

Abstract
McLeod syndrome (MLS) is a rare, X-linked, late-onset, disease involving hematological, brain, and neuromuscular systems, caused by mutations in XK that result in either defective XK or complete loss of XK protein. Acanthocytosis of erythrocytes is a typical feature. We report novel mutations in two patients who exhibited typical clinical characteristics of MLS. The coding and flanking intronic regions of XK were amplified by PCR, sequenced, and compared with the normal XK sequence. XK protein, and its complexed partner protein, Kell, were assessed by Western blot analysis. Patient 1 was found to have a single base insertion, 605insA at 175Ile creating a frame shift within the coding sequence of XK. Patient 2 had a single base substitution in the 3′ splice sequence of intron 2 (IVS2–2a>g). In both cases mutations resulted in the absence of XK protein.
Keywords: McLeod syndrome, Neuroacanthocytosis, XK protein, Kell protein, Acanthocytosis, McLeod blood group, Kx erythrocyte antigen, Kell blood group antigens
1. Introduction
McLeod neuroacanthocytosis syndrome (MLS) is a rare X-linked disease caused by mutations in the XK resulting in absent or defective XK protein [1,2]. About 30 different mutations of XK have been reported [3–5]. Mutations are evenly distributed throughout XK and range from point mutations in the coding region or in introns near or at splice junctions, to large deletions of XK that extend into neighboring loci. It is generally agreed that genotype and phenotype do not correlate [4,6–8]. MLS is a multisystem disorder with central nervous system (CNS), neuromuscular, and hematologic manifestations. Central nervous system symptoms include chorea, psychiatric abnormalities, peripheral neuropathy with a reflexia, cognitive decline and seizures. Neuromuscular manifestations include a sensorimotor axonopathy myopathy and cardiomyopathy. Hematological symptoms include a chronic hemolytic state due to the red blood cell acanthocytosis [6,10]. The disease, which is variable from patient to patient, progresses slowly and the majority of the cognitive and/or behavioral disorders occur by the fifth decade. There are similarities at the clinical and neuropathological levels between MLS, chorea-acanthocytosis and Huntington’s disease, all of which involve neurodegeneration of basal ganglia [6,9,10]. Central nervous system symptoms of MLS resemble Huntington’s disease (HD) or chorea-acanthocytosis (ChAc) and include a chorea, psychiatric abnormalities, cognitive decline, and generalized seizures. On the macro- and micro-structural neuropathological level, MLS displays caudate nucleus atrophy, with non-specific changes of neuronal loss and astrogliosis in the caudate nucleus, putarnen and pallidum. In the ChAc additional involvement of the substantia nigra and thalamus may be observed. In HD there is more widespread pathology with neuronal intranuclear inclusions. Importantly cortical pathology is marked in HD, less so in ChAc, and is often not noted in MLS [11].
The XK family is located at Xp21.1. XK has 3 exons and a large 3′ untranslated region [12, 13]. The gene product is predicted to be a 50 KDa polypeptide composed of 444 amino acids. Computer generated topology shows 10 transmembrane regions indicative of a putative membrane transporter. XK is expressed ubiquitously, but is found mainly in nervous tissue, heart and red cells [2,13,14]. In red blood cells XK is covalently linked to Kell glycoprotein through a disulfide bond forming a complex on the red cell membrane [15–18]. There is a correlation between expression of Kell and XK, thus XK levels are reduced when Kell is absent and vice versa.
The function of XK is not known. However, studies of an ion transporting system in red blood cells of Kel knockout mice showed that the Kell/XK complex is involved in ion flux, implying that Kell, XK, or tile Kell/XK complex may be associated with ion flux [15]. Kell is an endothelin-3-converting enzyme expressed mainly on erythroid cells [19,20], but not in brain [13, 14]. Kell is highly polymorphic and has over 30 different antigens, while XK has a single antigen, known as Kx.
MLS is confirmed (i) serologically – by the absence of Kx antigen and by reduced levels of all Kell antigens, known as the McLeod phenotype, (ii) biochemically – by the absence of XK and reduced amounts of Kell protein on red cells [3], or (iii) molecularly – by identification of mutation of XK.
This report describes two novel mutations found in two unrelated individuals with MLS clinical characteristics. In patient 1 we confirmed a frame shift mutation and in patient 2 we found a mutation in the 3′ acceptor site of the RNA splice site between exons 1 and 2, which did not permit normal splicing. Both mutations resulted in the absence of XK as confirmed by serological and biochemical analyses.
2. Patients and methods
2.1. Serology
Anticoagulated blood samples were received from two unrelated males with suspected McLeod syndrome based upon their neurological presentation. Informed consent was obtained prior to collection of blood samples. Washed red blood cells from these samples were tested by standard hemagglutination tests with anti-K, -k, -Kpa, and -Kpb (Immucor, Norcross, GA) according to the manufacturer’s directions. The patients’ red blood cells were also tested with anti-Kx/Km (single donor source in-house antibody).
2.2. Gene characterization
DNA from blood samples was isolated as previously described [21,22]. The three XK exons and the exon/intron boundaries were amplified by PCR. The Herculase PCR system was employed for amplification of exon 1 and conventional taq polymerase PCR was used for amplification of exons 2 and 3 as previously described [22]. The conditions of Herculase PCR were as follows: initial denaturation at 98 °C for 3 min followed by 25 cycles of amplification at 98 °C for 40 s, 65 °C for 30 s, and 72 °C for 1 min. The final cycle was extended at 72 °C for 7 min for completion of PCR. PCR products were separated in 0.8% agarose gel electrophoresis and the targeted amplicons were isolated using GENECLEAN SPIN Kit (MP Biomedicals, Solon, OH) and sequenced with the sequencing primers listed below (Genewiz Inc., South Plainfield, NJ). Mutations were identified by comparing the sequences with wild type XK sequences.
2.3. Primer sequences [13]
PCR 1, for amplification of 700 bp which includes exon 1, part of the adjacent promoter region, and the exon 1/intron 1 junction, XKEX1F: 5′-ACCAGGTCCAGTTCCAGAGCCCAGGCC-3′, XKIN1R: 5′-TTTGACGGAACCTAACAGGAATTCAGTGTG-3′.
PCR 2, for amplification of 600 bp which includes exon 2 and portions of flanking introns including RNA splice sites.BFP2: 5′-AGGACTGCATACTGAGAACTGGTC-3′, RP2: 5′-CCCTGCCTAGAATGCAGAGTCATA-3′.
PCR 3, for amplification of 1.2 kbp which includes exon 3 and portions of flanking introns including RNA splice sites. BFP3: 5′-AACTGGAAGTCAGGCTGTGCACAT-3′, RP3: 5′-GGCCAGTAATGCCTAGAAGAACAC-3′.
Sequencing PCR1, FOR: XKIN0F: 5′-CGGTTTGGGGCTGGGCATGCTGGGA- 3′, REV: XKIN1Ra: 5′-ATGATTCGTCGCGTTCCTGACTG-3′.
Sequencing PCR2, FOR: Seq 2.1, 5′-AGATCAGGCTACAGGACCGAGGCT-3′, REV: Seq 2.2, 5′-GAGCATAGTAACTAGAACACAGATTC-3′.
Sequencing PCR3, FOR1: Seq 3.1, 5′-TGACAGAGTGYCYGCYGCAGTAG-3′, FOR2: XK1202F, 5′-CACCCTTGCAAAAAGCTCTTTTCTTCC-3′, REV1: XK1040R, 5′-ACACCAGTAGCTGGTACCAATTATG-3′, REV2: XK1510R, 5′-GCCTTGTTCCCTGTGTAGGGCTCATTAT-3′.
2.4. Western blot analysis
Red blood cell ghosts were prepared by standard method [23] (red blood cells used for this evaluation were kept in liquid nitrogen prior the analysis). Western blot using red blood cell ghosts was carried out by 4–12% gradient PAGE using precast gels (Invitrogen, Carlsbad, CA). XK was detected using a polyclonal rabbit antibody raised against an internal peptide of XK (aa404–444); Kell was detected using a rabbit polyclonal antibody raised against human Kell isolated from red blood cell membranes.
3. Case reports
3.1. Patient 1
Patient 1 was a 65 year old male with disinhibited behavior and a tendency for jocularity. He had generalized chorea involving both upper and lower extremities proximally and distally, as well as the trunk and face. He had involuntary jaw opening movements without tongue protrusion, and slurred speech with partially compromised intelligibility. Repetitive movements were normal. Mild slowness was noted on the Luria test. There was mild rigidity in the upper extremities. Deep tendon reflexes were absent throughout. The behavioral changes, such as aggressiveness and impulsivity, had started 10 years earlier. Subsequently he developed involuntary limb and facial movements, impaired gait, falls, lack of coordination, speech and swallowing difficulties, cognitive dysfunction and dysesthesia in palms and soles. Electromyography (EMG) and nerve conduction study revealed a peripheral neuropathy of sensory axonal type in the lower extremities. Huntington’s disease (HD) genetic test was negative. MRI of the brain showed generalized atrophy and two foci of abnormal signal in left parietal periventricular white matter of nonspecific etiology. No atrophy of the caudate nucleus was noted. Electroencephalography (EEG) was normal. His red blood cell count was low at 3.72 × 106/μL (normal 4.7–6.1 × 106/μL). Hemoglobin and hematocrit were normal. Creatine phosphokinase (CPK) was 1914 U/L (normal ≤ 200 U/L). Fresh blood smear revealed 20–30% acanthocytosis. Liver function tests were abnormal, with AST levels reaching 107 IU/L (normal 10 to 34 IU/L). No cardiomyopathy was observed. Neuropsychological evaluation performed 1 year previously showed frontal subcortical dementia with significant disinhibition and executive function impairment. He had 3 paternal half siblings (2 males, 1 female), a full sister and two young adult sons, all unaffected.
3.2. Patient 2
Patient 2 was a 52 year old male with grossly normal mentation and alertness, normal short and long term memory, fluent language, intact calculation and abstract thinking. No depression or other pervading mood disturbances were reported. Chorea began in early childhood and he was diagnosed with McLeod syndrome at age 52. He had generalized tonic–clonic seizures that began 5 years previously. Speech pattern was slightly choreic with irregular volume control and intermittent pauses during long sentences. There was moderate generalized chorea affecting the trunk and extremities, which was accentuated by anxiety when asked to perform other tasks such as finger tapping and walking. Repetitive movements were normal. There was very mild rigidity in his arms with reinforcement. Light touch, vibratory, proprioceptive, pinprick, and temperature sensation were diminished distally in a stocking/glove distribution. Deep tendon reflexes were diminished in his arms and knees and absent at the ankles, and toes were downgoing. Posture and balance were intact. His gait showed a normal base, speed and stride, with a slight increase in chorea while walking, and difficulty performing tandem gait. He subsequently reported impaired concentration and attention, leading to inability to work. HD genetic test was negative. Ophthalmologic examination was negative for Kayser–Fleischer rings. Laboratory workup revealed the presence of acanthocytes on peripheral blood smear. CPK was persistently elevated >3000 U/L Nerve conduction studies revealed a sensorimotor axonal neuropathy. His EEG showed slowing in the right temporal lobe. Seizures were controlled on levetiracetam. His 54 year old brother had similar hyperkinetic movements and seizures without cardiomyopathy. His mother did not have chorea but had coronary artery disease and mitral valve prolapsed. The brother and mother were both found to have acanthocytes on peripheral blood smear, however detailed molecular genetic tests were not performed. The presence of acanthocytes in mother’s red blood cells may be explained as a result of possible skewed X-chromosome inactivation [24].
4. Results
On serological analyses both patients were found to be k−k+w, Kp (a−b+w), and Kx−, confirming the McLeod phenotype.
The clinical evaluation of both patients confirmed neuromuscular and central nervous system manifestations typical of McLeod syndrome.
PCR analyses and sequencing of DNA samples showed that patient 1 had an insertion of A at nt position 605 within the Ile codon at position 175 (605insA, 175Ile) in exon 3. This created a reading frame shift leading to a premature termination at a stop codon 23 codons downstream from the mutation in the same exon of XK (Fig. 1A). Patient 2 had an “A” to “G” mutation in intervening sequence 2 of XK within the 3′ RNA splice acceptor consensus sequence (IVS2–2a>g) (Fig. 1B). This strongly indicated aberrant mRNA splicing.
Fig. 1.
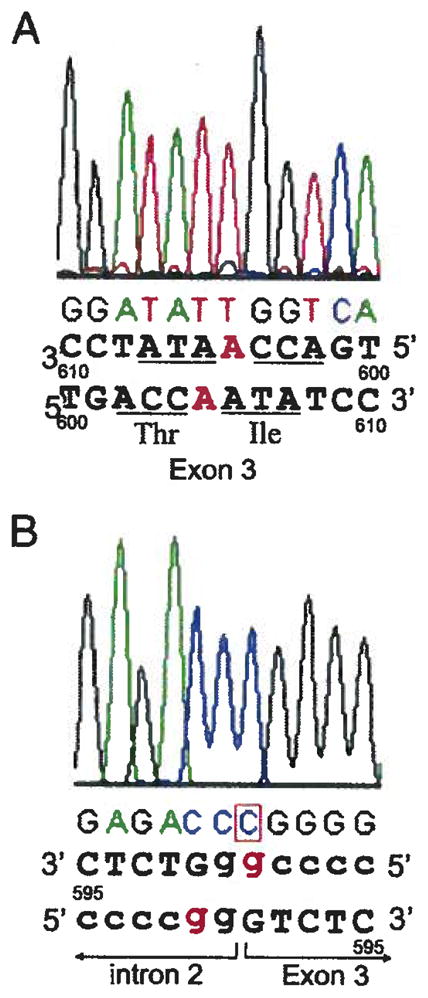
A: Reverse sequencing result of partial exon 3 off XK of patient 1 DNA showing a single nucleotide insertion at nt 605 (605insA) which changes the Ile at amino acid 175. The inserted nucleotide, “A” is Written in red. B: Reverse sequencing result of the boundary region of intron 2 and exon 3 of XK of patient 2 DNA showing the IVS2–A>G at the Splice acceptor consensus sequence, …ag, at 3′ of the intron 2. The mutated nucleotide, “G”, is written in red.
XK protein levels were evaluated by Western blot using anti-XK antibodies (Fig. 2A). No XK protein was detected in the red cell membranes of patient 1 in reducing conditions (lane 2 of right panels of both short and long exposure), as predicted based on PCR data. Similarly, patient 2’s red cell membranes also lacked XK protein, indicating the mutation did not permit normal splicing (lane 3 of right panels both short and long exposure). Since there was no XK present in red cell plasma membranes of patients 1 and 2, no Kell/XK complex was detected in non-reducing conditions (left panels, lane 2 for patient 1 and lane 3 for patient 2 of both short and long exposure). The control (lane 1) shows XK from normal red cell ghosts for comparison. The status of Kell glycoprotein was also analyzed by Western blot using anti-Kell antibodies. We found that under reducing conditions Kell protein levels were decreased as expected (Fig. 2B right panel of long exposure, lane 2 for patient 1 and lane 3 for patient 2) and there was no Kell/XK complex detectable in the non-reducing conditions (lane 2 for patient 1 and lane 3 for patient 2 in left panels of both short and long exposure) indicating that there was no XK present to form Kell/XK complex. The control (lane 1) shows XK from normal red cell ghosts for comparison.
Fig. 2.

A: Western blot of patients 1 and 2 red cell ghosts probed with anti-XK antibodies under non-reducing and reducing conditions with short and long exposure. Left panel (non-reducing conditions) showing absence of Kell/XK complex in samples of both patient 1 (lane 2) and patient 2 (lane 3). Normal red cell ghosts were analyzed similarly for comparison (lane 1 in both conditions). The over exposed panel also does not show presence of Kell/XK complex in either sample. Right panel (reducing conditions) showing absence of XK in patient 1 (lane 2) and patient 2 (lane 3). The same blot was exposed for longer time to confirm absence of XK. The protein of about 150 kDa in size marked as asterisk is present only in patient samples and is unknown. B: Western blot results of patients 1 and 2 red cell ghosts probed with anti-Kell antibodies under non-reducing and reducing conditions with short and long exposure. Lane 1 normal red cells: lane 2, patient 1 red cells. Lane 3, patient 3, red cells. Left panel (non-reducing conditions), showing absence of Kell/XK complex in lanes 2 and 3 indicating absence of XK. Right panel (reducing conditions) showing reduced amount of Kell in lanes 2 and 3 of long exposure.
5. Discussion
In patient 1, the single nucleotide insertion in exon 3 changed the Ile at 175 resulting in a reading frame shift and creating a stop codon resulting in premature termination and loss of XK protein, as confirmed by Western blot (Fig. 2A). In normal red cells, XK/Kell is seen as a complex in non-reducing conditions because XK and Kell are linked by a disulfide bond, which is preserved in this experimental condition [17]. We observed a reduced amount of Kell in the red cell membranes when XK was absent, as shown by the Western blot under reducing conditions (Fig. 2B long exposure). One other example of a single nucleotide insertion in XK resulting in the same observations has been previously reported [25].
The single nucleotide mutation in patient 2 changes A to G at the 3′ splice acceptor site of the IVS2. As predicted, this gt…/…gg, non-canonical splice sequence did not permit normal RNA splicing [26,27], also confirmed by Western blot (Fig. 2A, lanes 3). There have been 4 reported cases of MLS caused by mutations at splice sites and one case involving an intron 2 splice acceptor site that changed G to A [13,28,29]. The other three cases involved splice donor sites, one involving intron 1, and the other two involving intron 2 [1,30].
6. Conclusions
We report here two novel mutations in two cases of McLeod syndrome; 605insA at 175Ile in exon3 (patient 1) and IVS2–2a>g in XK (patient 2). In each case no normal XK, and hence no normal XK/Kell complex, was detected either by serological test or by Western blot, confirming McLeod phenotype. Both cases were clinically typical, with generalized chorea, peripheral neuropathy, elevated CPK, gait impairment and cognitive symptoms.
References
- 1.Russo DC, Lee S, Reid ME, Redman CM. Point mutations causing the McLeod phenotype. Transfusion. 2002;42:287–93. doi: 10.1046/j.1537-2995.2002.00049.x. [DOI] [PubMed] [Google Scholar]
- 2.Lee S, Sha Q, Wu X, Calenda G, Peng J. Expression profiles of mouse Kell, XK, and XPLAC mRNA. J Histochem Cytochem. 2007;55:365–74. doi: 10.1369/jhc.6A7126.2006. [DOI] [PubMed] [Google Scholar]
- 3.Lee S. The value of DNA analysis for antigens of the Kell and Kx blood group systems. Transfusion. 2007;47:32S–9S. doi: 10.1111/j.1537-2995.2007.01308.x. [DOI] [PubMed] [Google Scholar]
- 4.Walker RH, Danek A, Uttner I, Offner R, Reid M, Lee S. McLeod phenotype without the McLeod syndrome. Transfusion. 2007;47:299–305. doi: 10.1111/j.1537-2995.2007.01106.x. [DOI] [PubMed] [Google Scholar]
- 5.Arnaud L, Salachas F, Lucien N, Maisonobe T, Le Pennec PY, Babinet J, et al. Identification and characterization of a novel XK splice site mutation in a patient with McLeod syndrome. Transfusion. 2009;49:479–84. doi: 10.1111/j.1537-2995.2008.02003.x. [DOI] [PubMed] [Google Scholar]
- 6.Walker RH, Jung HH, Dobson-Stone C, Rampoldi L, Sano A, Tison F, et al. Neurologic phenotypes associated with acanthocytosis. Neurology. 2007;68:92–8. doi: 10.1212/01.wnl.0000250356.78092.cc. [DOI] [PubMed] [Google Scholar]
- 7.Klempir J, Roth J, Zarubova K, Pisacka M, Spackova N, Tilley L. The McLeod syndrome without acanthocytes. Parkinsonism Relat Disord. 2008;14:364–6. doi: 10.1016/j.parkreldis.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 8.Walker RH, Jung HH, Tison F, Lee S, Danek A. Phenotypic variation among brothers with the McLeod neuroacanthocytosis syndrome. Mov Disord. 2007;22:244–8. doi: 10.1002/mds.21224. [DOI] [PubMed] [Google Scholar]
- 9.Valko PO, Hanggi J, Meyer M, Jung HH. Evolution of striatal degeneration in McLeod syndrome. Eur J Neurol. 2010;17:612–8. doi: 10.1111/j.1468-1331.2009.02872.x. [DOI] [PubMed] [Google Scholar]
- 10.Walker RH, Danek A, Dobson-Stone C, Guerrini R, Jung HH, Lafontaine AL, et al. Developments in neuroacanthocytosis: expanding the spectrum of choreatic syndromes. Mov Disord. 2006;21:1794–805. doi: 10.1002/mds.21108. [DOI] [PubMed] [Google Scholar]
- 11.Hewer E, Danek A, Schoser BG, Miranda M, Reichard R, Castiglioni C, et al. McLeod myopathy revisited: more neurogenic and less benign. Brain. 2007;130:3285–96. doi: 10.1093/brain/awm269. [DOI] [PubMed] [Google Scholar]
- 12.Ho MF, Monaco AP, Blonden LA, van Ommen GJ, Affara NA, Ferguson-Smith MA, et al. Fine mapping of the McLeod locus (XK) to a 150–380-kb region in Xp21. Am J Hum Genet. 1992;50:317–30. [PMC free article] [PubMed] [Google Scholar]
- 13.Ho M, Chelly J, Carter N, Danek A, Crocker P, Monaco AP. Isolation of the gene for McLeod syndrome that encodes a novel membrane transport protein. Cell. 1994;77:869–80. doi: 10.1016/0092-8674(94)90136-8. [DOI] [PubMed] [Google Scholar]
- 14.Claperon A, Hattab C, Armand V, Trottier S, Bertrand O, Ouimet T. The Kell and XK proteins of the Kell blood group are not co-expressed in the central nervous system. Brain Res. 2007;1147:12–24. doi: 10.1016/j.brainres.2007.01.106. [DOI] [PubMed] [Google Scholar]
- 15.Zhu X, Rivera A, Golub MS, Peng J, Sha Q, Wu X, et al. Changes in red cell ion transport, reduced intratumoral neovascularization, and some mild motor function abnormalities accompany targeted disruption of the Mouse Kell gene (Kel) Am J Hematol. 2009;84:492–8. doi: 10.1002/ajh.21453. [DOI] [PubMed] [Google Scholar]
- 16.Khamlichi S, Bailly P, Blanchard D, Goossens D, Cartron JP, Bertrand O. Purification and partial characterization of the erythrocyte Kx protein deficient in McLeod patients. Eur J Biochem. 1995;228:931–4. [PubMed] [Google Scholar]
- 17.Lee S, Russo D, Redman CM. The Kell blood group system: Kell and XK membrane proteins. Semin Hematol. 2000;37:113–21. doi: 10.1016/s0037-1963(00)90036-2. [DOI] [PubMed] [Google Scholar]
- 18.Russo D, Redman C, Lee S. Association of XK and Kell blood group proteins. J Biol Chem. 1998;273:13, 950–6. doi: 10.1074/jbc.273.22.13950. [DOI] [PubMed] [Google Scholar]
- 19.Lee S, Lin M, Mele A, Cao Y, Farmar J, Russo D, et al. Proteolytic processing of big endothelin-3 by the kell blood group protein. Blood. 1999;94:1440–50. [PubMed] [Google Scholar]
- 20.Sha Q, Redman CM, Lee S. Endothelin-3-converting enzyme activity of the KEL1 and KEL6 phenotypes of the Kell blood group system. J Biol Chem. 2006;281:7180–2. doi: 10.1074/jbc.M507776200. [DOI] [PubMed] [Google Scholar]
- 21.John SW, Weitzner G, Rozen R, Scriver CR. A rapid procedure for extracting genomic DNA from leukocytes. Nucleic Acids Res. 1991;19:408. doi: 10.1093/nar/19.2.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee S, Wu X, Reid M, Redman C. Molecular basis of the K:6,−7 [Is(a+b−)] phenotype in the Kell blood group system. Transfusion. 1995;35:822–5. doi: 10.1046/j.1537-2995.1995.351096026362.x. [DOI] [PubMed] [Google Scholar]
- 23.Schwoch G, Passow H. Preparation and properties of human erythrocyte ghosts. Mol Cell Biochem. 1973;2:197–218. doi: 10.1007/BF01795474. [DOI] [PubMed] [Google Scholar]
- 24.Jung HH, Hergersberg M, Kneifel S, Alkadhi H, Schiess R, Weigell-Weber M, et al. McLeod syndrome: a novel mutation, predominant psychiatric manifestations, and distinct striatal imaging findings. Ann Neurol. 2001;49:384–92. [PubMed] [Google Scholar]
- 25.Starling A, Schlesinger D, Kok F, Passos-Bueno MR, Vainzof M, Zatz M. A family with McLeod syndrome and calpainopathy with clinically overlapping diseases. Neurology. 2005;65:1832–3. doi: 10.1212/01.wnl.0000187073.58307.41. [DOI] [PubMed] [Google Scholar]
- 26.Burset M, Seledtsov IA, Solovyev VV. Analysis of canonical and non-canonical splice Sites in mammalian genomes. Nucleic Acids Res. 2000;28:4364–75. doi: 10.1093/nar/28.21.4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jackson IJ. A reappraisal of non-consensus mRNA splice sites. Nucleic Acids Res. 1991;19:3795–8. doi: 10.1093/nar/19.14.3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Danek A, Rubio JP, Rampoldi L, Ho M, Dobson-Stone C, Tison F, et al. McLeod neuroacanthocytosis: genotype and phenotype. Ann Neurol. 2001;50:755–64. doi: 10.1002/ana.10035. [DOI] [PubMed] [Google Scholar]
- 29.Swash M, Schwartz MS, Carter ND, Heath R, Leak M, Rogers KL. Benign X-linked myopathy with acanthocytes (McLeod syndrome). Its relationship to X-linked muscular dystrophy. Brain. 1983;106(Pt 3):717–33. doi: 10.1093/brain/106.3.717. [DOI] [PubMed] [Google Scholar]
- 30.Daniels GL, Weinauer F, Stone C, Ho M, Green CA, Jahn-Jochem H, et al. A combination of the effects of rare genotypes at the XK and KEL blood group loci results in absence of Kell system antigens from the red blood cells. Blood. 1996;88:4045–50. [PubMed] [Google Scholar]