

Abstract
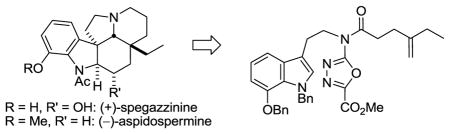
Divergent total syntheses of (+)-spegazzinine (1) and (−)-aspidospermine (2) and their extensions to the synthesis of C19-epi-aspidospermine and C3-epi-spegazzinine are detailed, confirming the relative stereochemistry and establishing the absolute configuration of (+)-spegazzinine. A powerful intramolecular [4 + 2]/[3 + 2] cycloaddition cascade of a 1,3,4-oxadiazole provided the pentacyclic skeleton and all the requisite stereochemistry of the natural products in a single reaction that forms three rings, four C–C bonds, and five stereocenters.
The Aspidosperma alkaloids continue to generate considerable interest due to their structural diversity, stereochemical complexity, central biosynthetic role in plants, and interesting biological properties. One member, spegazzinine (1, Figure 1), was first isolated in 1956 from Aspidosperma chakensis Spegazzini by Djerassi.1 It has since been shown to be a potent inhibitor of mitochondrial photophosphorylation in spinach and oxidative phosphorylation in rat liver mitochondria.2 Aspidospermine (2), first isolated from the bark of Aspidosperma quebrancho3 in the late 1800’s and from many other plant sources since,4 exhibits a wide range of biological activities, including diuretic, vasoconstriction, hypertensive,5 and respiratory stimulant properties.6,7 While spegazzinine has not yet been prepared by total synthesis, aspidospermine continues to be an attractive synthetic target8 as a result of its prominent position among the Aspidosperma alkaloids. However, all reported syntheses of aspidospermine proceed through 3, relying on a late-stage Fischer indole synthesis using o-methoxyphenylhydrazine originally disclosed by Stork and Dolfini nearly 50 years ago.8a
Figure 1.

Natural products and intermediate ketone 3
The stereochemistry of the C3 alcohol in 1 was assigned9 based on an 8 Hz coupling constant of C2-H with its neighboring C3-H in the 1H NMR of the closely related natural product spegazzinidine, which differs from 1 only by incorporation of an additional C16 hydroxyl group. The large coupling constant was interpreted to be indicative of a trans diaxial relationship between the two hydrogens, although this assignment and that of spegazzinidine have not been confirmed.
In recent efforts targeting additional key members of the Aspidosperma alkaloids including minovine,10 fendleridine,11 vindorosine and vindoline,12 and their extension to the total synthesis of vinblastine13 and related natural products including vincristine,14 and key analogues,15 we developed a powerful intramolecular [4 + 2]/[3 + 2] cycloaddition cascade of 1,3,4-oxadiazoles that provides the pentacyclic core and all the stereochemistry of the natural products in a single step (Figure 2).16 Herein, we report the extension of these studies to the total synthesis of 1 and 2. This strategy was viewed as especially suited for spegazzinine, whose C3 alcohol can be directly introduced upon reduction of a stable cyanohydrin formed after reductive oxido bridge cleavage of a nitrile derived from such a cycloaddition product. An added bonus of the approach is that late stage modification of a route needed to explore the spegazzinine C3 alcohol stereochemistry also permits a divergent17 synthesis of aspidospermine, entailing removal of the C3 alcohol in an approach fundamentally different from all prior reports.
Figure 2.

Cycloaddition cascade
The key cycloaddition is initiated by an intramolecular Diels–Alder reaction of a 1,3,4-oxadiazole with a tethered dienophile.18,19 Loss of N2 from the initial cycloadduct generates a 1,3-dipole, which is stabilized by the complementary substitution at the dipole termini. The regioselectivity of the subsequent 1,3-dipolar cycloaddition is dictated by the tether, but is reinforced by the intrinsic polarity of the reacting partners and provides an endo indole [3 + 2] cycloaddition, where the dipolarophile is sterically directed to the face opposite the newly formed six-membered ring.16,20 Four C–C bonds, three rings, five stereocenters, and the complete natural product skeleton are assembled in a single transformation.
The precursor tryptamine 6 to the key cycloaddition substrate 4 was prepared from 7-benzyloxyindole and modeled on a route first disclosed by Corey (Supporting Information Scheme 1).21 Tryptamine 6 was treated with 1,1-carbonyldiimidazole (CDI) to afford urea 7 (Scheme 1). Addition of methyl oxalyl hydrazide (8) to 7 (HOAc–THF, 40 °C, 16 h) furnished 9, which was converted to the 1,3,4-oxadiazole 10 (70%, 2 steps) upon treatment with TsCl and Et3N. Coupling of 10 and 4-ethyl-4-pentenoic acid (11) provided 4 (87%).
Scheme 1.

The key intramolecular [4 + 2]/[3 + 2] cycloaddition was accomplished by warming a solution of 4 at 180 °C in o-dichlorobenzene (o-DCB) to provide 5 as a single diastereomer in yields as high as 71% (Scheme 2). The stereochemistry of 5 was confirmed upon X-ray analysis22 of 15 and was in accordance with expectations. Treatment of 5 with NH3–MeOH cleanly provided the primary amide, and its dehydration upon treatment with trifluoroacetic anhydride (TFAA) and pyridine afforded nitrile 12 (85%).
Scheme 2.

Reductive oxido bridge opening of 12 was accomplished with NaCNBH3 in HCl–MeOH to provide the stable cyanohydrin 13 as a single diastereomer, resulting from convex face hydride reduction of the intermediate N-acyliminium ion. A number of reducing reagents were screened for the further reduction of 13, and the most direct for the synthesis of 1 proved to be NaBH4, which provided a 3.5:1 mixture of 15:14 in 66% (88% brsm). Larger or alternative reducing reagents failed to improve upon the ratio, favoring alcohol 15.22 However, increased yields (90%) of 15 were achieved through the use of N-selectride, but required inversion of the C3 alcohol stereochemistry to access 1. Mitsunobu inversion of the C3 alcohol in 15 could be accomplished to provide the chloroacetate (3 equiv chloroacetic acid,23 4 equiv Ph3P, 4 equiv DIAD, toluene, 1 h, 56%), with competitive alcohol elimination accounting for the remainder of the reaction product. Hydrolysis of the chloroacetate with K2CO3 in aqueous MeOH provided 14, bearing the requisite C3 alcohol stereochemistry for spegazzinine.
The amide carbonyl of 14 was reductively removed with LiAlH4 (63%) to provide the tertiary amine, which was globally deprotected upon treatment with H2–Pd/C to afford phenol 16 (Scheme 3). Due to the polarity of 16, the crude material was carried forward into the N-acetylation reaction, which after treatment with K2CO3 in MeOH completed the first total synthesis of spegazzinine (1). Resolution of racemic 1 by chiral phase chromatography provided both natural (+)-1 and ent-(−)-1, the former of which proved identical in all respects to an authentic sample of (+)-spegazzinine generously provided by GSK, establishing its identity and confirming the relative stereochemistry of the natural product.
Scheme 3.

With ample amounts of 15 in hand, we also prepared C3-epi-spegazzinine (18) featuring the C3 alcohol diastereomer (Scheme 3). Reduction of 15 with LiAlH4 provided the amine, which was globally deprotected with H2–Pd/C and subsequently acetylated to provide C3-epi-spegazzinine (18). The 1H NMR of 18 differed significantly from that of the natural product, further confirming the stereochemical assignment for synthetic 1. Most notably, the C2-H coupling constant in 18 is 5.5 Hz, significantly smaller than the 7.8 Hz coupling constant observed for synthetic 1 and consistent with the cis relationship of the C2/C3 hydrogens.
In order to determine the absolute configuration of (+)-spegazzinine, racemic 5 was resolved by chiral phase chromatography to provide (+)-5 and (−)-5 (Supporting Information Scheme 2). Each enantiomer was carried forward independently to the thioamide of 13, where crystallization and X-ray analysis of the natural enantiomer24 determined their absolute configuration. Elaboration of this intermediate to 1 defined the absolute configuration of (+)-spegazzinine to be as shown in Figure 1.
The attractive feature of the approach was its potential late stage modification to provide aspidospermine, simply requiring removal of the C3 alcohol. Capitalizing on the observation that olefin 19 formed competitive with Mitsunobu alcohol displacement, the divergent synthesis of 2 proved straightforward (Scheme 4). Mitsunobu activation of 15 was optimized (3 equiv 4-nitrobenzoic acid, 4 equiv Ph3P, 4 equiv DIAD, toluene) to afford exclusively the elimination product 19 (95% by LCMS). LiAlH4 reduction of the amide carbonyl followed by reduction of the double bond and benzyl deprotection effected by H2– Pd/C provided phenol 20. N-Acetylation provided 21, which upon treatment with TMSCH2N2 afforded racemic aspidospermine. Resolution of 2 by chiral phase chromatography provided both natural (−)- and ent-(+)-aspidospermine (2), completing its total synthesis. Remarkably, and to the best of our knowledge, this constitutes the first total synthesis of aspidospermine that does not rely on the Stork late-stage Fischer indole synthesis to provide the natural product.
Scheme 4.

In the course of our studies, we also observed an unprecedented stereochemical outcome for the reduction of an oxido bridge in the cascade cycloadducts. S-Methylation of thioamide 22, prepared by treatment of 12 with Lawesson’s reagent, with Meerwein’s reagent followed by reduction of the S-methyl thioimidate with NaBH4 provided alcohol 23 as the major product (Scheme 5). Not only was the C3 ketone released and reduced under these conditions, but hydride delivery to the intermediate iminium ion occurred from the bottom (concave) face, providing predominantly the thermodynamically less stable and unnatural stereochemistry at C19.25 This is in contrast to the treatment of 12 with NaCNBH3 under the acidic conditions described earlier, which affords exclusively 13 containing the natural C19 diastereomer present in the Aspidosperma alkaloids. Since the thioamide 22, like 12, also undergoes top (convex) face reduction with NaCNBH3 (96%) under acidic conditions where the cyanohydrin is stable (Figure 3), its atypical behavior under basic conditions suggest that the cyanohydrin collapses to the ketone prior to iminium ion reduction, and hydride delivery now occurs predominantly from the increasingly more accessible bottom face to both the iminium ion and the ketone to produce 23.
Scheme 5.

Figure 3.

Proposed route for the formation of 23
Alcohol 23 was converted to the methyl dithiocarbonate, which was deoxygenated under Barton–McCombie conditions to afford 25 (Scheme 5). The benzyl groups were removed by treatment with Na and t-BuOH in THF–NH3,12 cleanly affording phenol 26. N-Acetylation, followed by phenol methylation provided C19-epi-aspidospermine (28) whose unnatural C19 stereochemistry and unusual trans-fused 6,5-ring system were confirmed by X-ray analysis.25
Herein, the divergent total syntheses of (−)-aspidospermine (2) and (+)-spegazzinine (1), and their extension to the synthesis of C19-epi-aspidospermine and C3-epi-spegazzinine are reported, constituting the first total synthesis of (+)-spegazzinine, and the first reported total synthesis of (−)-aspidospermine that does not rely on a late-stage Fischer indole synthesis. The pentacyclic skeleton and all the requisite stereochemistry of the natural products were assembled by a tandem intramolecular [4 + 2]/[3 + 2] cycloaddition cascade of a 1,3,4-oxadiazole that forms three rings, four C–C bonds, and five stereocenters including three contiguous quaternary centers in a single operation. Further exploration of the cascade cycloaddition reactions of 1,3,4-oxadiazoles and their applications are in progress and will be reported in due course.
Supplementary Material
Acknowledgments
We gratefully acknowledge the financial support of the National Institutes of Health (CA042056) and the Skaggs Institute for Chemical Biology. We thank Dr. Kogure (TSRI) and Erin Anderson (TSRI) for assistance, Dr. Raj Chadha (TSRI) for the X-ray crystal structures, and GlaxoSmithKline (Dr. Don Hertzog) for an authentic sample of 1. J.P.L. is a Skaggs Fellow and recipient of a BMS graduate fellowship.
Footnotes
Supporting Information Available. Full experimental details are provided. This material is available via the Internet free of charge at http://pubs.acs.org.
References
- 1.Orazi OO, Corral RA, Holker JSE, Djerassi C. J Org Chem. 1956;21:979. [Google Scholar]
- 2.(a) Roveri OA, Vallejos RH. Biochim Biophys Acta. 1974;333:187. doi: 10.1016/0005-2728(74)90003-6. [DOI] [PubMed] [Google Scholar]; (b) Andreo CS. Arch Biochem Biophys. 1978;186:416. doi: 10.1016/0003-9861(78)90454-x. [DOI] [PubMed] [Google Scholar]
- 3.Fraude G. Ber Deut Chem Ges. 1878;11:2189. [Google Scholar]
- 4.Saxton JE. In: The Alkaloids. Cordell AG, editor. Vol. 51. Academic Press; San Diego: pp. 2–197. [Google Scholar]
- 5.Floriani F. An Farm Bioquim. 1931;2:215. [Google Scholar]
- 6.Banerjee JN, Lewis JJ. J Pharm Pharmacol. 1955;7:46. doi: 10.1111/j.2042-7158.1955.tb12004.x. [DOI] [PubMed] [Google Scholar]
- 7.Witkop B. J Am Chem Soc. 1948;70:3712. doi: 10.1021/ja01191a048. [DOI] [PubMed] [Google Scholar]
- 8.(a) Stork G, Dolfini JE. J Am Chem Soc. 1963;85:2872. [Google Scholar]; (b) Ban Y, Sato Y, Inoue I, Nagai M, Oishi T, Terashima M, Yonemitsu O, Kanaoka Y. Tetrahedron Lett. 1965;6:2261. doi: 10.1016/s0040-4039(00)70368-6. [DOI] [PubMed] [Google Scholar]; (c) Kuehne ME, Bayha C. Tetrahedron Lett. 1966;7:1311. [Google Scholar]; (d) Ban Y, Iijima I, Inoue I, Akagi M, Oishi T. Tetrahedron Lett. 1969;10:2067. doi: 10.1016/s0040-4039(01)88087-4. [DOI] [PubMed] [Google Scholar]; (e) Ban Y, Iijima I. Tetrahedron Lett. 1969;10:2523. doi: 10.1016/s0040-4039(01)88557-9. [DOI] [PubMed] [Google Scholar]; (f) Stevens RV, Fitzpatrick JM, Kaplan M, Zimmerman RL. J Chem Soc, Chem Commun. 1971:857. [Google Scholar]; (g) Martin SF, Desai SR, Phillips GW, Miller AC. J Am Chem Soc. 1980;102:3294. [Google Scholar]; (h) Pearson AJ. Tetrahedron Lett. 1981;22:4033. [Google Scholar]; (i) Pearson AJ, Rees DC. J Chem Soc Perkin Trans I. 1982:2467. [Google Scholar]; (j) Wu PL, Chu M, Fowler FW. J Org Chem. 1988;53:963. [Google Scholar]; (k) Meyers AI, Berney D. J Org Chem. 1989;54:4673. [Google Scholar]; (l) Iyengar R, Schildknegt K, Aubé J. Org Lett. 2000;2:1625. doi: 10.1021/ol005913c. [DOI] [PubMed] [Google Scholar]; (m) Fukuda Y, Shindo M, Shishido K. Org Lett. 2003;5:749. doi: 10.1021/ol034020s. [DOI] [PubMed] [Google Scholar]; (n) Iyengar R, Schildknegt K, Morton M, Aube J. J Org Chem. 2005;70:10645. doi: 10.1021/jo051212n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Pearson WH, Aponick A. Org Lett. 2006;8:1661. doi: 10.1021/ol0602506. [DOI] [PMC free article] [PubMed] [Google Scholar]; (p) Coldham I, Burrell AJM, White LE, Adams H, Oram N. Angew Chem Int Ed. 2007;46:6159. doi: 10.1002/anie.200701943. [DOI] [PubMed] [Google Scholar]; (q) Burrell AJM, Coldham I, Watson L, Oram N, Pilgram CD, Martin NG. J Org Chem. 2009;74:2290. doi: 10.1021/jo8019913. [DOI] [PubMed] [Google Scholar]
- 9.Djerassi C, Brewer HW, Budzikiewicz H, Orazi OO, Corral RA. J Am Chem Soc. 1962;84:3480. doi: 10.1007/BF02153843. [DOI] [PubMed] [Google Scholar]
- 10.Yuan ZQ, Ishikawa H, Boger DL. Org Lett. 2005;7:741. doi: 10.1021/ol050017s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Campbell EL, Zuhl AM, Liu CM, Boger DL. J Am Chem Soc. 2010;132:3009. doi: 10.1021/ja908819q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Wolkenberg SE, Boger DL. J Org Chem. 2002;67:7361. doi: 10.1021/jo020437k. [DOI] [PubMed] [Google Scholar]; (b) Choi Y, Ishikawa H, Velcicky J, Elliott GI, Miller MM, Boger DL. Org Lett. 2005;7:4539. doi: 10.1021/ol051975x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Elliott GI, Velcicky J, Ishikawa H, Li Y, Boger DL. Angew Chem Int Ed. 2006;45:620. doi: 10.1002/anie.200503024. [DOI] [PubMed] [Google Scholar]; (d) Ishikawa H, Elliott GI, Velcicky J, Choi Y, Boger DL. J Am Chem Soc. 2006;128:10596. doi: 10.1021/ja061256t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ishikawa H, Boger DL. Hetereocycles. 2007;72:95. [Google Scholar]; (f) Kato D, Sasaki Y, Boger DL. J Am Chem Soc. 2010;132:3685. doi: 10.1021/ja910695e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Sasaki Y, Kato D, Boger DL. J Am Chem Soc. 2010;132:13533. doi: 10.1021/ja106284s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ishikawa H, Colby DA, Boger DL. J Am Chem Soc. 2008;130:420. doi: 10.1021/ja078192m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishikawa H, Colby DA, Seto S, Va P, Tam A, Kakei H, Rayl TJ, Hwang I, Boger DL. J Am Chem Soc. 2009;131:4904. doi: 10.1021/ja809842b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Va P, Campbell EL, Robertson WM, Boger DL. J Am Chem Soc. 2010;132:8489. doi: 10.1021/ja1027748. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tam A, Gotoh H, Robertson WM, Boger DL. Biorg Med Chem Lett. 2010;20:6408. doi: 10.1016/j.bmcl.2010.09.091. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gotoh H, Duncan KK, Robertson WM, Boger DL. ACS Med Chem Lett. 2011;2:948. doi: 10.1021/ml200236a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Wilkie GD, Elliott GI, Blagg BSJ, Wolkenberg SE, Soenen DR, Miller MM, Pollack S, Boger DL. J Am Chem Soc. 2002;124:11292. doi: 10.1021/ja027533n. [DOI] [PubMed] [Google Scholar]; (b) Elliott GI, Fuchs JR, Blagg BSJ, Ishikawa H, Tao H, Yuan ZQ, Boger DL. J Am Chem Soc. 2006;128:10589. doi: 10.1021/ja0612549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boger DL, Brotherton CE. J Org Chem. 1984;49:4050. [Google Scholar]
- 18.(a) Boger DL. Tetrahedron. 1983;39:2869. [Google Scholar]; (b) Boger DL. Chem Rev. 1986;86:781. [Google Scholar]
- 19.Margetic D, Troselj P, Johnston MR. Mini-Rev Org Chem. 2011;8:49. [Google Scholar]
- 20.(a) Padwa A, Price AT. J Org Chem. 1995;60:6258. [Google Scholar]; (b) Padwa A, Price AT. J Org Chem. 1998;63:556. doi: 10.1021/jo971424n. [DOI] [PubMed] [Google Scholar]
- 21.He F, Bo Y, Altom JD, Corey EJ. J Am Chem Soc. 1999;121:6771. [Google Scholar]
- 22.The X-ray structure of 15 has been deposited with the Cambridge Crystallographic Data Centre (CCDC850758).
- 23.(a) Saiah M, Bessodes M, Antonakis K. Tetrahedron Lett. 1992;33:4317. [Google Scholar]; (b) Hughes DL, Reamer RA. J Org Chem. 1996;61:2967. doi: 10.1021/jo952180e. [DOI] [PubMed] [Google Scholar]
- 24.The X-ray structure of thioamide (−)-33 has been deposited with the Cambridge Crystallographic Data Centre (CCDC868362).
- 25.Walser A, Djerassi C. Helv Chim Acta. 1965;48:391. [Google Scholar]
- 26.The X-ray structure of 28 has been deposited with the Cambridge Crystallographic Data Centre (CCDC850757).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.