson PD, Clarkson P, Karas RH. Statin-associated myopathy. JAMA. 2003;289(13):1681–1690. doi: 10.1001/jama.289.13.1681. [DOI] [PubMed] [Google Scholar]
- 12.Waters DD. Safety of high-dose atorvastatin therapy. Am J Cardiol. 2005;96(5A):69F–75F. doi: 10.1016/j.amjcard.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 13.Kennedy SP, Barnas GP, Schmidt MJ, Glisczinski MS, Paniagua AC. Efficacy and tolerability of once-weekly rosuvastatin in patients with previous statin intolerance. J Clin Lipidol. 2011;5(4):308–315. doi: 10.1016/j.jacl.2011.03.454. [DOI] [PubMed] [Google Scholar]
- 14.Thompson PD, Clarkson PM, Rosenson RS. An assessment of statin safety by muscle experts. Am J Cardiol. 2006;97(8A):69C–76C. doi: 10.1016/j.amjcard.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 15.Piro RM, Di Cunto F. Computational approaches to disease gene prediction: rationale, classification and successes. FEBS J. 2012 doi: 10.1111/j.1742-4658.2012.08471.x. [DOI] [PubMed] [Google Scholar]
- 16.Abd TT, Jacobson TA. Statin-induced myopathy: a review and update. Expert Opin Drug Saf. 2011;10(3):373–387. doi: 10.1517/14740338.2011.540568. [DOI] [PubMed] [Google Scholar]
- 17.Harper CR, Jacobson TA. The broad spectrum of statin myopathy: from myalgia to rhabdomyolysis. Curr Opin Lipidol. 2007;18(4):401–408. doi: 10.1097/MOL.0b013e32825a6773. [DOI] [PubMed] [Google Scholar]
- 18.Psaty BM, Vandenbroucke JP. Opportunities for enhancing the FDA guidance on pharmacovigilance. JAMA. 2008;300(8):952–954. doi: 10.1001/jama.300.8.952. [DOI] [PubMed] [Google Scholar]
- 19.Law M, Rudnicka AR. Statin safety: a systematic review. Am J Cardiol. 2006;97(8A):52C–60C. doi: 10.1016/j.amjcard.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 20.Wilke RA, Lin DW, Roden DM, et al. Identifying genetic risk factors for serious adverse drug reactions: current progress and challenges. Nat Rev Drug Discov. 2007;6(11):904–916. doi: 10.1038/nrd2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chan J, Hui RL, Levin E. Differential association between statin exposure and elevated levels of creatine kinase. Ann Pharmacother. 2005;39(10):1611–1616. doi: 10.1345/aph.1G035. [DOI] [PubMed] [Google Scholar]
- 22.Mcclure DL, Valuck RJ, Glanz M, Murphy JR, Hokanson JE. Statin and statin-fibrate use was significantly associated with increased myositis risk in a managed care population. J Clin Epidemiol. 2007;60(8):812–818. doi: 10.1016/j.jclinepi.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 23.Molokhia M, Mckeigue P, Curcin V, Majeed A. Statin induced myopathy and myalgia: time trend analysis and comparison of risk associated with statin class from 1991–2006. PLoS ONE. 2008;3(6):e2522. doi: 10.1371/journal.pone.0002522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Graham DJ, Staffa JA, Shatin D, et al. Incidence of hospitalized rhabdomyolysis in patients treated with lipid-lowering drugs. JAMA. 2004;292(21):2585–2590. doi: 10.1001/jama.292.21.2585. [DOI] [PubMed] [Google Scholar]
- 25.Garcia-Rodriguez LA, Masso-Gonzalez EL, Wallander MA, Johansson S. The safety of rosuvastatin in comparison with other statins in over 100,000 statin users in UK primary care. Pharmacoepidemiol Drug Saf. 2008;17(10):943–952. doi: 10.1002/pds.1603. [DOI] [PubMed] [Google Scholar]
- 26.Wilke RA, Reif DM, Moore JH. Combinatorial pharmacogenetics. Nat Rev Drug Discov. 2005;4(11):911–918. doi: 10.1038/nrd1874. [DOI] [PubMed] [Google Scholar]
- 27.Mareedu RK, Modhia FM, Kanin EI, et al. Use of an electronic medical record to characterize cases of intermediate statin-induced muscle toxicity. Prev Cardiol. 2009;12(2):88–94. doi: 10.1111/j.1751-7141.2009.00028.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cannon CP, Braunwald E, Mccabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350(15):1495–1504. doi: 10.1056/NEJMoa040583. [DOI] [PubMed] [Google Scholar]
- 29.Larosa JC, Grundy SM, Waters DD, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med. 2005;352(14):1425–1435. doi: 10.1056/NEJMoa050461. [DOI] [PubMed] [Google Scholar]
- 30.De Lemos JA, Blazing MA, Wiviott SD, et al. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: phase Z of the A to Z trial. JAMA. 2004;292(11):1307–1316. doi: 10.1001/jama.292.11.1307. [DOI] [PubMed] [Google Scholar]
- 31.Pedersen TR, Faergeman O, Kastelein JJ, et al. High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction: the IDEAL study: a randomized controlled trial. JAMA. 2005;294(19):2437–2445. doi: 10.1001/jama.294.19.2437. [DOI] [PubMed] [Google Scholar]
- 32.Schech S, Graham D, Staffa J, et al. Risk factors for statin-associated rhabdomyolysis. Pharmacoepidemiol Drug Saf. 2007;16(3):352–358. doi: 10.1002/pds.1287. [DOI] [PubMed] [Google Scholar]
- 33.Krishnan GM, Thompson PD. The effects of statins on skeletal muscle strength and exercise performance. Curr Opin Lipidol. 2010;21(4):324–328. doi: 10.1097/MOL.0b013e32833c1edf. [DOI] [PubMed] [Google Scholar]
- 34.Egan A, Colman E. Weighing the benefits of high-dose simvastatin against the risk of myopathy. N Engl J Med. 2011;365(4):285–287. doi: 10.1056/NEJMp1106689. [DOI] [PubMed] [Google Scholar]
- 35.Lee E, Ryan S, Birmingham B, et al. Rosuvastatin pharmacokinetics and pharmacogenetics in white and Asian subjects residing in the same environment. Clin Pharmacol Ther. 2005;78(4):330–341. doi: 10.1016/j.clpt.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 36.Liao JK. Safety and efficacy of statins in Asians. Am J Cardiol. 2007;99(3):410–414. doi: 10.1016/j.amjcard.2006.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baye TM, Wilke RA. Mapping genes that predict treatment outcome in admixed populations. Pharmacogenomics J. 2010;10(6):465–477. doi: 10.1038/tpj.2010.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Albert MA, Glynn RJ, Fonseca FA, et al. Race, ethnicity, and the efficacy of rosuvastatin in primary prevention: the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) trial. Am Heart J. 2011;162(1):106–114. e102. doi: 10.1016/j.ahj.2011.03.032. [DOI] [PubMed] [Google Scholar]
- 39.Baye TM, Tiwari HK, Allison DB, Go RC. Database mining for selection of SNP markers useful in admixture mapping. BioData Min. 2009;2(1):1. doi: 10.1186/1756-0381-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eichler EE, Flint J, Gibson G, et al. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. 2010;11(6):446–450. doi: 10.1038/nrg2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomson G. Significance levels in genome scans. Adv Genet. 2001;42:475–486. doi: 10.1016/s0065-2660(01)42037-2. [DOI] [PubMed] [Google Scholar]
- 42.Neuvonen PJ, Niemi M, Backman JT. Drug interactions with lipid-lowering drugs: mechanisms and clinical relevance. Clin Pharmacol Ther. 2006;80(6):565–581. doi: 10.1016/j.clpt.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 43.Neuvonen PJ, Backman JT, Niemi M. Pharmacokinetic comparison of the potential over-the-counter statins simvastatin, lovastatin, fluvastatin and pravastatin. Clin Pharmacokinet. 2008;47(7):463–474. doi: 10.2165/00003088-200847070-00003. [DOI] [PubMed] [Google Scholar]
- 44.Kaspera R, Naraharisetti SB, Tamraz B, et al. Cerivastatin in vitro metabolism by CYP2C8 variants found in patients experiencing rhabdomyolysis. Pharmacogenet Genomics. 2010;20(10):619–629. doi: 10.1097/FPC.0b013e32833ecace. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prueksaritanont T, Ma B, Yu N. The human hepatic metabolism of simvastatin hydroxy acid is mediated primarily by CYP3A, and not CYP2D6. Br J Clin Pharmacol. 2003;56(1):120–124. doi: 10.1046/j.1365-2125.2003.01833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prueksaritanont T, Zhao JJ, Ma B, et al. Mechanistic studies on metabolic interactions between gemfibrozil and statins. J Pharmacol Exp Ther. 2002;301(3):1042–1051. doi: 10.1124/jpet.301.3.1042. [DOI] [PubMed] [Google Scholar]
- 47.Geisel J, Kivisto KT, Griese EU, Eichelbaum M. The efficacy of simvastatin is not influenced by CYP2D6 polymorphism. Clin Pharmacol Ther. 2002;72(5):595–596. [PubMed] [Google Scholar]
- 48.Mulder AB, Van Den Bergh FA, Vermes I. Response to “The efficacy of simvastatin is not influenced by CYP2D6 polymorphism” by Geisel et al. Clin Pharmacol Ther. 2003;73(5):475. doi: 10.1016/s0009-9236(03)00054-7. [DOI] [PubMed] [Google Scholar]
- 49.Mulder AB, Van Lijf HJ, Bon MA, et al. Association of polymorphism in the cytochrome CYP2D6 and the efficacy and tolerability of simvastatin. Clin Pharmacol Ther. 2001;70(6):546–551. doi: 10.1067/mcp.2001.120251. [DOI] [PubMed] [Google Scholar]
- 50.Nordin C, Dahl ML, Eriksson M, Sjoberg S. Is the cholesterol-lowering effect of simvastatin influenced by CYP2D6 polymorphism? Lancet. 1997;350(9070):29–30. doi: 10.1016/S0140-6736(05)66238-1. [DOI] [PubMed] [Google Scholar]
- 51.Frudakis TN, Thomas MJ, Ginjupalli SN, Handelin B, Gabriel R, Gomez HJ. CYP2D6*4 polymorphism is associated with statin-induced muscle effects. Pharmacogenet Genomics. 2007;17(9):695–707. doi: 10.1097/FPC.0b013e328012d0a9. [DOI] [PubMed] [Google Scholar]
- 52.Wilke RA, Moore JH, Burmester JK. Relative impact of CYP3A genotype and concomitant medication on the severity of atorvastatin-induced muscle damage. Pharmacogenet Genomics. 2005;15(6):415–421. doi: 10.1097/01213011-200506000-00007. [DOI] [PubMed] [Google Scholar]
- 53.Jemal M, Ouyang Z, Chen BC, Teitz D. Quantitation of the acid and lactone forms of atorvastatin and its biotransformation products in human serum by high-performance liquid chromatography with electrospray tandem mass spectrometry. Rapid Commun Mass Spectrom. 1999;13(11):1003–1015. doi: 10.1002/(SICI)1097-0231(19990615)13:11<1003::AID-RCM597>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 54.Riedmaier S, Klein K, Hofmann U, et al. UDP-glucuronosyltransferase (UGT) polymorphisms affect atorvastatin lactonization in vitro and in vivo. Clin Pharmacol Ther. 2010;87(1):65–73. doi: 10.1038/clpt.2009.181. [DOI] [PubMed] [Google Scholar]
- 55.Perera MA, Innocenti F, Ratain MJ. Pharmacogenetic testing for uridine diphosphate glucuronosyltransferase 1A1 polymorphisms: are we there yet? Pharmacotherapy. 2008;28(6):755–768. doi: 10.1592/phco.28.6.755. [DOI] [PubMed] [Google Scholar]
- 56.Matsushima S, Maeda K, Kondo C, et al. Identification of the hepatic efflux transporters of organic anions using double-transfected Madin-Darby canine kidney II cells expressing human organic anion-transporting polypeptide 1B1 (OATP1B1)/multidrug resistance-associated protein 2, OATP1B1/multidrug resistance 1, and OATP1B1/breast cancer resistance protein. J Pharmacol Exp Ther. 2005;314(3):1059–1067. doi: 10.1124/jpet.105.085589. [DOI] [PubMed] [Google Scholar]
- 57.Ho RH, Tirona RG, Leake BF, et al. Drug and bile acid transporters in rosuvastatin hepatic uptake: function, expression, and pharmacogenetics. Gastroenterology. 2006;130(6):1793–1806. doi: 10.1053/j.gastro.2006.02.034. [DOI] [PubMed] [Google Scholar]
- 58.Pasanen MK, Neuvonen M, Neuvonen PJ, Niemi M. SLCO1B1 polymorphism markedly affects the pharmacokinetics of simvastatin acid. Pharmacogenet Genomics. 2006;16(12):873–879. doi: 10.1097/01.fpc.0000230416.82349.90. [DOI] [PubMed] [Google Scholar]
- 59.Mwinyi J, Johne A, Bauer S, Roots I, Gerloff T. Evidence for inverse effects of OATP-C (SLC21A6) 5 and 1b haplotypes on pravastatin kinetics. Clin Pharmacol Ther. 2004;75(5):415–421. doi: 10.1016/j.clpt.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 60.Niemi M. Transporter pharmacogenetics and statin toxicity. Clin Pharmacol Ther. 2010;87(1):130–133. doi: 10.1038/clpt.2009.197. [DOI] [PubMed] [Google Scholar]
- 61.Ho RH, Kim RB. Transporters and drug therapy: implications for drug disposition and disease. Clin Pharmacol Ther. 2005;78(3):260–277. doi: 10.1016/j.clpt.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 62.Fiegenbaum M, Da Silveira FR, Van Der Sand CR, et al. The role of common variants of ABCB1, CYP3A4, and CYP3A5 genes in lipid-lowering efficacy and safety of simvastatin treatment. Clin Pharmacol Ther. 2005;78(5):551–558. doi: 10.1016/j.clpt.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 63.Keskitalo JE, Zolk O, Fromm MF, Kurkinen KJ, Neuvonen PJ, Niemi M. ABCG2 polymorphism markedly affects the pharmacokinetics of atorvastatin and rosuvastatin. Clin Pharmacol Ther. 2009;86(2):197–203. doi: 10.1038/clpt.2009.79. [DOI] [PubMed] [Google Scholar]
- 64.Keskitalo JE, Pasanen MK, Neuvonen PJ, Niemi M. Different effects of the ABCG2 c.421C>A SNP on the pharmacokinetics of fluvastatin, pravastatin and simvastatin. Pharmacogenomics. 2009;10(10):1617–1624. doi: 10.2217/pgs.09.85. [DOI] [PubMed] [Google Scholar]
- 65.Tomlinson B, Hu M, Lee VW, et al. ABCG2 polymorphism is associated with the low-density lipoprotein cholesterol response to rosuvastatin. Clin Pharmacol Ther. 2010;87(5):558–562. doi: 10.1038/clpt.2009.232. [DOI] [PubMed] [Google Scholar]
- 66.Robey RW, To KK, Polgar O, et al. ABCG2: a perspective. Adv Drug Deliv Rev. 2009;61(1):3–13. doi: 10.1016/j.addr.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Generaux GT, Bonomo FM, Johnson M, Doan KM. Impact of SLCO1B1 (OATP1B1) and ABCG2 (BCRP) genetic polymorphisms and inhibition on LDL-C lowering and myopathy of statins. Xenobiotica. 2011;41(8):639–651. doi: 10.3109/00498254.2011.562566. [DOI] [PubMed] [Google Scholar]
- 68.Chen YC, Chen YD, Li X, et al. The HMG-CoA reductase gene and lipid and lipoprotein levels: the multi-ethnic study of atherosclerosis. Lipids. 2009;44(8):733–743. doi: 10.1007/s11745-009-3314-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chien KL, Wang KC, Chen YC, et al. Common sequence variants in pharmacodynamic and pharmacokinetic pathway-related genes conferring LDL cholesterol response to statins. Pharmacogenomics. 2010;11(3):309–317. doi: 10.2217/pgs.09.160. [DOI] [PubMed] [Google Scholar]
- 70.Donnelly LA, Doney AS, Dannfald J, et al. A paucimorphic variant in the HMG-CoA reductase gene is associated with lipid-lowering response to statin treatment in diabetes: a GoDARTS study. Pharmacogenet Genomics. 2008;18(12):1021–1026. doi: 10.1097/FPC.0b013e3283106071. [DOI] [PubMed] [Google Scholar]
- 71.Krauss RM, Mangravite LM, Smith JD, et al. Variation in the 3-hydroxyl-3-methylglutaryl coenzyme a reductase gene is associated with racial differences in low-density lipoprotein cholesterol response to simvastatin treatment. Circulation. 2008;117(12):1537–1544. doi: 10.1161/CIRCULATIONAHA.107.708388. [DOI] [PubMed] [Google Scholar]
- 72.Mangravite LM, Medina MW, Cui J, et al. Combined influence of LDLR and HMGCR sequence variation on lipid-lowering response to simvastatin. Arterioscler Thromb Vasc Biol. 2010;30(7):1485–1492. doi: 10.1161/ATVBAHA.110.203273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Medina MW, Gao F, Ruan W, Rotter JI, Krauss RM. Alternative splicing of 3-hydroxy-3-methylglutaryl coenzyme A reductase is associated with plasma low-density lipoprotein cholesterol response to simvastatin. Circulation. 2008;118(4):355–362. doi: 10.1161/CIRCULATIONAHA.108.773267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Polisecki E, Muallem H, Maeda N, et al. Genetic variation at the LDL receptor and HMG-CoA reductase gene loci, lipid levels, statin response, and cardiovascular disease incidence in PROSPER. Atherosclerosis. 2008;200(1):109–114. doi: 10.1016/j.atherosclerosis.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Baker SK, Tarnopolsky MA. Statin-associated neuromyotoxicity. Drugs Today (Barc) 2005;41(4):267–293. doi: 10.1358/dot.2005.41.4.908565. [DOI] [PubMed] [Google Scholar]
- 76.Gambelli S, Dotti MT, Malandrini A, et al. Mitochondrial alterations in muscle biopsies of patients on statin therapy. J Submicrosc Cytol Pathol. 2004;36(1):85–89. [PubMed] [Google Scholar]
- 77.Phillips PS, Haas RH, Bannykh S, et al. Statin-associated myopathy with normal creatine kinase levels. Ann Intern Med. 2002;137(7):581–585. doi: 10.7326/0003-4819-137-7-200210010-00009. [DOI] [PubMed] [Google Scholar]
- 78.Vladutiu GD, Simmons Z, Isackson PJ, et al. Genetic risk factors associated with lipid-lowering drug-induced myopathies. Muscle Nerve. 2006;34(2):153–162. doi: 10.1002/mus.20567. [DOI] [PubMed] [Google Scholar]
- 79.Peters BJ, Klungel OH, Visseren FL, De Boer A, Maitland-Van Der Zee AH. Pharmacogenomic insights into treatment and management of statin-induced myopathy. Genome Med. 2009;1(12):120. doi: 10.1186/gm120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Oh J, Ban MR, Miskie BA, Pollex RL, Hegele RA. Genetic determinants of statin intolerance. Lipids Health Dis. 2007;6:7. doi: 10.1186/1476-511X-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nawarskas JJ. HMG-CoA reductase inhibitors and coenzyme Q10. Cardiol Rev. 2005;13(2):76–79. doi: 10.1097/01.crd.0000154790.42283.a1. [DOI] [PubMed] [Google Scholar]
- 82.Young JM, Florkowski CM, Molyneux SL, et al. Effect of coenzyme Q(10) supplementation on simvastatin-induced myalgia. Am J Cardiol. 2007;100(9):1400–1403. doi: 10.1016/j.amjcard.2007.06.030. [DOI] [PubMed] [Google Scholar]
- 83.Johnson TE, Zhang X, Bleicher KB, et al. Statins induce apoptosis in rat and human myotube cultures by inhibiting protein geranylgeranylation but not ubiquinone. Toxicol Appl Pharmacol. 2004;200(3):237–250. doi: 10.1016/j.taap.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 84.Matzno S, Yasuda S, Juman S, et al. Statin-induced apoptosis linked with membrane farnesylated Ras small G protein depletion, rather than geranylated Rho protein. J Pharm Pharmacol. 2005;57(11):1475–1484. doi: 10.1211/jpp.57.11.0014. [DOI] [PubMed] [Google Scholar]
- 85.Sacher J, Weigl L, Werner M, Szegedi C, Hohenegger M. Delineation of myotoxicity induced by 3-hydroxy-3-methylglutaryl CoA reductase inhibitors in human skeletal muscle cells. J Pharmacol Exp Ther. 2005;314(3):1032–1041. doi: 10.1124/jpet.105.086462. [DOI] [PubMed] [Google Scholar]
- 86.Sakamoto K, Honda T, Yokoya S, Waguri S, Kimura J. Rab-small GTPases are involved in fluvastatin and pravastatin-induced vacuolation in rat skeletal myofibers. FASEB J. 2007;21(14):4087–4094. doi: 10.1096/fj.07-8713com. [DOI] [PubMed] [Google Scholar]
- 87.Itagaki M, Takaguri A, Kano S, Kaneta S, Ichihara K, Satoh K. Possible mechanisms underlying statin-induced skeletal muscle toxicity in L6 fibroblasts and in rats. J Pharmacol Sci. 2009;109(1):94–101. doi: 10.1254/jphs.08238fp. [DOI] [PubMed] [Google Scholar]
- 88.Gianfagna F, Cugino D, Santimone I, Iacoviello L. From candidate gene to genome-wide association studies in cardiovascular disease. Thromb Res. 2011 doi: 10.1016/j.thromres.2011.11.014. [DOI] [PubMed] [Google Scholar]
- 89.Mccarthy MI, Hirschhorn JN. Genome-wide association studies: potential next steps on a genetic journey. Hum Mol Genet. 2008;17(R2):R156–165. doi: 10.1093/hmg/ddn289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Antonarakis SE, Beckmann JS. Mendelian disorders deserve more attention. Nat Rev Genet. 2006;7(4):277–282. doi: 10.1038/nrg1826. [DOI] [PubMed] [Google Scholar]
- 91.Doerge RW. Mapping and analysis of quantitative trait loci in experimental populations. Nat Rev Genet. 2002;3(1):43–52. doi: 10.1038/nrg703. [DOI] [PubMed] [Google Scholar]
- 92.Holland JB. Genetic architecture of complex traits in plants. Curr Opin Plant Biol. 2007;10(2):156–161. doi: 10.1016/j.pbi.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 93.Pereira NL, Weinshilboum RM. The impact of pharmacogenomics on the management of cardiac disease. Clin Pharmacol Ther. 2011;90(4):493–495. doi: 10.1038/clpt.2011.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fan JB, Chee MS, Gunderson KL. Highly parallel genomic assays. Nat Rev Genet. 2006;7(8):632–644. doi: 10.1038/nrg1901. [DOI] [PubMed] [Google Scholar]
- 95.Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy--a genomewide study. N Engl J Med. 2008;359(8):789–799. doi: 10.1056/NEJMoa0801936. [DOI] [PubMed] [Google Scholar]
- 96.Voora D, Shah SH, Spasojevic I, et al. The SLCO1B1*5 genetic variant is associated with statin-induced side effects. J Am Coll Cardiol. 2009;54(17):1609–1616. doi: 10.1016/j.jacc.2009.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Donnelly LA, Doney AS, Tavendale R, et al. Common nonsynonymous substitutions in SLCO1B1 predispose to statin intolerance in routinely treated individuals with type 2 diabetes: a go-DARTS study. Clin Pharmacol Ther. 2011;89(2):210–216. doi: 10.1038/clpt.2010.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Peters B, editor. Methodological approaches to the pharmacogenomics of statins. 2010. [Google Scholar]
- 99.Wilke RA, Xu H, Denny JC, et al. The emerging role of electronic medical records in pharmacogenomics. Clin Pharmacol Ther. 2011;89(3):379–386. doi: 10.1038/clpt.2010.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Roden DM, Johnson JA, Kimmel SE, et al. Cardiovascular pharmacogenomics. Circ Res. 2011;109(7):807–820. doi: 10.1161/CIRCRESAHA.110.230995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Isackson PJ, Ochs-Balcom HM, Ma C, et al. Association of common variants in the human eyes shut ortholog (EYS) with statin-induced myopathy: Evidence for additional functions of EYS. Muscle Nerve. 2011 doi: 10.1002/mus.22115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Marciante KD, Durda JP, Heckbert SR, et al. Cerivastatin, genetic variants, and the risk of rhabdomyolysis. Pharmacogenet Genomics. 2011;21(5):280–288. doi: 10.1097/FPC.0b013e328343dd7d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Emily M, Mailund T, Hein J, Schauser L, Schierup MH. Using biological networks to search for interacting loci in genome-wide association studies. Eur J Hum Genet. 2009;17(10):1231–1240. doi: 10.1038/ejhg.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kasarskis A, Yang X, Schadt E. Integrative genomics strategies to elucidate the complexity of drug response. Pharmacogenomics. 2011;12(12):1695–1715. doi: 10.2217/pgs.11.115. [DOI] [PubMed] [Google Scholar]
- 105.Neale BM, Sham PC. The future of association studies: gene-based analysis and replication. Am J Hum Genet. 2004;75(3):353–362. doi: 10.1086/423901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cantor RM, Lange K, Sinsheimer JS. Prioritizing GWAS results: A review of statistical methods and recommendations for their application. Am J Hum Genet. 2010;86(1):6–22. doi: 10.1016/j.ajhg.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Baye TM, Butsch Kovacic M, Biagini Myers JM, et al. Differences in candidate gene association between European ancestry and African American asthmatic children. PLoS ONE. 2011;6(2):e16522. doi: 10.1371/journal.pone.0016522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ashley EA, Butte AJ, Wheeler MT, et al. Clinical assessment incorporating a personal genome. Lancet. 2010;375(9725):1525–1535. doi: 10.1016/S0140-6736(10)60452-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bansal V, Libiger O, Torkamani A, Schork NJ. Statistical analysis strategies for association studies involving rare variants. Nat Rev Genet. 2010;11(11):773–785. doi: 10.1038/nrg2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lettre G, Palmer CD, Young T, et al. Genome-wide association study of coronary heart disease and its risk factors in 8,090 African Americans: the NHLBI CARe Project. PLoS Genet. 2011;7(2):e1001300. doi: 10.1371/journal.pgen.1001300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kwan T, Benovoy D, Dias C, et al. Genome-wide analysis of transcript isoform variation in humans. Nat Genet. 2008;40(2):225–231. doi: 10.1038/ng.2007.57. [DOI] [PubMed] [Google Scholar]
- 112.Clark TA, Schweitzer AC, Chen TX, et al. Discovery of tissue-specific exons using comprehensive human exon microarrays. Genome Biol. 2007;8(4):R64. doi: 10.1186/gb-2007-8-4-r64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Xing Y, Ouyang Z, Kapur K, Scott MP, Wong WH. Assessing the conservation of mammalian gene expression using high-density exon arrays. Mol Biol Evol. 2007;24(6):1283–1285. doi: 10.1093/molbev/msm061. [DOI] [PubMed] [Google Scholar]
- 114.Laaksonen R, Katajamaa M, Paiva H, et al. A systems biology strategy reveals biological pathways and plasma biomarker candidates for potentially toxic statin-induced changes in muscle. PLoS ONE. 2006;1:e97. doi: 10.1371/journal.pone.0000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Laaksonen R. On the mechanisms of statin-induced myopathy. Clin Pharmacol Ther. 2006;79(6):529–531. doi: 10.1016/j.clpt.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 116.Relling MV, Klein TE. CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network. Clin Pharmacol Ther. 2011;89(3):464–467. doi: 10.1038/clpt.2010.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jacobson TA. Statin safety: lessons from new drug applications for marketed statins. Am J Cardiol. 2006;97(8A):44C–51C. doi: 10.1016/j.amjcard.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 118.Pasternak RC, Smith SC, Jr, Bairey-Merz CN, Grundy SM, Cleeman JI, Lenfant C. ACC/AHA/NHLBI clinical advisory on the use and safety of statins. J Am Coll Cardiol. 2002;40(3):567–572. doi: 10.1016/s0735-1097(02)02030-2. [DOI] [PubMed] [Google Scholar]
- 119.Grundy SM. The issue of statin safety: where do we stand? Circulation. 2005;111(23):3016–3019. doi: 10.1161/CIRCULATIONAHA.105.557652. [DOI] [PubMed] [Google Scholar]
- 120.Prisant LM, Downton M, Watkins LO, et al. Efficacy and tolerability of lovastatin in 459 African-Americans with hypercholesterolemia. Am J Cardiol. 1996;78(4):420–424. doi: 10.1016/s0002-9149(96)00330-x. [DOI] [PubMed] [Google Scholar]
- 121.Jacobson TA, Chin MM, Curry CL, et al. Efficacy and safety of pravastatin in African Americans with primary hypercholesterolemia. Arch Intern Med. 1995;155(17):1900–1906. [PubMed] [Google Scholar]
- 122.Puccetti L, Ciani F, Auteri A. Genetic involvement in statins induced myopathy. Preliminary data from an observational case-control study. Atherosclerosis. 2010;211(1):28–29. doi: 10.1016/j.atherosclerosis.2010.02.026. [DOI] [PubMed] [Google Scholar]
- 123.Bruckert E, Hayem G, Dejager S, Yau C, Begaud B. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients--the PRIMO study. Cardiovasc Drugs Ther. 2005;19(6):403–414. doi: 10.1007/s10557-005-5686-z. [DOI] [PubMed] [Google Scholar]
- 124.Hedenmalm K, Alvan G, Ohagen P, Dahl ML. Muscle toxicity with statins. Pharmacoepidemiol Drug Saf. 2010;19(3):223–231. doi: 10.1002/pds.1895. [DOI] [PubMed] [Google Scholar]
- 125.Sinzinger H, O’grady J. Professional athletes suffering from familial hypercholesterolaemia rarely tolerate statin treatment because of muscular problems. Br J Clin Pharmacol. 2004;57(4):525–528. doi: 10.1111/j.1365-2125.2004.02044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rowan C, Brinker AD, Nourjah P, et al. Rhabdomyolysis reports show interaction between simvastatin and CYP3A4 inhibitors. Pharmacoepidemiol Drug Saf. 2009;18(4):301–309. doi: 10.1002/pds.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ray GM. Antiretroviral and statin drug-drug interactions. Cardiol Rev. 2009;17(1):44–47. doi: 10.1097/CRD.0b013e3181903b7f. [DOI] [PubMed] [Google Scholar]
- 128.Schneck DW, Birmingham BK, Zalikowski JA, et al. The effect of gemfibrozil on the pharmacokinetics of rosuvastatin. Clin Pharmacol Ther. 2004;75(5):455–463. doi: 10.1016/j.clpt.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 129.Ishikawa C, Ozaki H, Nakajima T, et al. A frameshift variant of CYP2C8 was identified in a patient who suffered from rhabdomyolysis after administration of cerivastatin. J Hum Genet. 2004;49(10):582–585. doi: 10.1007/s10038-004-0188-6. [DOI] [PubMed] [Google Scholar]
- 130.Ozaki H, Ishikawa CT, Ishii T, et al. Clearance rates of cerivastatin metabolites in a patient with cerivastatin-induced rhabdomyolysis. J Clin Pharm Ther. 2005;30(2):189–192. doi: 10.1111/j.1365-2710.2005.00633_1.x. [DOI] [PubMed] [Google Scholar]
- 131.Morimoto K, Oishi T, Ueda S, Ueda M, Hosokawa M, Chiba K. A novel variant allele of OATP-C (SLCO1B1) found in a Japanese patient with pravastatin-induced myopathy. Drug Metab Pharmacokinet. 2004;19(6):453–455. doi: 10.2133/dmpk.19.453. [DOI] [PubMed] [Google Scholar]
- 132.Brunham LR, Lansberg PJ, Zhang L, et al. Differential effect of the rs4149056 variant in SLCO1B1 on myopathy associated with simvastatin and atorvastatin. Pharmacogenomics J. 2011 doi: 10.1038/tpj.2010.92. [DOI] [PubMed] [Google Scholar]
- 133.Furihata T, Satoh N, Ohishi T, et al. Functional analysis of a mutation in the SLCO1B1 gene (c.1628T>G) identified in a Japanese patient with pravastatin-induced myopathy. Pharmacogenomics J. 2009;9(3):185–193. doi: 10.1038/tpj.2009.3. [DOI] [PubMed] [Google Scholar]